

Abstract
Insulin resistance (IR) and hyperinsulinemia are hallmarks of the metabolic syndrome, as are central adiposity, dyslipidemia, and a predisposition to type 2 diabetes, atherosclerotic cardiovascular disease, hypertension, and certain cancers. Regular exercise and calorie restriction have long been known to increase insulin sensitivity and decrease the prevalence of these disorders. The subsequent identification of AMP-activated protein kinase (AMPK) and its activation by exercise and fuel deprivation have led to studies of the effects of AMPK on both IR and metabolic syndrome–related diseases. In this review, we evaluate this body of literature, with special emphasis on the hypothesis that dysregulation of AMPK is both a pathogenic factor for these disorders in humans and a target for their prevention and therapy.
Introduction
The fuel-sensing enzyme AMP-activated protein kinase (AMPK) was first described as an enzyme activated by changes in the AMP/ATP ratio that could both increase cellular ATP generation (e.g., fatty acid oxidation) and diminish ATP use for less critical processes (e.g., fatty acid, triglyceride, and protein synthesis) (1). In addition to glucose transport, lipid and protein synthesis, and fuel metabolism, AMPK regulates a wide array of other physiological events, including cellular growth and proliferation, mitochondrial function and biogenesis, and factors that have been linked to insulin resistance (IR), including inflammation, oxidative and ER stress, and autophagy (Figure 1A). Furthermore, AMPK does so by phosphorylating both key enzymes and transcriptional activators and coactivators.
Figure 1. AMPK actions and putative linkage between decreased AMPK activity and metabolic syndrome–associated diseases.
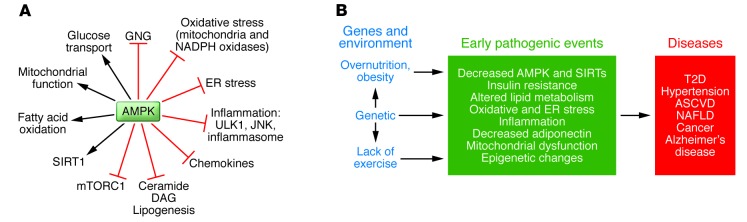
(A) Effects of AMPK activation. In addition to activating processes that produce ATP and diminish its consumption, AMPK inhibits inflammation, ER and oxidative stress, and activates autophagy, all of which appear to be involved in the pathogenesis of IR. Where studied, SIRT1 can produce many of the same effects as AMPK (see also text and Figure 3). The above-listed actions of AMPK and others have been extensively reviewed (5, 6, 67). GNG, gluconeogenesis; ULK1, UNC-51–like kinase 1; JNK, JUN-activated kinase. (B) Proposed link between AMPK and IR in the setting of the metabolic syndrome. It has long been held that the combination of overnutrition (obesity), inactivity, and indeterminate genetic factors predispose humans to the metabolic syndrome and associated disorders. Based on studies of the offspring of patients with metabolic syndrome–associated disorders, hyperinsulinemia and IR may antedate such diseases as hypertension, type 2 diabetes, and ASCVD by many years (reviewed in ref. 28). Likewise, studies in both experimental animals and humans have implicated oxidative and ER stress and low-grade inflammation and decreased adiponectin in the pathogenesis of these disorders. An emerging body of evidence, predominantly but not exclusively from animal models, suggests that dysregulation of AMPK, and probably sirtuins, could both contribute to these abnormalities and be a target for their prevention and therapy (13). One possibility is that such dysregulation of AMPK and sirtuins causes epigenetic changes (methylation, acetylation, etc.) that could contribute to the diseases (151). NAFLD, nonalcoholic fatty liver disease; T2D, type 2 diabetes.
Here, we examine 2 hypotheses suggested by more recent studies: (a) dysregulation of AMPK plays an important role in the pathogenesis of IR and metabolic syndrome–associated diseases in humans and experimental animals; and (b) strategies that activate AMPK can be harnessed for the prevention and treatment of these abnormalities. These hypotheses emanated from associations between the metabolic syndrome and some downstream targets of AMPK, such as glucose transport and lipogenesis (2–8). In addition, exercise (9) and electrically induced contractions (10) were shown to activate AMPK. These observations, coupled with epidemiological evidence that diseases associated with the metabolic syndrome (e.g., type 2 diabetes, hypertension, atherosclerotic cardiovascular disease [ASCVD], and even certain cancers) are less prevalent in physically active people (11–13) and the demonstration that regular exercise improves whole-body insulin action (11, 12), suggest a central role for AMPK in regulating insulin sensitivity. Such studies also raise the possibility that pharmacological AMPK activators as well as exercise could be used for ameliorating IR in type 2 diabetes (4, 8).
In model systems, sustained decreases in AMPK activity accompany IR, whereas AMPK activation increases insulin sensitivity (5, 6, 13). In addition, decreases in AMPK activity accompanying IR were described in adipose tissue of humans with Cushing’s syndrome, an effect attributable to high levels of glucocorticoids (14), and in a subgroup of very obese patients undergoing bariatric surgery who were insulin resistant (15, 16). The latter comprise approximately 75% of bariatric surgery patients and show a greater predisposition to metabolic syndrome–associated diseases than do the remaining 25% of such patients who are equally obese, but less hyperinsulinemic and more insulin sensitive (17–19).
Insulin resistance in physiology and disease
Studies with a perfused rat hindquarter preparation demonstrated that insulin-stimulated glucose uptake in skeletal muscle is reduced in fed versus fasted rats (20) and in sedentary versus recently exercised rats (21), suggesting that the fed and sedentary rats are essentially more insulin resistant. Such IR is physiological, rather than disease associated, and dynamically responds to changes in nutritional and physical activity.
In contrast, in patients with the metabolic syndrome, IR and hyperinsulinemia are sustained and are associated with impaired insulin action and a predisposition to multiple diseases (Figure 1B) (22–24). Whether IR plays an active role in the pathogenesis of these diseases or defends against a “glucolipotoxic” insult that may harm insulin-sensitive tissues (e.g., by increasing oxidative stress) is presently under debate (25, 26). A diagnosis of the metabolic syndrome is based on measurements of plasma glucose (less than 100 mg/dl) and other parameters such as triglycerides, HDL cholesterol, blood pressure, and waist circumference; abnormalities in 3 of the 5 of risk factors are required for a metabolic syndrome diagnosis (24, 27). Interestingly, individuals with the metabolic syndrome typically have a decreased capacity for exercise (28) and show evidence of low-grade inflammation (29), similar to patients with obesity and type 2 diabetes (24, 29). Furthermore, oxidative and ER stress, mitochondrial dysfunction, altered lipid metabolism, and dysregulation of AMPK and a closely related group of fuel-sensing molecules, the sirtuins, have also been described in both humans and experimental animals with metabolic syndrome–associated diseases and appear to promote their pathogenesis (22, 24).
