

Abstract
Rigosertib (ON 01910.Na), a synthetic novel benzyl styryl sulfone, was administered to 28 patients with advanced cancer in a Phase I trial in order to characterize its pharmacokinetic profile, determine the dose-limiting toxicities (DLT), define the recommended phase II dose (RPTD) and to document any antitumor activity. Patients with advanced malignant neoplasms refractory to standard therapy were given escalating doses of rigosertib (50, 100, 150, 250, 325, 400, 650, 850, 1,050, 1,375, 1,700 mg/m2/24h) as a 3-day continuous infusion (CI) every 2 weeks. An accelerated Fibonacci titration schedule with specified decreases for toxicities was used for escalation until grade ≥2 toxicity occurred. Intrapatient dose escalation was allowed if toxicity was grade ≤2 and the disease remained stable. Plasma pharmacokinetics (PK) and urinary PK assessments were studied in the 1st and 4th cycles. Twenty-nine patients (12 men and 17 women; age 36-87 y with a median of 63 y) were registered, but one died before study drug was given. Twenty-eight patients received a median of 3 cycles of therapy. Most common grade ≥2 toxicities attributable to rigosertib included fatigue, anorexia, vomiting and constipation. DLTs included muscular weakness, hyponatremia, neutropenia, delirium and confusional state. Risk factors for severe toxicities include pre-existing neurological dysfunction or advanced gynecologic cancer after pelvic surgery. Rigosertib pharmacokinetics showed rapid plasma distribution phases and urinary excretion. Elevations in plasma Cmax and AUC due to decreases in plasma clearance were associated with acute grade ≥3 toxicities. Of 22 evaluable patients, 9 (41%) achieved a best overall response of stable disease; all other patients (n=13; 59%) progressed. The median progression-free survival time was 50 days (95% confidence interval [CI]: 37-80 days). Nine (41%) patients survived for over 1 y. In summary, prolonged IV infusions of rigosertib were generally well tolerated. Nine (41%) patients achieved stable disease and 9 (41%) patients survived for over 1 year. The RPTD appears to be 850 mg/m2/24hr CI x 3 days. (ClinicalTrials.gov identifier: NCT01538537).
Keywords: Rigosertib, ON01910.Na, phase 1 study, polo-like kinase, phosphatidylinositol-3-kinase
Introduction
Rigosertib (ON 01910.Na) is a novel synthetic benzyl styryl sulfone (Figure 1A) that is cytotoxic against a variety of human tumor cell lines in vitro and inhibits growth of tumor xenografts in nude mice [1-3]. While the compound was originally considered to be a novel Polo-like kinase 1 (Plk1) inhibitor [1], a direct effect on Plk1 could not be confirmed in subsequent studies [4]. More recent studies indicate that rigosertib appears to inhibit the interaction of Plk 1 with the Raf family of kinases, thus affecting its activation (Divakar & Reddy, unpublished data). In addition, rigosertib inhibits the phosphatidylinositol-3 kinase (PI3K) pathway [5]. Available data show that the drug produces three major abnormalities in tumor cells: (a) abnormal cell division including irregular chromosomal segregation and cytokinesis; (b) G2/M arrest and apoptosis in many tumor cells (in contrast, normal fibroblasts were arrested in G1), and (c) decreased expression of Cdc25C and cyclin D1 [1,5].
Figure 1.

A. Chemical structure and IUPAC name of the compounds used in the study. Rigosertib, ON 01910.Na, Sodium (E)-2-(2-methoxy-5-((2,4,6-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate. B. ON 01500, a potential metabolite. C. Chemical structures of Temazepam and D. ON 01370 used as internal standards for drug assay.
Our in vitro cell culture studies showed that the cell kill effect of rigosertib was exposure time-dependent, rather than drug concentration-dependent [6]. Because of this finding, we examined the clinical effects of the drug administered as a prolonged intravenous (IV) infusion. Rigosertib was administered as a 3-day continuous infusion (CI) cycle. The cycle was repeated every 2 weeks. We found the drug to be well tolerated in most patients, whereas in others, the drug was profoundly toxic.
Patients and methods
This study was conducted on inpatients in the Clinical Research Center (CRC) at the Mount Sinai Hospital, New York. Trained research nurses implemented all aspects of the protocol. The study was approved by the Institutional Review Board of Icahn School of Medicine at Mount Sinai.
Patient eligibility
Selection criteria included (a) histologically confirmed malignancy that was metastatic or unresectable and for which standard curative or palliative measures did not exist or were no longer effective; (b) at least 4 weeks since the last dose of potentially myelosuppressive treatment (at least 6 weeks for nitrosoureas or mitomycin C). Subjects had to have recovered from reversible drug toxicities (except alopecia, stable residual neuropathy, and residual hand-foot syndrome). Patients with prior doxorubicin chemotherapy were excluded if more than 450 mg/m2 had been given; (c) prior radiotherapy patients were eligible after 4 weeks had elapsed and the maximal area of hematopoietically active bone marrow treated was less than 25%; (d) age >18 y; (e) ECOG performance status <2; (f) patients must have had nearly normal organ and bone marrow function as defined by: Hgb >10 gm/dL, WBC >4,000/μL, absolute neutrophil count >1,500/μL, platelets >100,000/μL, total bilirubin and creatinine within normal institutional limits, AST(SGOT)/ALT(SGPT) <2.5 x institutional upper limit of normal (ULN) (for hepatoma patients, bilirubin ≤3 mg/dL, AST(SGOT)/ALT(SGPT) <5 × institutional ULN). Patients with ascites were eligible, if manageable with diuretic agents alone. Patients with prior esophageal bleeding were eligible after 6 months if varices had been sclerosed or banded; (g) sexually active patients had to agree to use adequate contraception throughout the study; (h) ability to understand and willingness to sign a written informed consent document.
Exclusion Criteria included major surgery within the past 14 days; use of any other investigational agent or chemotherapy, radiotherapy, hormonal treatment, or immunotherapy while on the study; clinical evidence of central nervous system metastasis (except for asymptomatic brain metastases that had been resected or irradiated and clinically stable); history of allergic reactions to compounds of similar chemical or biologic composition as rigosertib; major third space fluid accumulation (serum sodium <134 mEq/L); severe liver dysfunction (Child-Pugh Class C or uncompensated Class B with encephalopathy and prothrombin time >1.5 × ULN); uncontrolled intercurrent illness including HIV infection; psychiatric illness/social situations that would limit compliance with study requirements; and pregnancy or lactation.
Clinical trial material
The clinical preparation was a sterile, stable, pure, water-soluble solid that is formulated into a final drug product, “ON 01910.Na Concentrate,” a solution of 75 mg/mL in a polyethylene glycol (PEG 400) vehicle, packaged in a sealed glass vial that is diluted prior to IV administration. The concentrate was available in two formulations: 100% PEG 400 was used for the first 21 patients, and 50% PEG 400 in phosphate buffer for patients #22–29.