AMPK structure and regulation
AMPK is a heterotrimer consisting of a catalytic subunit (α) and 2 regulatory subunits (β and γ). Isoforms of each subunit have been identified (2 α and β isoforms, and 3 γ isoforms) (Figure 2A). The γ subunit contains 4 cystathionine β-synthase (CBS) domains, which form 4 potential adenine nucleotide–binding sites. Structural and in vitro binding studies have revealed that the γ subunit in fact binds 3 nucleotides. One molecule of AMP is bound in a nonexchangeable manner at site 4, with 2 additional exchangeable nucleotide-binding sites (sites 1 and 3) (30). Initial studies suggested that when a cell is energetically stressed, the increased AMP concentration causes AMP to displace ATP from the exchangeable sites, resulting in a modest (2- to 5-fold) allosteric activation of AMPK. Displacement of ATP from the exchangeable sites also protects the enzyme against dephosphorylation of the phosphothreonine 172 residue on its α subunit, leading to an even greater (100- to 1,000-fold) increase in activity. Recent studies (30) suggest that displacement of ATP by ADP rather than AMP may be primarily responsible for this secondary and larger activation. In addition, ADP and AMP have been reported to promote phosphorylation of threonine 172 (31), providing another potential tier of regulation.
Figure 2. Subunit structure and regulation of AMPK.

(A) Structure of AMPK. Schematic representation of AMPK highlighting important regions within each of its 3 subunits, as described in the main text (adapted from ref. 64). AID, autoinhibitory domain; CBM, carbohydrate-binding module; CBS, cystathionine-β synthase. (B) Regulation of AMPK. LKB1 and CaMKKβ activate AMPK by phosphorylation of threonine 172 (T172) within the kinase domain of the α subunit. AMPK is returned to an inactive form by dephosphorylation catalyzed by the action of protein phosphatases (PPase). Binding of ADP and AMP to the γ subunit of AMPK protects against dephosphorylation, maintaining the kinase in an active conformation, although recent studies suggest that ADP is likely to be the important physiological regulator of this process. ADP and AMP have also been reported to promote LKB1-mediated phosphorylation of AMPK, whereas calcium directly activates CaMKKβ. In addition, AMP causes a modest allosteric activation of AMPK. Finally, glycogen (and branched-chain carbohydrates) bind to the glycogen-binding domain within the β subunit, allosterically inhibiting AMPK. P, phosphorylation of T172.
The β subunit contains a carbohydrate-binding module (also referred to as a glycogen-binding domain) that is found in a number of enzymes involved in carbohydrate metabolism. Glycogen, as well as branched-chain sugars, inhibit AMPK presumably by binding to this domain. Interestingly, this domain is also implicated in the mechanism of AMPK activation by small-molecule activators (32). At least 2 protein kinases catalyze the activating phosphorylation of the α subunit of AMPK on threonine 172: the tumor suppressor liver kinase B1 (LKB1), which does so in response to changes in energy state, and calcium calmodulin–dependent protein kinase kinase β (CAMKKβ), which is activated by increases in intracellular Ca2+ (Figure 2B) (33, 34).
AMPK and insulin resistance
Effects of exercise
As already noted, exercise increases whole-body insulin sensitivity and diminishes plasma insulin levels in humans. In addition, both epidemiological and randomized, prospective studies showed that regular exercise diminishes the likelihood that humans will develop ASCVD (35, 36) and type 2 diabetes (28, 37–39). Thus, the benefits of exercise are widely attributed to its effect on reducing whole-body IR and hyperinsulinemia. Although this explanation, which assumes a systemic effect of exercise on insulin action, still has merit, the discovery in rodents that exercise acutely increases AMPK activity in adipose tissue (40) and aortic endothelium and media (41, 42), as well as liver (40) and muscle, raises the possibility that AMPK activation in specific and underappreciated tissues contributes to its beneficial effects. For instance, in rodent aorta, treadmill running increases the activity or expression of 3 known AMPK regulators, LKB1, CAMKKβ, and SIRT1, and it concurrently activates endothelial nitric oxide synthase (eNOS) (41), an enzyme generally thought to protect against atherogenesis in experimental animals (43).
AMPK activators — pharmacological and hormonal
Metformin.
In 2001, metformin, a widely used antidiabetic drug that modestly increases insulin sensitivity, was reported to act as an AMPK activator (44). An extended follow-up study (45) showed that metformin use was associated with a significant decrease in the incidence of myocardial infarction and a 36% decrease in all-cause mortality. Furthermore, multiple studies have suggested that the incidence of many cancers is diminished in diabetic patients treated with metformin (46), although this conclusion has been questioned (47). Metformin does not act directly on LKB1 or AMPK, but rather appears to activate AMPK by modestly inhibiting complex I of the mitochondrial electron transport chain and thus decreasing the cellular energy state (48). In liver, this decreased energy state is sufficient to diminish glucose production, even in mice lacking hepatic AMPK, indicating AMPK-independent effects of metformin. Moreover, metformin-induced accumulation of AMP, which inhibits adenylate cyclase and reduces cAMP levels and PKA activity, may be an AMPK-independent mechanism by which metformin antagonizes glucagon signaling (49). Metformin has been shown to activate AMPK in many tissues, including adipose, skeletal muscle, and heart, although, to date, evidence that genetic downregulation of AMPK prevents its biological actions has been obtained only in cultured cells.
Adiponectin.
AMPK is also activated by the adipokine adiponectin (50, 51). Notably, diminished serum adiponectin levels are associated with IR and appear to be a strong predictor of type 2 diabetes and cardiovascular disease in obese humans (52–54). Conversely, elevated adiponectin levels are associated with increased insulin sensitivity, a lower incidence of type 2 diabetes that is independent of obesity, and a decreased risk of coronary heart disease (52–54). Adiponectin also induces extracellular calcium influx through the adiponectin receptor 1 (55) and may have effects independent of AMPK.