Treatment plan, definition of dose-limiting toxicities, MTD and dose-escalation plan
In 28-day repeated dosing studies in rats, the no-observed-adverse-effect-level (NOAEL) of regosertib was 30 mg/kg/day or 180 mg/m2/day. In dog studies, the initial distribution phases occurred with half-lives ranging from 17 to 40 min, while the terminal elimination phase ranged from 2.7 to 5.8 h. The Cmax and AUCinf values increased with increasing doses (Onconova Investigator’s Brochure 16th edition July 11, 2012). The starting dose of 50 mg/m2/24h x 3 days was chosen based on animal toxicological studies, less than one third of the NOAEL in rats for daily administration. Intra-patient dose escalation was allowed, provided that previously untreated patients had already been entered at the new dose level and that a patient at his or her lower dose level had only grade (Gr) 0-2 toxicity and had no disease progression. In the absence of toxicity, rigosertib doses were escalated every 2 weeks. Dose escalation followed a Fibonacci scheme with an initial accelerated dose-escalation phase in which 1-patient cohorts received rigosertib every 2 weeks until drug-related Gr 2 toxicity (according to the National Cancer Institute Common Toxicity Criteria for Adverse Events (CTCAE) v. 3.) [7], excluding alopecia, occurred, at which time 2 additional patients were added to subsequent cohorts. If none of the 3 patients in the cohort experienced Dose-limiting Toxicities (DLTs), the dose was escalated by a half-Fibonacci step. DLT was defined as any drug-related Gr 4 hematologic toxicity (Gr 4 neutropenia or thrombocytopenia lasting ≥5 days), other Gr ≥3 toxicities or Gr 2 hemorrhage. If a DLT was seen in the first patient of a cohort, dosing went back a half-step. The next dose level occurred if no DLT was reported in the 3 patients or if no more than 1 DLT occurred in an expanded cohort of 6 patients. If a DLT was seen in 1 of the 3 patients, 3 additional patients were enrolled in the cohort. If DLTs were seen in 2 of 6 patients in a cohort, dose escalation was stopped and the Maximally Administered Dose (MAD) was deemed to have been reached. The maximally tolerated dose (MTD) was defined as the highest feasible dose tested in which fewer than 33% of patients experienced DLT attributable to the study drug, with at least 6 patients treated at that dose level and assessable for toxicity. The MTD was also considered to be the Recommended Phase II Dose (RPTD).
Pharmacokinetic (PK) sampling and analytic assays
PK studies were carried out during the first cycle in all enrolled patients, and in those patients who reached a fourth cycle. Serum was collected for PK studies in patients up to the 325 mg/m2/24h dose level. Plasma was collected at the 400 mg/m2/24h dose level and above. No differences in drug concentrations were found between serum and plasma. A “Heparin lock” was placed in the arm contralateral to the arm in which the drug was infused. After discarding the first 2 mL of blood to clear the dead space, 5 mL blood was collected in each of 2 “green top” tubes with heparin anticoagulant before initiation of infusion, 1 h, 3 h, 6 h, 24 h and 48 h and 10 min before termination at 72 h during infusion. After termination of infusion, 2 mL blood samples were collected in “green top” tubes at 10, 20 and 30 min and 1, 3, 6, 24, 48 and 72 h. Samples were centrifuged at 1000 xg at 4°C for 10 min in the research ward within 30 min, and the sera or plasmas were stored frozen (-20°C) until the following morning when they were transferred to a lab and stored at -75°C freezer. The last three specimens were often collected on an ambulatory basis.
Rigosertib and its potential metabolite ON 01500 (Figure 1B) were measured by a validated high-performance liquid chromatography/tandem mass spectrometry (LC-MS/MS) assay [8,9]. Briefly 0.1 mL samples were extracted by 0.5 mL acetonitrile solution spiked with internal standard (temazepam, Figure 1C). After vortexing for 1 min, the mixture centrifuged at 1000 xg for 10 min, the supernatant removed, and the residue dried in TurboVap (Biotage, Charlotte, NC) at ~37°C. The residues were then reconstituted in 1.0 mL solution of 30% acetonitrile/70% H2O. The solutions were vortexed and centrifuged at 1000 xg for 5 min. The supernatant was transferred to autosampler vials for LC-MS/MS analysis. The HPLC system consisted of LC-10ADVP pump and SIL-HTA autosampler (Shimadzu, Columbia, MD), which was interfaced to an API 4000 Tandem Mass Spectrometer (AB Sciex, Foster City, CA) by a Turbo Ion Spray® source. The extracts were separated on a BDS Hypersil C18 column (100 x 3.00 mm, 3 μm; Thermo Fisher Scientific, Pittsburgh, PA) with a prefilter (0.5 μm; Supelco, Bellefonte, PA) by isocratic elution at 55% mobile phase B (mobile phase A: 10 mM ammonium acetate and mobile phase B: 0.1% formic acid in acetonitrile) at a flow rate 0.5 mL/min for 5 min. Positive ions were detected in the MRM mode with ion transitions at 452.1 → 194.2 for rigosertib; 394.0 → 136.0 for ON 01500 and 301.3 → 255.1 for the internal standard. The PK parameters were calculated based on the Non Compartmental Analysis using WinNonlin version 5.2 (Pharsight Corp, Cary, NC).
Early urine sample collection was faulty. After patient #7, urine was collected at zero time, during 0-4 h, 4-8 h, 8-24 h, 24-48 h, 48-72 h, 72-96 h and 96-120 h. Urine was collected in 4 liter bottles and alkalinized with sodium bicarbonate to pH8.0-9.0. Total fractionated urine volume and pH were recorded and a 10 ml fraction frozen at -75°C until analysis. To 60 µL aliquots of urine, 15 μL of internal standard (ON 01370, C19H22O6S, MW 378.44 Da, Onconova Therapeutics, Figure 1D) and 75 µL of cold (-4°C) acetonitrile were added. Each sample was vortexed for 1 min, followed by centrifugation (at about 9,000 xg) at 4°C for 30 min. The clear supernatants were transferred into new vials. Ten μL aliquots were injected into the liquid chromatograph/mass spectrometer (LC/MS).
The HPLC system consisted of a Model 1525 µ HPLC pump, and Model 717 Plus auto sampler, (both from Waters, Milford, MA). The column was Altima C18 7.5 × 4.6 mm i.d., 5 μm particle size (Grace & Co., Deerfield, IL), operated at atmospheric temperature. The mobile phase was composed of 50% of acetonitrile: H2O (v/v) with 0.1% formic acid, delivered isocratically at a flow rate of 0.5 mL/min. Run time was 3 min. The mass spectrometer was a Model QuattroLC triple quadrupole instrument (Waters, Milford, MA) equipped with an electrospray ionization source (ESI), operated in the positive mode. Temperature for desolvation was 250°C. Ion source temperature was 100°C. Optimal cone voltage and collision energy were 55 V and 27 eV for rigosertib, and 15 V and 20 eV for ON 01370, respectively. Dwell time was 0.1 sec. Nitrogen was used as the nebulizer and argon, the collision gas, pressure was 3.0 × 10-5 mbar. Data acquisition and processing for both chromatography and mass spectrometry were performed using the MassLynx® Software v 4.0 (Waters). For quantification, the mass spectrometer was operated in the selected reaction mode, monitoring transitions of m/z 474.0 → 216.0 for rigosertib, and m/z 379.0 → 194.0 for the internal standard. The peak areas of the analyte and internal standard were determined in arbitrary units in each analysis and each calibration sample, using the MassLynx® software. Concentrations of rigosertib were determined from plotted calibration curves using conventional methods.
Statistical methods
The following populations were studied: Intent-to-treat (ITT), which included all enrolled patients (n=29); Safety Evaluable, which included all enrolled patients who received at least 1 dose of rigosertib (n=28); and Efficacy Evaluable, which included all enrolled patients who received at least 1 dose of rigosertib and either had at least 1 post-baseline tumor reassessment or discontinued treatment prior to the first scheduled tumor assessment because of disease progression or death (n=22).
Demographic data were displayed, and summary statistics were used to describe the study population. Safety data were tabulated for all patients. These data included clinical and laboratory parameters and adverse events (AEs). AEs were tabulated by body system, severity and relation to treatment. Descriptive statistics were used to summarize the demographic and baseline characteristics and safety parameters (AEs, laboratory parameters, ECOG performance status). Treatment-emergent AEs (TEAEs) were summarized using the Medical Dictionary for Regulatory Activities (MedDRA), by system organ class (SOC) and preferred term (PT), by maximum severity (using the CTCAE v. 3) and by relationship to treatment. Efficacy data were tabulated, and classified by tumor type.
Results
Patient characteristics
Table 1 shows patient characteristics. The ITT population included 17 (59%) female and 12 (41%) male patients of whom 79% were White, 10% Black, 7% Hispanic, and 3% Asian. Most (79%) of the patients had a baseline ECOG performance status of 1. A wide variety of cancers were represented. All but one patient (pancreas cancer with liver metastasis) had received one or more regimens of chemotherapy appropriate for their cancers. Five patients had had prior hormone therapy, which included tamoxifen, anastrozole, letrozole, exemestane and testosterone. Ten patients had had prior targeted therapy, which included erlotinib, cetuximab, bevacizumab, bortezomib and sorafenib. A total of 29 patients were registered, one patient with squamous cell carcinoma of thyroglossal cyst died before treatment was initiated, and the remaining 28 patients were analyzed.
Table 1.
Patient characteristics
| Characteristic | No. of Patients (N=29) |
|---|---|
| Sex | |
| Men | 12 |
| Women | 17 |
| Age, years | |
| Range (y) | 36–87 |
| Median (y) | 63 |
| Eastern Cooperative Oncology Group performance status | |
| 0 | 3 (10.3%) |
| 1 | 23 (79.3%) |
| 2 | 3 (10.3%) |
| Primary tumor site | |
| Colorectal | 5 (17.2%) |
| Liver | 3 (10.3%) |
| Pancreas | 3 (10.3%) |
| Ovary | 3 (10.3%) |
| Corpus uteri | 3 (10.3%) |
| Hematopoietic system | 3 (10.3%) |
| Breast | 2 (6.9%) |
| Lung | 2 (6.9%) |
| Connective & soft tissue | 2 (6.9%) |
| Osteosarcoma | 1 (6.9%) |
| Urinary bladder | 1 (3.4%) |
| SCC of thyroglossal cyst | 1 (3.4%) |
| Prior surgery (excluding biopsy only) | 21 (72.4%) |
| Prior chemotherapy | 28 (96.5%) |
| Prior radiotherapy | 9 (31.0%) |
| Prior hormone therapy | 5 (17.2%) |
| Prior targeted therapy | 10 (34.4%) |
Process of dose-escalation and toxicities
Table 2 summarizes the dose escalation scheme. The total of 28 patients received a total of 115 cycles of treatment (median: 3 cycles; range: 1-19) and 11 dose levels (50-1,700 mg/m2/24h) were explored. Because intrapatient dose escalation was allowed, 2 patients received 2 dose levels, 1 patient 3 dose levels and another 1 patient 4 dose levels. A total of 35 patient-dose levels ranging 1 to 6 cycles were analyzed. Since no definitive cumulative effects were recognized and major toxicities occurred usually only from the first cycle, toxicities were analyzed on a patient-dose level basis at the maximum attained.
Table 2.
Dose levels of rigosertib administered
| Dose level | Drug dose mg/m2/24h x 3 days (mg/m2/cycle) | #pt receiving Initial cycle | Total cycles/dose level | #pts at each dose level | #cycles per dose level in individual patient |
|---|---|---|---|---|---|
| Level 1 | 50 (150) | 1 | 6 | 1 | 6 |
| 2 | 100 (300) | 1 | 8 | 2 | 4, 4 |
| 3 | 150 (450) | 2 | 7 | 2 | 1, 6 |
| 4 | 250 (750) | 1 | 11 | 3 | 4, 4, 3 |
| 5 | 325 (975) | 2 | 12 | 3 | 5, 5, 2 |
| 6 | 400 (1,200) | 4 | 13 | 4 | 1, 3, 6, 3 |
| 7 | 650 (1,950) | 1 | 7 | 2 | 4, 3 |
| 8 | 850 (2,550) | 2 | 11 | 3 | 3, 5, 3 |
| 9 | 1,050 (3,150) | 5 | 18 | 6 | 1, 3, 6, 1, 6, 1 |
| 10 | 1,375 (4,125) | 8 | 21 | 8 | 3, 2, 3, 3, 3, 3, 1, 3 |
| 11 | 1,700 (5,100) | 1 | 1 | 1 | 1 |
| Total | 28 | 115 | 35 |
Note: (1) Intrapatient dose escalation was allowed: two pts received two dose levels, one pt 3 dose levels and one pt 4 dose levels. (2) Study drug interruption: In one patient with pancreas cancer, second cycle of treatment (650 mg/m2/24h) was interrupted after 24 h infusion because of left sided flank pain. A CT scan showed PD. In another patient with hepatoma, first cycle of treatment with 1,050 mg/m2/24h was interrupted on day 3 after because of CNS toxicity.
Doses were initially escalated according to a Fibonacci escalation scheme, with one patient per each dose level until Gr 2 toxicity was observed. The first patient, who received 50 mg/m2/24h x 3 days, did not experience Gr ≥2 toxicity. She remained on treatment for 9 months with 3 dose escalations. Her lung cancer tumor marker remained essentially stable while on rigosertib, whereas it had risen sharply on the preceding treatment. Two patients were treated at the 100 mg/m2/24h dose level and experienced no Gr ≥2 toxicity. Two patients were treated at the 150 mg/m2/24h dose level. One patient with myelodysplastic syndrome/leukemic phase was treated with permission from the sponsor. In this patient the response was evaluated by the International Working Group (IWG) response criteria [10] and her clinical course is described briefly at the end of this section. The single patient who was treated at the 250 mg/m2/24h dose level did not experience any Gr ≥2 toxicity.
The first patient treated at the 400 mg/m2/24h dose level experienced a Serious Adverse Event (SAE): a 43 yr old white man with hepatoma (Patient #6) received 400 mg/m2/24h x 3 days. When he returned for the 2nd cycle, he was found to have acute renal failure associated with a methicillin-resistant Staphylococcus aureus infection. He improved on antibiotics. On the assumption that this SAE might have been due to the study drug, subsequent patients were treated at one half the Fibonacci de-escalation, i.e. at 325 mg/m2/24h. A subsequent MRI showed that his hepatoma had extended into the inferior vena cava causing renal failure. His renal “toxicity” was now deemed unrelated to the study drug. Subsequent doses at 325 mg/m2/24h, 400 mg/m2/24h and 650 mg/m2/24h were uneventful. Two patients who were treated at the 400 mg/m2/24h had preexisting lymphopenia, and the values did not worsen after rigosertib administration. The single patient treated at the 650 mg/m2/24h dose level did not experience any grade ≥2 toxicity. Doses were then escalated to 1,050 mg/m2/24h x 3 days.
The first patient (Patient #13) who received 1,050 mg/m2/24h x 3 days developed the second SAE. A 58 y old man with small cell lung cancer, extensive stage, with stable CNS metastasis and hemiparesis tolerated the 3-day infusion of the study drug well but fell the next day due to weakness. His clinical and imaging studies were unchanged. Hgb was 9.2 g/dL, WBC 5,900/µL and PLT 60,000/µL. He was alert and stable. There was no tumor progression. The Gr 3 musculoskeletal weakness constituted a study drug-related DLT in this already compromised patient. Treatment was discontinued.
The Fibonacci increment was thus deescalated by one half step. We entered 3 patients at the 850 mg/m2/24h dose level without major (Gr 3-4) side effects. Patients with compromised bone marrow function or preexisting hemiparesis appeared to be at high risk for drug toxicity and were, therefore, subsequently excluded.
At 1,050 mg/m2/24h, two more patients entered without encountering Gr ≥2 toxicity. At a half Fibonacci step up to 1,375 mg/m2/24h, one patient had thrombocytopenia to 37,000/µL during gram negative sepsis, but thrombocytopenia did not develop during a third cycle when infection was under control. Two additional patients tolerated 1,375 mg/m2/24h well.