Thiazolidinediones and other AMPK activators.
In addition to metformin, another group of type 2 diabetes drugs, the thiazolidinediones (TZDs), were found to be AMPK activators (56). Although TZDs can activate AMPK in some tissues by lowering their energy state (57, 58), studies in adiponectin knockout mice strongly suggested the TZDs’ ability to increase hepatic insulin sensitivity in vivo is predominantly adiponectin mediated (59, 60). TZDs are also well-established activators of the peroxisome proliferator-activated receptors (PPARs). As such, the adiponectin-independent mechanism of TZD-induced changes could be PPAR mediated (61). In type 2 diabetes prevention trials, metformin and TZDs independently diminished progression from impaired glucose tolerance to type 2 diabetes by 31% (38, 39) and 70% (62), respectively, versus a 70% decrease induced by lifestyle changes (diet and exercise) (38, 39). Despite these results in preventing progression to diabetes, the use of TZDs has been curtailed recently because of potential side effects (63, 64).
As reviewed elsewhere (65–67), many additional pharmacological AMPK activators have been identified or are in development, including several that directly activate AMPK (32). Likewise, some compounds already in use for improving glycemic control, such as GLP-1 receptor activators and DPP IV inhibitors (68), or in therapeutic trial, such as salicylates (69), have been shown to increase AMPK activity in a number of tissues in addition to their other actions.
Studies in genetically obese and fat-fed rodents
Decreased AMPK activity occurs in many genetic rodent models with a metabolic syndrome phenotype, including ob/ob mice (leptin deficient), fa/fa rats (leptin receptor deficient) and the male ZDF rat (leptin receptor deficient with a mutation in the insulin promoter) (13). In all of these rodents, therapy with the AMPK activator AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside) improved IR and glucose homeostasis (70–73). In the ZDF rat, AICAR completely prevented the development of diabetes as well as ectopic lipid deposition and degranulation of the pancreatic islet β cells (74), effects also observed in rats exercised prior to anticipated diabetes onset (75). In addition to AICAR and exercise, other AMPK activators, such as TZDs (76), metformin (77), and polyphenols, (78) have similar effects in preventing metabolic syndrome phenotypes in rodents, suggesting a common mechanism of action. Whether all the benefits of these compounds are AMPK mediated remains to be determined, but some have been tested and are discussed here.
Studies in rodents genetically deficient in whole-body, liver, and skeletal muscle AMPK
The picture that emerges from studies in rodents deficient in AMPK activity and fed a high-fat diet to produce IR is less clear. Transgenic overexpression of an inactive α2 subunit (α2i TG) and AMPK knockout models have been generated that would reduce or eliminate AMPK activity in muscle (α2, β1/β2, γ3), liver (β1), or the whole body (α2 or α1). In general, in rodents challenged with a high-fat diet, the effects have been equivocal, with some demonstrating that elimination of AMPK in muscle or liver plays little role in generating or exacerbating IR (reviewed in ref. 79), whereas others clearly show an increase in IR compared with control rodents (80, 81). Until the reasons for the differing results are resolved, the question of whether a decrease in AMPK activity predisposes to IR and the metabolic syndrome will remain controversial.
From a treatment perspective, an intriguing set of observations has recently been made by 2 groups regarding the necessity of AMPK. Um and colleagues (82) demonstrated that resveratrol, a polyphenol found in red wine and a known AMPK activator (78), prevents IR in wild-type mice placed on a high-fat diet, but not in whole-body AMPK α2 knockout mice. Likewise, under very similar conditions, Jelenik et al. (83) found that hepatic IR is no longer alleviated by dietary n-3 polyunsaturated fatty acid supplementation when AMPKα2 is not present. Whether other compounds or dietary supplements require the presence of AMPK for their anti-IR properties remains to be determined.
The AMPK-SIRT1 cycle
Sirtuins are a group of histone/protein deacetylases that are regulated by changes in the cellular redox state (NAD+/NADH ratio) and increases in nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme for NAD synthesis. Sirtuins have been evaluated intensively because of their apparent role in combating aging (84, 85). SIRT1, the most studied member of this family, responds to overfeeding, starvation, changes in energy expenditure and exercise (13), as well as to adiponectin (54), much as AMPK does, although with somewhat different timing (13). SIRT1 can activate AMPK by deacetylating the upstream kinase LKB1, which promotes LKB1 translocation from the nucleus to the cytosol, where it is activated and in turn phosphorylates and activates AMPK (86–88). Likewise, AMPK can activate SIRT1 by increasing the NAD/NADH ratio or the expression/activity of NAMPT (89). Collectively, these findings suggest the existence of an AMPK-SIRT1 cycle that links the cell’s energy and redox states (13). In addition, AMPK and SIRT1 (and most likely other sirtuins) act on common transcriptional activators and coactivators, including the mitochondrial master regulator PGC1α and members of the FoxO family. Finally, both AMPK and SIRT1 activators can decrease atherosclerosis and prevent diabetes in experimental animals (13, 90).
Beneficial actions of AMPK
AMPK activation is associated with a wide array of beneficial effects on metabolic syndrome–associated diseases. Its potential benefits in pancreatic β cells, liver, and muscle have been reviewed elsewhere (6, 7, 13, 91). Here, we will focus on adipose tissue and 2 cell types that until recently have received less attention in the context of IR, macrovascular endothelium, and leukocytes.
Vascular endothelium.
Early studies demonstrated that AMPK and IR are linked in the vascular endothelium (92). Human umbilical vein endothelial cells (HUVECs) incubated for 24 hours in high-glucose medium become insulin resistant and show decreased mitochondrial membrane potential, abnormalities that were prevented when AMPK was activated by various means (92–94). Subsequent studies showed that pharmacological AMPK activators and genetic AMPK overexpression could prevent inflammation (i.e., NF-κB pathway transactivation), oxidative stress, and apoptosis induced by incubation with the fatty acid palmitate or the inflammatory cytokine TNF-α (95, 96).