We then entered at the next full Fibonacci level, 1,700 mg/m2/24h a 73 year-old woman (Patient #21) who had papillary serous adenocarcinoma of the peritoneum. Her initial serum sodium was 137 mEq/L. After drug infusion, on day 5, she developed involuntary violent tremors (Gr 3) of her arms and hands, ataxia and inability to walk, as well as Gr 3 fatigue. Her serum sodium level was found to be 112 mEq/L (Gr 4 hyponatremia). These were considered DLTs and SAEs (Tables 4, 5A, 5B). The tremor lasted for 5 h, and then improved spontaneously. Serum osmolality was 244 mOsm/kg, urine osmolality was 201 mOsm/kg, and urinary sodium was 59 mEq/L suggesting drug-induced syndrome of inappropriate secretion of anti-diuretic hormone (SIADH): She was treated with fluid restriction, but the nadir serum Na reached 110 mEq/L on day 7. She was given a hypertonic (3%) saline infusion and demeclocycline (300 mg twice a day). She remained afebrile and conscious, with auditory and visual hallucinations on days 8 (serum Na 121 mEq/L) and 9 (serum Na 125 mEq/L), respectively. On day 10, she had sinus tachycardia of 107 beats/min. CT angiogram was negative for pulmonary embolism, and troponin level was zero. Patient was discharged home on day 11 (serum Na 129 mEq/L). With Gr 3 neuromuscular toxicities and ataxia, Gr 4 hyponatremia, and Gr 3 psychosis (hallucination), we reached clear-cut DLT (Table 5A).
Table 4.
Treatment-emergent adverse events (TEAEs) attributed to rigosertib treatment-Laboratory
| Dose Levels | Drug dose mg/m2 x 3 d | #Pt | Anemia | Leucopenia | Lymphocytopenia | Thrombocytopenia | Hyponatremia | Hypokalemia | Hypophosphatemia | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||||||
| Grade | Grade | Grade | Grade | Grade | Grade | Grade | |||||||||||||||||
|
| |||||||||||||||||||||||
| 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | |||
| 1 | 50 | 1 | 1 | ||||||||||||||||||||
| 2 | 100 | 2 | 1a | ||||||||||||||||||||
| 3 | 150 | 2 | |||||||||||||||||||||
| 4 | 250 | 3 | |||||||||||||||||||||
| 5 | 325 | 3 | |||||||||||||||||||||
| 6 | 400 | 4 | (1b) | (2d) | |||||||||||||||||||
| 7 | 650 | 2 | |||||||||||||||||||||
| 8 | 850 | 3 | |||||||||||||||||||||
| 9 | 1,050 | 6 | 1 | 2 | |||||||||||||||||||
| 10 | 1,375 | 8 | 1c | 1c | 2c | 1 | 2 | 1c | 1 | 1 | 1 | 2 | 3 | ||||||||||
| 11 | 1,700 | 1 | 1 | ||||||||||||||||||||
| Total | 35 | ||||||||||||||||||||||
Non-TEAEs are shown in parenthesis or not counted.
patient with myelodysplastic syndrome/leukemic phase with preexisting anemia worsened.
patient with hepatoma had disease progression with large tumor thrombosis in the inferior vena cana and right renal vein.
as a part of multi-organ toxicity.
Both patients had preexisting lymphopenia and the values did not worsen after treatment.
Additional toxicities: Acute renal failure (n=3), one Gr 2 at 400 mg/m2/24h dose level, related to disease progression (non-TEAE); second and third, Gr 2 and Gr 3 each at 1,375 mg/m2/24h dose level as a part of multi-organ toxicity; diabetic ketoacidosis (n=1) Gr 2 at 1,375 mg/m2/24h dose level.
Table 5A.
Dose-limiting toxicities (DLTs)
| Patient ID | Dose level | Drug dose (mg/m2/24h) | Age | Gender | MedDRA | Gradea | Onset Cycle Dayb | Duration Daysc | Relation to Therapy | Outcomed |
|---|---|---|---|---|---|---|---|---|---|---|
| 0013 | 9 | 1,050 | 58 | Male | Muscle weakness | 3 | Cycle 1 Day 6 | 10 | Possible | 1 |
| 0029 | 9 | 1,050 | 58 | Male | Confusional state | 3 | Cycle 1 Day 3 | 8 | Probable | 2 |
| 0025 | 10 | 1,375 | 76 | Female | Hyponatremia | 4 | Cycle 3 Day 11 | 16 | Probable | 1 |
| 0026 | 10 | 1,375 | 64 | Female | Confusional state | 3 | Cycle 1 Day 3 | 17 | Possible | 1 |
| Delirium | 3 | Cycle 1 Day 5 | 15 | Possible | 1 | |||||
| Leukopenia (neutropenia) | 4 | Cycle 1 Day 7 | 8 | Possible | 1 | |||||
| 0021 | 11 | 1,700 | 73 | Female | Hyponatremia | 4 | Cycle 1 Day 5 | 12 | Probable | 2 |
Grade 1=Mild, 2=Moderate, 3=Severe, 4=Life-threatening, 5=Fatal.
Day relative to the first dose of study medication.
Duration of DLT calculated in days from the onset date to the resolution date.
Outcome: 1=Resolved, 2=Resolved with residual effects.
Table 5B.
Serious adverse events (SAEs)
| Patient ID | Dose level | Drug dose (mg/m2/24h) | Age | Gender | MedDRA | Gradea | Onset Cycle Dayb | Duration Daysc | Relation to Therapy | Outcomed |
|---|---|---|---|---|---|---|---|---|---|---|
| 0003 | 3 | 150 | 74 | Female | Febrile netropenia | 3 | Cycle 1 Day 4 | 12 | Possible | 1 |
| 0013 | 9 | 1,050 | 58 | Male | Muscle weakness | 3 | Cycle 1 Day 6 | 10 | Possible | 1 |
| 0029 | 9 | 1,050 | 58 | Male | Confusional state | 3 | Cycle 1 Day 3 | 8 | Probable | 2 |
| 0025 | 10 | 1,375 | 76 | Female | Hyponatremia | 3 | Cycle 1 Day 9 | 1 | Possible | 1 |
| Anemia | 3 | Cycle 2 Day 8 | 2 | Possible | 1 | |||||
| Leukopenia (neutropenia) | 3 | Cycle 2 Day 6 | 7 | Probable | 1 | |||||
| Anemia | 4 | Cycle 2 Day 9 | 1 | Possible | 1 | |||||
| Thrombocytopenia* | 2 | Cycle 2 Day 8 | 1 | Possible | 1 | |||||
| Hyponatremia | 4 | Cycle 3 Day 11 | 16 | Probable | 1 | |||||
| 0026 | 10 | 1,375 | 64 | Female | Confusional state | 3 | Cycle 1 Day 3 | 17 | Possible | 1 |
| Delirium | 3 | Cycle 1 Day 5 | 15 | Possible | 1 | |||||
| Anemia | 4 | Cycle 1 Day 6 | 2 | Possible | 1 | |||||
| Leukopenia (neutropenia) | 4 | Cycle 1 Day 7 | 8 | Probable | 1 | |||||
| Thrombocytopenia | 3 | Cycle 1 Day 9 | 11 | Probable | 1 | |||||
| Nausea* | 2 | Cycle 1 Day 3 | 17 | Possible | 1 | |||||
| Vomiting* | 2 | Cycle 1 Day 2 | 18 | Possible | 1 | |||||
| Headache* | 2 | Cycle 1 Day 2 | 9 | Possible | 1 | |||||
| Lethargy* | 2 | Cycle 1 Day 6 | 4 | Possible | 1 | |||||
| 0021 | 11 | 1,700 | 73 | Female | Fatigue | 3 | Cycle 1 Day 5 | 4 | Possible | 1 |
| Hyponatremia | 4 | Cycle 1 Day 5 | 12 | Probable | 2 | |||||
| Tremors/shaking | 3 | Cycle 1 Day 5 | 1 | Possible | 1 |
Patient 0003 had myelodysplastic syndrome with leukemic phase. Her neutropenia was evaluated using IWG response criteria [10].
Grade 1=Mild, 2=Moderate, 3=Severe, 4=Life-threatening, 5=Fatal.
Day relative to the first dose of study medication.
Duration of DLT calculated in days from the onset date to the resolution date.
Outcome: 1=Resolved, 2=Resolved with residual effects.
As a part of multi-organ toxicities.
Five additional gynecological cancer patients were entered at one-step lower dose level of 1,375 mg/m2/24h. One of these patients (Patient #25) developed grade 4 hyponatremia (serum Na 114 mEq/L) after the 3 cycles of treatment. This was considered a DLT and SAE (Tables 5A, 5B). Another patient (Patient #26), a 64 yr-old with endometrial carcinoma, developed acute multiorgan dysfunction sequentially over a 16 day period constituting DLTs and SAEs including Gr 3 delirium and confusion, headache, nausea, vomiting, Gr 4 anemia, Gr 4 leucopenia and Gr 3 thrombocytopenia. On day 11, she developed ileus/small bowel obstruction (SBO). She was symptomatically treated, clinically improved and was sent home on day 19. She had a history of Lyme disease involving the CNS. Four of five patients developed small bowel obstruction. We concluded that this dose level, too, was beyond the MTD.
Consequently, we went back to the 2-step lower dose of 1,050 mg/m2/24h x 3 days and two more patients were entered: One patient (Patient #29) experienced Gr 3 confusional state deemed a DLT and SAE (Tables 5A, 5B). He had a diagnosis of metastatic hepatoma and former drug addiction. Thus, of a total of 6 patients treated at the dose of 1,050 mg/m2/24h dose level, two developed Gr 3 DLTs (muscle weakness and confusional state). At this point, the sponsor discontinued the study. Our data suggest that the RPTD of a 3-day infusion is probably 850 mg/m2/24h x 3 days.
One patient with myelodysplastic syndrome with leukemic phase received one cycle of rigosertib 150 mg/m2/24h daily x 3. Her baseline blood counts were Hgb of 10.0 g/dL, WBC of 1,900/μL (Blast 26%) and PLT of 42,000/μL. She had been whole blood- and PLT transfusion-dependent. On day 3, Hgb was 9.4 g/dL, WBC was 1,600/µL and PLT was 14,000/µL. She was PLT transfused. On day 4, WBC decreased to 800/μL (Blasts 54%), and she became febrile with temperature of 37.9°C. She was started on ciprofloxacin, and then cefepine plus vancomycin were added. On day 5, her Hgb dropped to 5.7 g/dL and RBC were transfused. She developed central venous access port infection and the port was removed. Urine- and blood cultures were negative, fibrinogen was 475 mg/dL and haptoglobin was 174 mg/dL. Blood counts returned to the base line by day 10. It was decided that the treatment was too toxic with little clinical benefit. She was taken off study and went to other treatment.
Safety analysis
Tables 3 and 4 summarize drug-related treatment-emergent adverse events (TEAEs) defined as all AEs that occurred after the first dose of study medication or within the 30-day post-treatment period in all evaluable patients by System Organ Class and Preferred Term. Patients with multiple TEAEs are counted once within a summary category but with events in more than one category were counted once within each category.
Table 3.
Treatment-emergent adverse events (TEAEs) attributed to rigosertib treatment-Clinical
| Dose Levels | Drug dose mg/m2 x 3 d | #Pt | Fatigue | Infection | Ileus/SBOe | Muscle cramps/twitching/tremors | Muscle weakness | CNS symptoms | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||||
| Grade | Grade | Grade | Grade | Grade | Grade | |||||||||||||||
|
| ||||||||||||||||||||
| 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | 1-2 | 3 | 4 | |||
| 1 | 50 | 1 | 1 | |||||||||||||||||
| 2 | 100 | 2 | ||||||||||||||||||
| 3 | 150 | 2 | 1c | |||||||||||||||||
| 4 | 250 | 3 | ||||||||||||||||||
| 5 | 325 | 3 | ||||||||||||||||||
| 6 | 400 | 4 | 1b | 1 | 2d | 1 | ||||||||||||||
| 7 | 650 | 2 | ||||||||||||||||||
| 8 | 850 | 3 | 2a | 2 | ||||||||||||||||
| 9 | 1,050 | 6 | 1a | 1d | 1g | 1h | 1i | |||||||||||||
| 10 | 1,375 | 8 | 5 | 1b | 2d | 3 | 1 | 3 | 1j | |||||||||||
| 11 | 1,700 | 1 | 1 | 1f | 1k | |||||||||||||||
| Total | 35 | |||||||||||||||||||
Only toxicities which included Gr ≥3 are shown.
One of two patients with pre-existing fatigue symptom worsened;
pre-existing fatigue symptom worsened;
patient with MDS/AL hospitalized with febrile neutropenia;
including Port-A-Cath infection;
SBO, small bowel obstruction, all had gynecological neoplasms with pelvic surgery;
Patient #21;
Patient #13;
Hallucination;
Confusion, loss of recent memory;
Lethargy, confusion and delirium;
ataxia.
Additional clinical toxicities (all Gr 1-2) include pyrexia (Gr 1, n=2), epiphora (Gr 1, n=2), anorexia (Gr 1-2, n=4), vomiting (Gr 1, n=1), constipation (Gr 1-2, n=4), diarrhea (Gr 1-2, n=3), myalgia (Gr 1-2, n=4), dizziness (Gr 1-2, n=3), headache (Gr 2, n=2), and hypertension (Gr 1, n=4).
All 28 evaluable patients experienced TEAEs, which were considered to be drug related in 25 patients (89%). These included 12 (43%) patients who experienced fatigue, 8 (29%) with constipation (among them 4 were secondary to ileus/SBO); 7 (25%) with infectious complications, and muscle cramps/twitching/tremors; 5 (18%) with vomiting (among them 4 were from ileus) and hyponatremia; 4 (14%) with anorexia, ileus/SBO, myalgia, hypertension, and CNS dysfunction; 3 (11%) with dizziness, diarrhea, anemia, hypokalemia, hypophosphatemia, leucopenia, lymphopenia, thrombocytopenia (among them one with gram negative sepsis), and acute renal failure, and two (7%) with pyrexia, headache and epiphora. Myelosuppression was seen only sporadically and was not dose-limiting. These events occurred most often at the 1,375 mg/m2/24h dose level (Tables 3 & 4). The onset of the vast majority of TEAEs occurred during the first 8 weeks of treatment and mostly during the first 4 weeks. The TEAE rate of occurrence, severity, and drug-relationship did not appear to increase with weeks on treatment, suggesting a lack of cumulative toxicity.
Tables 5A, 5B summarizes the DLTs and SAEs attributed to rigosertib treatment. Five patients experienced DLTs with onset during or after cycle 1. These included muscular weakness, confusional state, delirium, hyponatremia and neutropenia (Table 5A). DLTs were observed in the 1,050, 1,375, and 1,700 mg/m2/24h dose cohorts. Six patients experienced a total of 21 drug-related SAEs (Table 5B). In three patients toxicities occurred as multi-organ involvements. These included nausea and vomiting, fatigue, anemia, (febrile) neutropenia, thrombocytopenia, muscular weakness, tremor, confusional state, delirium, hyponatremia, and lethargy. The study was discontinued without formally defining the RPTD.
The majority of safety evaluable patients (n=19, 66%) came off study because of progression of disease. Four patients (14%) were taken off study because of investigator decision, 3 (10%) because they declined further treatment, and 2 (7%) because of drug-related TEAEs (Gr 3 febrile neutropenia and Gr 3 confusional state). For the 26 patients who had post-baseline ECOG scores, the maximum ECOG score during the study was 0 for 3 patients (12%), 1 for 20 patients (77%), and 2 for 3 patients (12%). One patient died of disease progression during the 30-day follow-up period. No patient died due to a TEAE. No deaths occurred during the study.