Another action of AMPK in endothelial cells in an elevated glucose setting is the phosphorylation of eNOS on serine 1177 (97). This modification enhances its ability to synthesize NO, a molecule that diminishes oxidative stress and possibly atherogenesis in the aorta (43). Likewise, statins have been shown to activate AMPK and increase NO synthesis in HUVECs and in whole aorta in mice (98), effects that may contribute to their antiatherosclerotic action not accounted for by cholesterol lowering (98, 99). Direct effects of AMPK on atherosclerosis have also been demonstrated in AMPK knockout models. For instance, loss of AMPKα2 in apoE-deficient mice exacerbates atherosclerosis when these mice are fed an atherogenic diet (100). Additionally, various AMPK activators (AICAR and polyphenols) have been shown to prevent atherosclerosis, with the natural polyphenol, berberine, working specifically through AMPK (101).
Macrophage activation and adipose tissue inflammation
Macrophage recruitment and activation (i.e., conversion or partial conversion of antiinflammatory M2 to proinflammatory M1 macrophages) are central features of chronic low-grade inflammatory diseases, including many associated with the metabolic syndrome. Adipose tissue of obese, insulin-resistant humans and experimental animals contain markedly increased numbers of proinflammatory macrophages, which are thought to be responsible both for removing dead adipocytes and for the inflammation that precedes IR (102–104). In addition, macrophage invasion and inflammation have been observed in the islets of humans and various rodents with both type 1 and type 2 diabetes (105).
AMPK was first implicated in macrophage regulation in studies showing that AMPK activators diminish inducible NOS (iNOS, also known as NOS3) synthesis in macrophages and adipocytes (106). Subsequently, Sag et al. (107) reported that treatment of macrophages with antiinflammatory cytokines, such as IL-10 and TGF-β, rapidly activated AMPK in these cells, whereas the proinflammatory stimulus LPS diminished AMPK activity. Likewise, inhibition of AMPK activity by RNAi or transfection of an inactive AMPK mutant enhanced LPS-induced increases in the inflammatory cytokines TNF-α and IL-6 and diminished IL-10 in these cells. More recently, Yang and coworkers (108) demonstrated that increased AMPK activity, caused by the expression of a constitutively active AMPKα1, inhibits both LPS and palmitate-induced NF-κB signaling in macrophages. They also observed that inactivating AMPK in macrophages in a macrophage/adipocyte coculture system inhibited both insulin signaling and glucose uptake in the adipocytes (i.e., it produced IR). Finally, the same investigators found that AMPK activation increased SIRT1 expression in macrophages and that this led to the deacetylation and downregulation of NF-κB (i.e., decreased inflammation).
The critical nature of AMPK in macrophages was recently underscored in a study examining wild-type mice that received transplanted bone marrow from AMPK β1–deficient mice and were then fed a high-fat diet (109). As a consequence of having virtually no AMPK activity in their macrophages, these mice had higher serum levels of inflammatory cytokines, more bone marrow–derived macrophage infiltration into fat pads with gene expression patterns consistent with M1-polarized cells, and increased systemic and hepatic IR.
Additional evidence of AMPK and SIRT1 linksto inflammation
Why changes in AMPK and SIRT1 exert effects on inflammation and other events that alter insulin sensitivity is incompletely understood. As recently reviewed (110), one factor may be their actions on fuel metabolism in immune cells. Increases in glucose uptake, glycolysis, and the activity of the pentose phosphate pathway (which generates NADPH), as well as decreases in AMPK and presumably sirtuin activity, have been observed in M1 macrophages and T helper cells (110), both of which are more numerous in inflamed adipose tissue (111). Conversely, noninflammatory cells, such as M2 macrophages, regulatory T cells, and quiescent memory T cells, have lower glycolytic rates and higher rates of oxidative metabolism, presumably related to their higher AMPK and SIRT1 activities. The infiltration of adipose tissue with various T cells is an early event that is critical in regulating macrophage recruitment and causing systemic IR (112). Perhaps related to these effects on inflammation, AMPK activation increases autophagy and, secondarily, mitochondrial function and prevents the inflammatory activation of adipose tissue macrophages. AMPK presumably has a similar effect on lymphocytes (112). As reviewed elsewhere, AMPK appears to act, at least in part, by phosphorylating and inhibiting ULK1, an enzyme implicated in regulating autophagy and the formation of NLR family pyrin domain–containing 3 (NLRP3) inflammasomes (5, 19, 113, 114). Upregulation of NLRP3 inflammasomes has recently been described in macrophages differentiated from monocytes obtained from the blood of obese individuals with recent-onset type 2 diabetes. Furthermore, when the patients were treated with metformin for 2 months, AMPK was activated and IL-1β maturation was decreased in the differentiated macrophages, suggesting inflammasome deactivation (115).
Inflammation and oxidative and ER stress: interactions with AMPK and sirtuins
An increasing body of work has linked inflammation to oxidative and ER stress in the pathogenesis of IR and cellular dysfunction in adipose tissue, liver, muscle, and pancreatic β cells (Figure 3). Notably, these cellular stress factors are reduced by genetic or pharmacological activation of AMPK or some sirtuins (13, 116, 117). Conversely, oxidative and ER stress and inflammation can diminish AMPK and SIRT1 activity (5, 118, 119). Whether an abnormality in one of these stressors or AMPK, SIRT1 or SIRT3, or yet other factors initiates IR and cellular damage in vivo is unclear and will require longitudinal time-course studies to sort out. What has been more clearly established is that regular exercise can chronically diminish oxidative and ER stress as well as inflammation in liver, muscle, and adipose tissue, suggesting that AMPK activation is a therapeutic target (120–122).
Figure 3. Proposed interrelations of AMPK and sirtuins 1 and 3 (SIRTs) with oxidative and ER stress and inflammation.