Besides the current study, a number of other phase I-II clinical trials of rigosertib have been conducted in which the drug was administered using similar dosing schedules and dose-intensities (Investigator Brochure Rigosertib Sodium, Version 16, July 11, 2012) [9,11-13]. These included Study 04-03 for patients with advanced solid tumors and Studies 04-05, 04-15, and 04-17 for patients with hematological malignancies (MDS and AML). We compared the frequencies of AEs reported for ≥5% of patients in each of these studies with those observed in the current study. In spite of substantial overlap in the rigosertib dose-intensity used in the different trials, hematologic, musculoskeletal and neurologic and psychiatric AEs in particular were seen more frequently in the current trial compared to the other 4 studies, while renal and bladder toxicity were more frequent in the trials for MDS and AML. Moreover, drug-related Gr ≥3 AEs were observed in over 30% of patients on the current trial, in 20% of the patients in the MDS/AML trials, and in 12.5% of patients on study 04-03 (Investigator Brochure Rigosertib Sodium, Version 16, July 11, 2012).
Efficacy analysis
The primary measure of drug activity was best overall response, which was evaluated every 2 cycles (4 weeks). Of 22 evaluable patients, 9 (41%) achieved a best overall response of stable disease (SD) during the study; all other patients (n=13; 59%) had progression of disease. No patients achieved a CR or PR. The tumor types and progression-free survival (PFS) duration of the 9 patients with SD are summarized in Table 6.
Table 6.
Patients who had stable disease (SD) or who lived more than one year
| Patient ID | Drug dose (mg/m2/24h) | Tumor type | Stable Disease - PFS | Overall survival (>1 y) |
|---|---|---|---|---|
| 0001 | 50 | Adenocarcinoma of the lung | 323 days (46.1 weeks) | 526 days (75.1 weeks) |
| 0002 | 100 | Breast carcinoma | - | 946 days (135.1 weeks) |
| 0004 | 150 | Adenocarcinoma of the colon | 80 days (11.4 weeks) | 1,142 days (163.1 weeks) |
| 0007 | 325 | Hepatocellular carcinoma | 67 days (9.6 weeks) | 848 days (121.1 weeks) |
| 0010 | 400 | Pancreatic carcinoma | 163 days (23.3 weeks) | 608 days (86.9 weeks) |
| 0014 | 850 | Breast carcinoma | 63 days (9.0 weeks) | 650 days (92.9 weeks) |
| 0015 | 850 | Adenocarcinoma of the colon | 80 days (11.4 weeks) | 526 days (75.1 weeks) |
| 0016 | 1,050 | Adenocarcinoma of the colon | 87 days (12.4 weeks) | - |
| 0018 | 1,375 | Osteosarcoma | - | 442 days (63.1 weeks) |
| 0023 | 1,375 | Carcinoma of the corpus uteri | 37 days (5.3 weeks) | 568 days (81.1 weeks) |
| 0025 | 1,375 | Malignant mixed Műllerian tumor | 46 days (6.6 weeks) | - |
Overall, 20 of the 22 patients (91%) progressed during the study. The median PFS time was 50 days (95% confidence interval [CI]: 37-80 days). The last observation occurred at 323 days, after which no patient was progression-free.
Of the 28 patients in the safety evaluable population, 21 (75%) died. The median overall survival (OS) time was 232 days (95% CI: 151-526 days). The last event recorded was at 1,142 days. Nine (41%) patients survived for ≥1 year. Seven of these patients had SD. The tumor types and their OS duration are listed in Table 6.
Pharmacokinetic (PK) analysis
Serum (or plasma) PK parameters are presented in Table 7. In the first patient PK blood was collected on two occasions at cycle 7 and cycle 11 with two different dose levels. Blood PK parameters during cycle 1 were determined in 18 patients and in 4 patients the blood collections were repeated at cycle 4. In one patient, the cycle 1 and cycle 4 doses were different. In the remaining 3 patients, rigosertib was administered at the same dose level. PK values for these 3 patients showed no accumulation of rigosertib at cycle 4. Three other patients who experienced both drug-related SAEs and acute DLTs (Tables 5A, 5B) had aberrantly high Cmax and AUC0-∞ as well as significant reductions in Plasma Clearance (Figure 2), consistent with a direct relationship between drug exposure and toxicity.
Table 7.
Serum and plasma rigosertib pharmacokinetic parameters
| Patient ID | Cycle # | Dose given (mg/m2/24h) | Lambda (1/h) | T1/2 (h) | Tmax (h) | Cmax (mg/L) | Cmax/dose | AUC0-∞ (mg.h/L) | AUC0-/dose | Vz (L/m2) | Clearance (L/h/m2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0001 | 7 | 100 | 20 | 3.47 | 48 | 0.78 | 2.6 | 49.3 | 164.2 | 30.5 | 6.1 |
| 0001 | 11 | 250 | 0.59 | 1.17 | 3 | 1.70 | 2.27 | 49.6 | 66.1 | 25.4 | 15.1 |
| 0002 | 4 | 100 | 0.16 | 4.26 | 3 | 1.23 | 4.1 | 82.9 | 276.2 | 22.2 | 3.6 |
| 0004 | 1 | 150 | 0.28 | 2.46 | 3 | 1.51 | 3.36 | 64.8 | 144.0 | 24.6 | 6.9 |
| 0004 | 4 | 150 | 0.22 | 3.09 | 6 | 1.23 | 2.73 | 51.2 | 113.7 | 39.2 | 8.8 |
| 0006 | 1 | 400 | 0.23 | 3 | 72 | 2.88 | 2.4 | 208.6 | 173.8 | 24.9 | 5.8 |
| 0007 | 1 | 325 | 0.29 | 2.4 | 6 | 3.52 | 3.61 | 174.3 | 178.8 | 19.4 | 5.6 |
| 0008 | 1 | 325 | 0.32 | 2.19 | 3 | 2.30 | 2.36 | 138.3 | 141.8 | 22.3 | 7.1 |
| 0009 | 1 | 400 | 0.12 | 5.82 | 3 | 3.94 | 3.28 | 184.0 | 153.4 | 54.8 | 6.5 |
| 0010 | 1 | 400 | 0.18 | 3.94 | 72 | 2.50 | 2.09 | 99.3 | 82.7 | 68.7 | 12.1 |
| 0011 | 4 | 400 | 0.29 | 2.38 | 48 | 2.79 | 2.33 | 180.0 | 150.0 | 22.9 | 6.7 |
| 0012 | 1 | 650 | 0.18 | 3.87 | 3 | 2.71 | 1.51 | 139.3 | 77.4 | 72.2 | 12.9 |
| 0013 | 1 | 1,050 | 0.44 | 1.56 | 72 | 12.57 | 3.99 | 628.1 | 199.4 | 11.3 | 5.0 |
| 0014 | 1 | 850 | 0.57 | 1.21 | 3 | 6.96 | 2.73 | 407.1 | 159.6 | 10.9 | 6.3 |
| 0014 | 4 | 850 | 0.59 | 1.18 | 72.2 | 5.55 | 2.18 | 289.0 | 113.3 | 15.1 | 8.8 |
| 0015 | 1 | 850 | 0.49 | 1.4 | 6 | 5.08 | 1.61 | 213.8 | 67.9 | 29.9 | 14.7 |
| 0015 | 4 | 1,050 | 0.57 | 1.22 | 3 | 6.14 | 1.95 | 229.1 | 72.7 | 24.1 | 13.7 |
| 0016 | 1 | 1,050 | 0.31 | 2.25 | 1 | 4.67 | 1.48 | 129.9 | 41.2 | 78.8 | 24.3 |
| 0016 | 4 | 1,050 | 0.56 | 1.24 | 48 | 5.64 | 1.79 | 316.2 | 100.4 | 17.8 | 10.0 |
| 0017 | 1 | 1,050 | 0.27 | 2.61 | 3 | 5.80 | 1.84 | 245.6 | 78.0 | 48.3 | 12.8 |
| 0019 | 1 | 1,375 | 0.61 | 1.13 | 6 | 6.21 | 1.5 | 362.8 | 87.9 | 18.6 | 11.4 |
| 0020 | 1 | 1,375 | 0.53 | 1.31 | 3 | 7.14 | 1.73 | 318.0 | 77.1 | 24.6 | 13.0 |
| 0021 | 1 | 1,700 | 0.63 | 1.1 | 72 | 14.48 | 2.84 | 649.3 | 127.3 | 12.4 | 7.9 |
| 0025 | 1 | 1,375 | 0.42 | 1.66 | 6 | 8.36 | 2.03 | 311.5 | 75.5 | 31.7 | 13.2 |
| 0026 | 1 | 1,375 | 0.48 | 1.44 | 72.2 | 35.62 | 8.63 | 1,134.5 | 275.0 | 7.5 | 3.6 |
| 0027 | 1 | 1,375 | 0.34 | 2.05 | 24 | 8.53 | 2.07 | 512.4 | 124.2 | 23.8 | 8.0 |
| Average | 0.38 | 2.27 | 2.7 | 131.6 | 29.4 | 9.4 | |||||
| SD | 0.16 | 1.19 | 1.5 | 62.2 | 19.1 | 4.6 | |||||
| CV% | 42.9 | 52.3 | 53.5 | 47.3 | 65.1 | 49.1 |
Lambda, Elimination rate constant; T1/2, Half life; Tmax, Time to reach maximum (peak) plasma concentration following drug administration; Cmax, Maximum (peak) plasma concentration following drug administration; Cmax/Dose, Dose normalized maximum (peak) plasma concentration; AUC0-∞, Area under the plasma concentration – time curve from time zero to infinity; AUC0-∞/Dose, Dose normalized area under the plasma concentration – time curve from time zero to infinity; Vz, Apparent volume of distribution during the terminal phase.