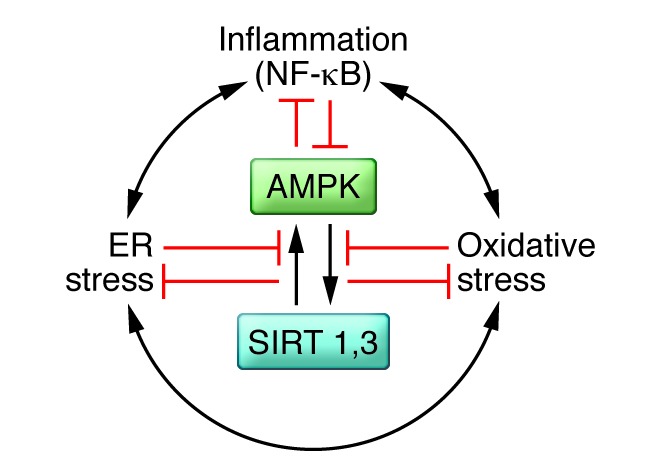
AMPK and SIRT1 both activate each other and diminish oxidative and ER stress and low-grade inflammation in various settings. Conversely, oxidative and ER stress and inflammation, which activate each other, appear to diminish AMPK and SIRT1. In principle, any of these factors could be targeted to combat IR and the development of metabolic syndrome–associated disorders; however, to date, the most success has been observed with therapies that target AMPK.
As reviewed elsewhere, AMPK exerts actions on metabolism, inflammation, and other parameters in liver, muscle, and pancreatic β cells, similar to its effects on adipose tissue and endothelium (3, 5, 13, 123, 124). In addition, AMPK exerts effects in specific regions of the CNS that regulate food intake, sympathetic nervous system activity, and circadian rhythms (2, 67). Whether and how dysregulation of AMPK in the CNS contributes to IR in the periphery have not been systematically studied; however, feeding rats and mice a high-fat diet for as little as 1 to 3 days has been shown to cause hypothalamic inflammatory signaling and subsequent gliosis (125). Furthermore, evidence of gliosis was found by MRI in the mediobasal hypothalamus of obese humans (125).
Insulin resistance and decreased AMPK in humans
The association of decreased AMPK activity and IR has been conclusively demonstrated in the adipose tissue of both very obese individuals undergoing bariatric surgery and patients with Cushing’s syndrome.
Cushing’s syndrome.
Korbonits and coworkers observed decreased AMPK activity in the visceral adipose tissue of patients with Cushing’s syndrome, most of whom had elevated plasma cortisol levels due to a functioning adrenal adenoma (14). The same investigators found that infusion of glucocorticoids into rodents also diminished AMPK activity (126). Individuals with Cushing’s syndrome are characterized by IR, increases in visceral fat, and a predisposition to both type 2 diabetes and ASCVD. Similar abnormalities are observed in patients treated with high doses of glucocorticoids for extended periods, although their effect on ASCVD is somewhat controversial (127). Interestingly, incubation of adipocytes with cortisol causes IR that is associated with increases in oxidative stress, but, as expected, not with inflammation (128).
Patients undergoing bariatric surgery.
Gauthier (15) and Xu (16) and their coworkers observed that AMPK activity is significantly diminished (30%–50%) in the adipose tissue of 75% of the severely obese individuals undergoing bariatric surgery who are insulin resistant, compared with the remaining 25% who are insulin sensitive. Such decreases in AMPK activity were observed in omental, subcutaneous abdominal, and epiploic fat and were accompanied by increased oxidative stress both in these depots and in plasma (129), as had been reported previously (130). In contrast, differences in gene expression were more depot specific. In general, increases in inflammatory genes such as IFNG, angiotensinogen (16), IL1B (131), IL8, and chemokines (132), accompanied by macrophage and lymphocyte infiltration (16, 131, 133) and decreases in the expression of genes for PGC1A (16) and SIRT1 (133) and enzymes related to β oxidation of fatty acids and the citric acid cycle (134), were observed in visceral adipose tissue of insulin-resistant compared with insulin-sensitive subjects. The strongest predictors of IR in these patients was macrophage infiltration of adipose tissue, together with decreased plasma adiponectin (133).
Implications of the findings in bariatric surgery patients.
An increasing body of evidence indicates that various bariatric surgery procedures reverse type 2 diabetes and other disorders associated with the metabolic syndrome, including dyslipidemia, hypertension, and polycystic ovary disease (19, 135). In addition, bariatric surgery diminishes long-term mortality (up to 20 years after surgery) from coronary heart disease by 30% (18) and the prevalence of solid tumors by 70% (5 years after surgery) (19). Interestingly, the effect on cardiovascular disease mortality was observed primarily in patients in the 2 highest quintiles of plasma insulin preoperatively (18) (i.e., the most insulin-resistant subjects). These patients also have low AMPK activity, increased oxidative and (presumably) ER stress, and inflammation in their adipose tissue (16).
Whether the decreases in AMPK and SIRT1 or the increases in oxidative and ER stress and inflammation disappear more rapidly following bariatric surgery remains to be determined. Also requiring study is why some obese people remain insulin sensitive, whereas the majority of them are insulin resistant. In addition to the possibilities already discussed, such factors as alterations in capillary density and permeability (136, 137), differences in collagen VI deposition (53, 138, 139), the release of LPS from bacteria by the gut microbiome (140–142), alterations of lipid droplet proteins such as FSP27 (CIDEC) that regulate the rates of lipid deposition and lipolysis in adipose tissue (19, 143), and events that cause an imbalance between nutrient load and mitochondrial function (144), need to be considered. With respect to the microbiome, AMPK activity is significantly increased in tissues of germ-free mice (145) and in mice treated with antibiotics (146), suggesting that AMPK suppression by factors released by bacteria of the gastrointestinal tract or other sites may be a normal occurrence. Finally, ER stress has not yet been compared in the adipose tissue of insulin-sensitive and -resistant obese patients. On the other hand, like oxidative stress and inflammation, ER stress diminishes over time after bariatric surgery (147).
Other tissues.
Efforts to determine whether decreased AMPK activity occurs in other tissues, most notably the skeletal muscle of humans with metabolic syndrome–associated disorders, have yielded mixed results. One group reported decreased AMPK activity in the muscle of obese, insulin-resistant patients, including some with type 2 diabetes (148), and another reported decreased AMPK activation by exercise in obese and diabetic patients (149). Others have not observed such effects, however (6, 22, 150).
Concluding remarks
The findings presented in this review strongly suggest a close link between dysregulation of AMPK and IR in both rodents and humans. AMPK activity is diminished in adipose tissue of very obese insulin-resistant people, in whom it is associated with increases in oxidative stress and more variable changes in gene expression. These changes are also associated with alterations in the release of numerous humoral factors that could contribute to IR and dysfunction in other tissues that lead to metabolic syndrome–associated diseases (Figure 1B).