Figure 2.
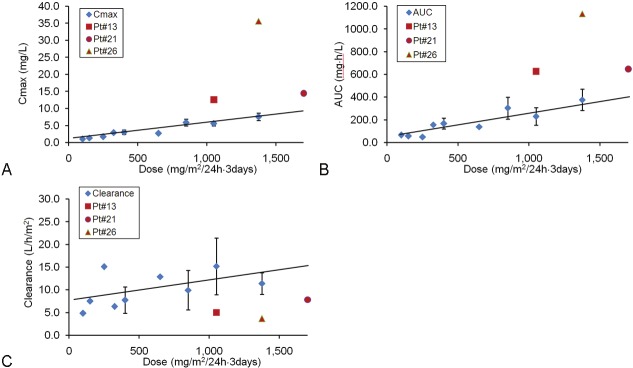
Analyses of plasma rigosertib pharmacokinetics. A. Cmax vs. Dose levels. B. AUC0-∞ vs. Dose levels. C. Clearance vs. Dose levels. Dose levels with >3 data points were shown as mean ± SD, dose levels with 2 data points were shown as mean ± the range. The least square regression line was drawn. Correlation coefficient values calculated by Excel “correl” function were: Dose vs. Cmax: 0.962; Dose vs. AUC0-∞: 0.921 and Dose vs. Clearance: 0.521. PK values of 3 patients who experienced SAEs and DLTs are shown in red marks.
Urinary excretion of rigosertib was evaluated in 16 patients (Table 8). Two patients (Patient #7 and Patient #15) had urine collection after the first and the 4th cycle of treatment. No cumulative effect was seen. Urinary rigosertib excretion was rapid in all patients and ceased soon after the end of infusion. The amount of drug excreted ranged from 3 to 15% of the total dose administered, with a mean of 9% and 97% of it occurring before 72 h. No significant amounts of ON 01500, the potential metabolite of ON 01910.Na, were detected in serum and urine specimens (detection limit ≤10 ng/mL).
Table 8.
Quantity of rigosertib found in collected urine from patients
| Pt#-cycle | Dose (mg/m2/24h) | Total drug administered (mg) | Quantity of drug (mg) during collection time (h) | Drug excreted, % of total administered | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 0-4 | 4-8 | 8-24 | 24-48 | 48-72 | 72-96 | 96-120 | Total | ||||
| 0007-1 | 325 | 2,048 | 2.7 | 2.8 | 12.7 | 36.7 | 32.5 | 0.1 | 0 | 87.5 | 4.3% |
| 0007-4 | 325 | 2,048 | 0.2 | 13.8 | 26.5 | 28.8 | 23.3 | 3.0 | * | 95.6 | 4.7% |
| 0010-1 | 400 | 2,580 | 11.1 | 16.6 | 52.2 | 83.0 | 66.0 | 4.8 | * | 233.9 | 9.1% |
| 0011-1 | 400 | 2,232 | 0.3 | 4.1 | 18.0 | 23.8 | 18.2 | 0.7 | 0 | 65.1 | 2.9% |
| 0012-1 | 650 | 4,641 | 15.8 | 23.7 | ** | 369.2 | 315.7 | 16.5 | 1.2 | (742.1) | (16.0%) |
| 0014-1 | 850 | 4,463 | 26.9 | 28.7 | 162.3 | 182.8 | 221.2 | 50.1 | ** | 672.0 | 15.1% |
| 0015-1 | 850 | 4,106 | 4.1 | 9.3 | 106.0 | 169.5 | 179.0 | 8.8 | *** | 476.7 | 11.6% |
| 0013-1 | 1,050 | 6,458 | 39.8 | 64.7 | 144.1 | 185.1 | 162.5 | 3.0 | 0.7 | 599.9 | 9.3% |
| 0015-4 | 1,050 | 5,072 | 17.7 | 8.6 | 62.0 | 138.4 | 147.1 | 20.6 | *** | 394.4 | 7.8% |
| 0016-1 | 1,050 | 6,030 | 66.4 | 30.3 | 119.0 | 143.6 | 162.2 | 11.1 | 1.2 | 533.8 | 8.9% |
| 0017-1 | 1,050 | 5,292 | 9.4 | 4.6 | 111.2 | 110.0 | 269.3 | 11.8 | 0 | 516.3 | 9.8% |
| 0019-1 | 1,375 | 8,164 | 90.3 | 85.7 | 121.0 | 224.6 | 35.8 | 3.7 | *** | 561.1 | 6.9% |
| 0020-1 | 1,375 | 7,178 | 59.0 | 46.5 | 118.0 | 214.1 | 181.3 | 112.5 | 0.6 | 732.0 | 10.2% |
| 0023-1 | 1,375 | 5,414 | 42.2 | 49.0 | 83.7 | 102.4 | *** | *** | *** | (277.3) | (5.1%) |
| 0024-1 | 1,375 | 6,681 | 88.8 | 59.1 | 384.0 | 311.8 | 131.7 | *** | *** | 975.4 | 14.6% |
| 0025-1 | 1,375 | 6,435 | 59.1 | 45.6 | 75.4 | 215.9 | 103.5 | 0.0 | 0 | 499.5 | 7.8% |
| 0027-1 | 1,375 | 8,224 | 1.2 | 51.6 | 204.4 | 555.5 | 256.2 | 3.8 | 6.3 | 1,079.0 | 13.1% |
| 0021-1 | 1,700 | 8,109 | 51.0 | 24.1 | 139.7 | 263.8 | 292.4 | 30.3 | *** | 801.3 | 9.9% |
Urine volume not recorded,
sample lost,
sample was not collected.
Numbers in parenthesis were based on insufficient urine collection during drug infusion periods. These data were not used for analysis.
Discussion
Rigosertib is a novel inhibitor of the Plk-1 and PI3 kinase pathways that disrupts the G2-M cell cycle transition and induces mitotic catastrophe and cell death in malignant cells. Recently, two reports have examined rigosertib’s mechanisms of action. One study reported that rigosertib caused a rapid decrease of cyclin D1 levels by blocking cyclin D1 mRNA translation through inhibition of the PI-3K/Akt/mTOR/eIF4E-BP signaling pathway, triggering cytochrome C-dependent apoptosis in mantle cell lymphoma cells [5]. The other research showed that rigosertib caused hyperphosphorylation of RanGAP1·SUMO1, which was sustained for more than 24 h and led to G2/M arrest and subsequent induction of apoptosis [14].