What is not yet clear is whether the decreases in AMPK and SIRT1 and possibly other sirtuins are primary events that promote IR and metabolic syndrome–associated disorders or if decreased AMPK and SIRT1 are components of adaptive responses to IR, oxidative and ER stresses, and inflammation. AMPK can decrease all of these factors, and these stressors in turn can diminish AMPK activity (Figure 3). Determining causality in humans may prove difficult; however, time-course studies in tissue and plasma from severely obese individuals after bariatric surgery and similar measurements in normal-weight offspring of people with metabolic syndrome–associated diseases, who often show early evidence of IR (28), might prove useful.
Another question is: why focus on AMPK when, hypothetically, oxidative and ER stress and inflammation could be therapeutic targets? A simplistic answer is that the safety and efficacy of therapies that activate AMPK are more established. Thus, AMPK can be activated by exercise and calorie restriction, whose safety and efficacy for preventing and, to some extent, treating metabolic syndrome–associated disorders such as type 2 diabetes, hypertension, and ASCVD, are reasonably established. In addition, perhaps fortuitously, pharmacological agents developed for the treatment of type 2 diabetes, such as metformin, TZDs, GLP1 agonists, and dipeptidyl peptidase IV (DPP IV) inhibitors, have all been shown to activate AMPK, and in several instances, have demonstrated utility in preventing the progression of impaired glucose tolerance to type 2 diabetes. Finally, the identification of potent and specific AMPK activators appears to be imminent. If so, we may soon be able to determine more directly the therapeutic utility of AMPK activation for the prevention and treatment of metabolic syndrome–associated disorders.
Acknowledgments
Supported by grants from the NIH (R01 DK019514, R01 DK067509, P01 HL068758, and R24 DK094749, to N.B. Ruderman), the Medical Research Council UK (to D. Carling), and the Canadian Institute of Health Research (to M. Prentki). M. Prentki is the recipient of a Canada Research Chair in diabetes and metabolism. The authors thank Jihana Mottley, Christina Nielsen-Campbell, Catherine Rucci, and Kathryn Spielman for their assistance in preparing the manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2013;123(7):2764–2772. doi:10.1172/JCI67227.
References
- 1.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 2.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Ruderman NB, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3(4):340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- 4.Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol. 1999;276(1 pt 1):E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- 5.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med. 2011;89(7):667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 7.Viollet B, Andreelli F. AMP-activated protein kinase and metabolic control. Handb Exp Pharmacol. 2011;(203):303–330. doi: 10.1007/978-3-642-17214-4_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277(1 pt 1):E1–E10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 9.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270(2 pt 1):E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 10.Vavvas D, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272(20):13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 11.Bjorntorp P, De Jounge K, Sjostrom L, Sullivan L. The effect of physical training on insulin production in obesity. Metabolism. 1970;19(8):631–638. doi: 10.1016/0026-0495(70)90020-X. [DOI] [PubMed] [Google Scholar]
- 12.Bjorntorp P, Fahlen M, Grimby G. Carbohydrate and lipid metabolism in middle-aged, physically well-trained men. Metabolism. 1972;21(11):1037–1044. doi: 10.1016/0026-0495(72)90034-0. [DOI] [PubMed] [Google Scholar]
- 13.Ruderman NB, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4):E751–E760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kola B, et al. Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing’s syndrome. J Clin Endocrinol Metab. 2008;93(12):4969–4973. doi: 10.1210/jc.2008-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gauthier MS, et al. Decreased AMP-activated protein kinase activity is associated wtih increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404(1):382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu XJ, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53(4):792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjöström L, et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol. 2009;10(7):653–662. doi: 10.1016/S1470-2045(09)70159-7. [DOI] [PubMed] [Google Scholar]
- 18.Sjöström L, et al. Bariatric surgery and long-term cardiovascular events. JAMA. 2012;307(1):56–65. doi: 10.1001/jama.2011.1914. [DOI] [PubMed] [Google Scholar]
- 19.Xu XJ, Pories WJ, Dohm LG, Ruderman NB. What distinguishes adipose tissue of severely obese humans who are insulin sensitive and resistant? Curr Opin Lipidol. 2013;24(1):49–56. doi: 10.1097/MOL.0b013e32835b465b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman MN, Ruderman NB. Insulin sensitivity of rat skeletal muscle: effects of starvation and aging. Am J Physiol. 1979;236(5):E519–E523. doi: 10.1152/ajpendo.1979.236.5.E519. [DOI] [PubMed] [Google Scholar]
- 21.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest. 1982;69(4):785–793. doi: 10.1172/JCI110517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418(2):261–275. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diabetes.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 24.Ruderman NB, Shulman GI. Textbook of Endocrinology. 2010. The metabolic syndrome. In: Jamieson JL, DeGroot LJ, eds. pp. 822–839. 6th ed. Philadelphia, Pennsylvania: Elsevier; [Google Scholar]
- 25.Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet. 2011;378(9786):169–181. doi: 10.1016/S0140-6736(11)60614-4. [DOI] [PubMed] [Google Scholar]
- 26.Nolan CJ, Ruderman NB, Prentki M. Intensive use of insulin in type 2 diabetes: the risk of causing harm. The Lancet Diabetes & Endocrinology. doi: 10.1016/S2213-8587(13)70027-5. In press. [DOI] [PubMed] [Google Scholar]
- 27.Alberti KG, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 28.Ruderman NB, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47(5):699–713. doi: 10.2337/diabetes.47.5.699. [DOI] [PubMed] [Google Scholar]
- 29.Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27(3):813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 30.Xiao B, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472(7342):230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakhill JS, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332(6036):1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 32.Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282(45):32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- 33.Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658–31665. doi: 10.1074/jbc.R112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stahmann N, Woods A, Carling D, Heller R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. . Mol Cell Biol. 2006;26(16):5933–5945. doi: 10.1128/MCB.00383-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.[No authors listed]. Surgeon General’s report on physical activity and health. From the Centers for Disease Control and Prevention. JAMA. 1996;276(7):522. doi: 10.1001/jama.1996.03540070018010. [DOI] [PubMed] [Google Scholar]
- 36.Paffenbarger RS, Jr, Wing AL, Hyde RT. Physical activity as an index of heart attack risk in college alumni. Am J Epidemiol. 1978;108(3):161–175. doi: 10.1093/oxfordjournals.aje.a112608. [DOI] [PubMed] [Google Scholar]
- 37.Frisch RE, Wyshak G, Albright TE, Albright NL, Schiff I. Lower prevalence of diabetes in female former college athletes compared with nonathletes. Diabetes. 1986;35(10):1101–1105. doi: 10.2337/diabetes.35.10.1101. [DOI] [PubMed] [Google Scholar]