In a previous phase I trial, Jimeno et al. demonstrated the safety and clinical activity of 2 h intravenous infusions of rigosertib in the treatment of patients with solid tumors [9]. Most notably, one patient with advanced ovarian cancer had an objective response after 4 cycles and remained progression free for 24 months. In the present report, we describe the toxicity and clinical activity of 72 h continuous IV infusions of rigosertib in a similar patient population. The choice of this dosing schedule was based on preclinical studies that showed that rigosertib’s in vitro cytotoxic effects were exposure time-dependent rather than drug-concentration dependent [6]. Moreover, animal PK studies had indicated that rigosertib is excreted rapidly with a distribution half-life of 17-40 min and a terminal elimination phase of 2.7-5.8 h. These data suggested that the compound best be administered as a continuous IV infusion. In addition, animal studies suggested that rigosertib did not appear to be strongly toxic. This prompted us to employ an accelerated dose escalation scheme until an instance of Gr 2 toxicity occurred. Because we encountered unexpected patterns of toxicity, the study was halted before the RPTD was formally defined. However, our results suggest that a rigosertib dose of 850 mg/m2/24h x 3 days every 2 weeks would be feasible and the toxicity profile acceptable for phase 2 development.
Overall the clinical activity of rigosertib administered as 3-day continuous IV infusions was modest. Of 22 evaluable patients with advanced refractory cancer and good performance status, 9 (41%) achieved SD and 9 patients (including 7 with SD) survived for ≥1 year. The initial phase I clinical trial of IV rigosertib conducted at Johns Hopkins University in 2008 reported one partial response associated with a significant decrease in CA-125 level in a patient with ovarian cancer that had relapsed from prior treatment [9]. This report prompted referral of 6 patients with refractory endometrial and ovarian cancers to our study. None of these patients responded to rigosertib treatment. Moreover, four of these patients developed Gr 3-4 ileus/SBO, suggesting the possibility that a history of pelvic surgery might be a risk factor for rigosertib toxicity.
The clinical activity observed in our study is comparable to that seen in the second phase I trial of rigosertib administered as a weekly continuous 24 h IV infusion for patients with solid tumors (Study 04-03) (Investigator Brochure Rigosertib Sodium, Version 16, July 11, 2012). Twelve (33.3%) of the 36 evaluable patients achieved SD. All other patients progressed. Nine patients (25%) were still alive at 1 year.
Cycle 1 pharmacokinetics of rigosertib in the current trial were also comparable to those seen in Study 04-03. In both studies, both Cmax and AUC0-∞ increased in a dose-proportional fashion and were of the same order of magnitude. Urinary excretion pattern of rigosertib was again in accord with other studies (Investigator Brochure Rigosertib Sodium, Version 16, July 11, 2012).
The impressive activity of rigosertib against human cancer cell lines in vitro [1], against human tumor xenografts in vivo [1-3] and in some clinical cancers [11,13] together with identification of new mechanisms of action, warrant further study of the biological activities of this class of compounds using this and other methods of administration.
Acknowledgments
This study was supported by the Grant Numbers M01 RR00071 and UL1RR029887 both from the National Center for Research Resources (NCRR), NIH, Bethesda, MD; Onconova Therapeutics, Inc. Newtown, PA; the T.J. Martell Foundation for Leukemia, Cancer and AIDS Research, New York, NY; and the Myra Shaw Cancer Research Fund, Brooklyn, NY. We are thankful for excellent service provided by staff nurses and administrators of the Clinical Research Center (CRC) at the Mount Sinai Hospital. We acknowledge the contributions of Steven Cosenza, Edward O’Rourke and Daniel Fox who assembled the rigosertib IND, Isabelle Darnis-Wilhelm who analyzed overall rigosertib safety data and Michael Reiss who provided medical writing services on behalf of Onconova, Inc.
Disclosure of conflict of interest
TO, LS, JR and JFH serve as consultants to Onconova Therapeutics, Inc. JFH receives grant support from Onconova Therapeutics, Inc. CR, MM and FW are employees of Onconova Therapeutics, Inc.
References
- 1.Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–86. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Jiang JD, Li Y, Wang YL, Reddy PE, Roboz J, Ohnuma T, Silverman L, Holland JF. Anticancer effects of ON-1910Na (Abst 5382) Proc Am Assoc Cancer Res. 2004;45:1241. [Google Scholar]
- 3.Jimeno A, Chan A, Cusatis G, Zhang X, Wheelhouse J, Solomon A, Chan F, Zhao M, Cosenza S, Reddy MVR, Rudek M, Kulesza P, Reddy EP, Hidalgo M. Evaluation of ON 01910. Na, a novel modulator of Polo-like kinase 1 (Plk1) pathway, and development of a cyclin-B1-based predictive assay in pancreatic cancer (Abst 5391) Proc Am Assoc Cancer Res. 2007:48. [Google Scholar]
- 4.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 5.Prasad A, Park IW, Allen H, Zhang X, Reddy MVR, Boominathan R, Reddy EP, Groopman JE. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2009;28:1518–28. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- 6.Preda A, Ohnuma T, Jiang JD, Holland JF, Reddy PE. Cross-resistance to ON 01910. Na among drug resistant human tumor cell lines (Abstract 4707) Proc Am Assoc Cancer Res. 2006;47:1106. [Google Scholar]
- 7.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89:1138–47. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Zhao M, Jimeno A, He P, Ramana Reddy MV, Hidalgo M, Donehower RC, Rudek MA. Validation and implementation of a liquid chromatography/tandem mass spectrometry assay to quantitate ON 01910. Na, a mitotic progression modulator, in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;856(1-2):198–204. doi: 10.1016/j.jchromb.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, Hidalgo M, Baker SD, Donehower RC. Phase I study of ON 01910. Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J. Clin. Oncol. 2008;26:5504–10. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chason BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, Pinto A, Beran M, de Witte TM, Stone RM, Mittelman M, Sanz GF, Gore SD, Schiffer CA, Kantarjian H. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–25. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 11.Olnes MJ, Shenoy A, Weinstein B, Pfannes L, Loeliger K, Tucker Z, Tian X, Kwak M, Wilhelm F, Yong AS, Maric I, Maniar M, Scheinberg P, Groopman J, Young NS, Sloand EM. Directed therapy for patients with myelodysplastic syndromes (MDS) by suppression of cyclin D1 with ON 01910. Na. Leuk Res. 2012;36:982–9. doi: 10.1016/j.leukres.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverman LR, Odchimar-Reissig R, Navada SC, Ohnuma T, Najfeld V, Wilhelm F, Reddy EP, Holland JF. Effects of a novel benzyl styryl sulfone derivative ON 01910. Na on the myelodysplastic syndrome (MDS) derived clone in patients relapsing following response to azacitidine (AzaC) therapy. Blood (ASH Annu Meet Abstr) 2009:114. Abstr nr 4839. [Google Scholar]
- 13.Seetharama M, Fan AC, Tran M, Xu L, Renschler JP, Felsher DW, Sridhara K, Wilhelm F, Greenberg PL. Treatment of higher risk myelodysplastic syndrome patients unresponsive to hypomethylating agents with ON 01910. Na. Leuk Res. 2012;36:98–103. doi: 10.1016/j.leukres.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oussenko IA, Holland JF, Reddy EP, Ohnuma T. Effect of ON 01910. Na, an anticancer mitotic inhibitor, on cell cycle progression correlates with RanGAP1 hyperphosphorylation. Cancer Res. 2011;71:4968–76. doi: 10.1158/0008-5472.CAN-10-1603. [DOI] [PubMed] [Google Scholar]