- 38.Knowler WC, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuomilehto J, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344(18):1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 40.Park H, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277(36):32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 41.Cacicedo JM, Gauthier MS, Lebrasseur NK, Jasuja R, Ruderman NB, Ido Y. Acute exercise activates AMPK and eNOS in the mouse aorta. Am J Physiol Heart Circ Physiol. 2011;301(4):H1255–H1265. doi: 10.1152/ajpheart.01279.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang QJ, McMillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD. Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: role of vascular signalling kinases. J Physiol. 2009;587(pt 15):3911–3920. doi: 10.1113/jphysiol.2009.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 44.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.[No authors listed]. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):854–865. doi: 10.1016/S0140-6736(98)07037-8. [DOI] [PubMed] [Google Scholar]
- 46.Soranna D, et al. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis. Oncologist. 2012;17(6):813–822. doi: 10.1634/theoncologist.2011-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevens RJ, et al. Cancer outcomes and all-cause mortality in adults allocated to metformin: systematic review and collaborative meta-analysis of randomised clinical trials. Diabetologia. 2012;55(10):2593–2603. doi: 10.1007/s00125-012-2653-7. [DOI] [PubMed] [Google Scholar]
- 48.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomas E, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99(25):16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi T, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 52.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26(3):439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 53.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55(6):1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- 54.Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17(2):185–196. doi: 10.1016/j.cmet.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. . Nature. 2010;464(7293):1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- 56.Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun. 2004;314(2):580–585. doi: 10.1016/j.bbrc.2003.12.120. [DOI] [PubMed] [Google Scholar]
- 57.Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277(28):25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 58.LeBrasseur NK, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291(1):E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 59.Nawrocki AR, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281(5):2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 60.Kubota N, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281(13):8748–8755. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 61.Kintscher U, Law RE. PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. Am J Physiol Endocrinol Metab. 2005;288(2):E287–E291. doi: 10.1152/ajpendo.00440.2004. [DOI] [PubMed] [Google Scholar]
- 62.DeFronzo RA, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011;364(12):1104–1115. doi: 10.1056/NEJMoa1010949. [DOI] [PubMed] [Google Scholar]
- 63.Tchernof A, Despres JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93(1):359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 64.Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445(1):11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 65.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9(5):407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 67.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Svegliati-Baroni G, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x. [DOI] [PubMed] [Google Scholar]
- 69.Hawley SA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336(6083):918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buhl ES, et al. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51(7):2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 71.Halseth AE, Ensor NJ, White TA, Ross SA, Gulve EA. Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose concentrations. Biochem Biophys Res Commun. 2002;294(4):798–805. doi: 10.1016/S0006-291X(02)00557-0. [DOI] [PubMed] [Google Scholar]
- 72.Song XM, et al. 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia. 2002;45(1):56–65. doi: 10.1007/s125-002-8245-8. [DOI] [PubMed] [Google Scholar]
- 73.Bergeron R, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol. 1999;276(5 pt 1):E938–E944. doi: 10.1152/ajpendo.1999.276.5.E938. [DOI] [PubMed] [Google Scholar]
- 74.Yu X, et al. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: prevention of diabetes and ectopic lipid deposition. Diabetologia. 2004;47(11):2012–2021. doi: 10.1007/s00125-004-1570-9. [DOI] [PubMed] [Google Scholar]
- 75.Pold R, et al. Long-term AICAR administration and exercise prevents diabetes in ZDF rats. Diabetes. 2005;54(4):928–934. doi: 10.2337/diabetes.54.4.928. [DOI] [PubMed] [Google Scholar]
- 76.de Souza CJ, Yu JH, Robinson DD, Ulrich RG, Meglasson MD. Insulin secretory defect in Zucker fa/fa rats is improved by ameliorating insulin resistance. Diabetes. 1995;44(8):984–991. doi: 10.2337/diabetes.44.8.984. [DOI] [PubMed] [Google Scholar]
- 77.Matthaei S, et al. In vivo metformin treatment ameliorates insulin resistance: evidence for potentiation of insulin-induced translocation and increased functional activity of glucose transporters in obese (fa/fa) Zucker rat adipocytes. Endocrinology. 1993;133(1):304–311. doi: 10.1210/en.133.1.304. [DOI] [PubMed] [Google Scholar]
- 78.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36(9):470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 79.O’Neill HM. AMPK and exercise: glucose uptake and insulin sensitivity. Diabetes Metab J. 2013;37(1):1–21. doi: 10.4093/dmj.2013.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dasgupta B, et al. The AMPK beta2 subunit is required for energy homeostasis during metabolic stress. Mol Cell Biol. 2012;32(14):2837–2848. doi: 10.1128/MCB.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fujii N, et al. Ablation of AMP-activated protein kinase alpha2 activity exacerbates insulin resistance induced by high-fat feeding of mice. Diabetes. 2008;57(11):2958–2966. doi: 10.2337/db07-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Um JH, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59(3):554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jelenik T, et al. AMP-activated protein kinase alpha2 subunit is required for the preservation of hepatic insulin sensitivity by n-3 polyunsaturated fatty acids. Diabetes. 2010;59(11):2737–2746. doi: 10.2337/db09-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sinclair DA, Guarente L. Unlocking the secrets of longevity genes. Sci Am. 2006;294(3):48–51. doi: 10.1038/scientificamerican0306-48. [DOI] [PubMed] [Google Scholar]
- 85.Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family’s role in aging and age-associated pathologies. J Clin Invest. 2013;123(3):973–979. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lan F, Cacicedo J, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hou X, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283(29):20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Price NL, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15(5):675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. . Nature. 2009;458(7241):1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zang M, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55(8):2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- 91.Prentki M, Madiraju SR. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol Cell Endocrinol. 2012;353(1–2):88–100. doi: 10.1016/j.mce.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 92.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51(1):159–167. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- 93.Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47(6):1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- 94.Kukidome D, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55(1):120–127. doi: 10.2337/diabetes.55.01.06.db05-0943. [DOI] [PubMed] [Google Scholar]
- 95.Cacicedo JM, Yagihashi N, Keaney JF, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324(4):1204–1209. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 96.Ruderman NB, et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans. 2003;31(pt 1):202–206. doi: 10.1042/BST0310202. [DOI] [PubMed] [Google Scholar]
- 97.Chen ZP, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443(3):285–289. doi: 10.1016/S0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 98.Sun W, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114(24):2655–2662. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- 99.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 100.Dong Y, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121(6):792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Q, Zhang M, Liang B, Shirwany N, Zhu Y, Zou MH. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: the role of uncoupling protein 2. PLoS One. 2011;6(9):e25436. doi: 10.1371/journal.pone.0025436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Inves. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 106.Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem. 2004;279(20):20767–20774. doi: 10.1074/jbc.M401390200. [DOI] [PubMed] [Google Scholar]
- 107.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181(12):8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285(25):19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Galic S, et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121(12):4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 111.Goossens GH, et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol Immunol. 2012;50(3):142–149. doi: 10.1016/j.molimm.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 112.Maciver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choi AMK, Nakahira K. Dampening insulin signaling by an NLRP3 ‘meta-flammasome’. Nat Immunol. 2011;12(5):379–380. doi: 10.1038/ni.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69(18):2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11(6):443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nogueiras R, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92(3):1479–1514. doi: 10.1152/physrev.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dolinsky VW, Chan AY, Robillard Frayne I, Light PE, Des Rosiers C, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation. 2009;119(12):1643–1652. doi: 10.1161/CIRCULATIONAHA.108.787440. [DOI] [PubMed] [Google Scholar]
- 119.Choudhury M, Qadri I, Rahman SM, Schroeder-Gloeckler J, Janssen RC, Friedman JE. C/EBPbeta is AMP kinase sensitive and up-regulates PEPCK in response to ER stress in hepatoma cells. Mol Cell Endocrinol. 2011;331(1):102–108. doi: 10.1016/j.mce.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.da Luz G, et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol. 2011;111(9):2015–2023. doi: 10.1007/s00421-010-1802-2. [DOI] [PubMed] [Google Scholar]
- 121.de Lemos ET, Oliveira J, Pinheiro JP, Reis F. Regular physical exercise as a strategy to improve antioxidant and anti-inflammatory status: benefits in type 2 diabetes mellitus. Oxid Med Cell Longev. 2012;2012:741545. doi: 10.1155/2012/741545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu J, et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab. 2011;13(2):160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.El-Assaad W, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144(9):4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 124.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 125.Thaler JP, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Christ-Crain M, et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. FASEB J. 2008;22(6):1672–1683. doi: 10.1096/fj.07-094144. [DOI] [PubMed] [Google Scholar]
- 127.La Montagna G, et al. Insulin resistance is an independent risk factor for atherosclerosis in rheumatoid arthritis. Diab Vasc Dis Res. 2007;4(2):130–135. doi: 10.3132/dvdr.2007.031. [DOI] [PubMed] [Google Scholar]
- 128.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 129.Uzun H, Konukoglu D, Gelisgen R, Zengin K, Taskin M. Plasma protein carbonyl and thiol stress before and after laparoscopic gastric banding in morbidly obese patients. Obes Surg. 2007;17(10):1367–1373. doi: 10.1007/s11695-007-9242-8. [DOI] [PubMed] [Google Scholar]
- 130.Tinahones FJ, et al. Oxidative stress in severely obese persons is greater in those with insulin resistance. Obesity (Silver Spring). 2009;17(2):240–246. doi: 10.1038/oby.2008.536. [DOI] [PubMed] [Google Scholar]
- 131.Barbarrosa N, et al. The obese healthy paradox: is inflammation the answer? Biochem J. 2010;430(1):141–149. doi: 10.1042/BJ20100285. [DOI] [PubMed] [Google Scholar]
- 132.Hardy OT, et al. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis. 2011;7(1):60–67. doi: 10.1016/j.soard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Klöting N, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299(3):E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 134.Qatanani M, et al. Inverse regulation of inflammation and mitochondrial function in adipose tissue defines extreme insulin sensitivity in morbidly obese patients. Diabetes. 2013;62(3):855–863. doi: 10.2337/db12-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Manco M. Obesity: Epidemiology, Pathophysiology, and Prevention. 2012. Bariatric surgery and reversal of metabolic disorders. In: Bagchi B, Preuss HG, eds. pp. 931–940. Boca Raton, Florida, USA: CRC Press; [Google Scholar]
- 136.Gealekman O, et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation. 2011;123(2):186–194. doi: 10.1161/CIRCULATIONAHA.110.970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goossens GH, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. 2011;124(1):67–76. doi: 10.1161/CIRCULATIONAHA.111.027813. [DOI] [PubMed] [Google Scholar]
- 138.Civitarese AE, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4(3):e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pasarica M, et al. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab. 2009;94(12):5155–5162. doi: 10.1210/jc.2009-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121(6):2126–2132. doi: 10.1172/JCI58109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152(4):673–684. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 142.Burcelin R, Garidou L, Pomie C. Immuno-microbiota cross and talk: the new paradigm of metabolic diseases. Semin Immunol. 2012;24(1):67–74. doi: 10.1016/j.smim.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 143.Puri V, et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci U S A. 2008;105(22):7833–7838. doi: 10.1073/pnas.0802063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gilliam LA, Neufer PD. Transgenic mouse models resistant to diet-induced metabolic disease: is energy balance the key? J Pharmacol Exp Ther. 2012;342(3):631–636. doi: 10.1124/jpet.112.192146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carvalho BM, et al. Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia. 2012;55(10):2823–2834. doi: 10.1007/s00125-012-2648-4. [DOI] [PubMed] [Google Scholar]
- 147.Gregor MF, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58(3):639–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55(8):2277–2285. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- 149.Sriwijitkamol A, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56(3):836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hojlund K, et al. AMPK activity and isoform protein expression are similar in muscle of obese subjects with and without type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;286(2):E239–E244. doi: 10.1152/ajpendo.00326.2003. [DOI] [PubMed] [Google Scholar]
- 151.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]