

Abstract
Hormone therapies for advanced prostate cancer target the androgen receptor (AR) ligand-binding domain (LBD), but these ultimately fail and the disease progresses to lethal castration-resistant prostate cancer (CRPC). The mechanisms that drive CRPC are incompletely understood, but may involve constitutively active AR splice variants that lack the LBD. The AR N-terminal domain (NTD) is essential for AR activity, but targeting this domain with small-molecule inhibitors is complicated by its intrinsic disorder. Here we investigated EPI-001, a small-molecule antagonist of AR NTD that inhibits protein-protein interactions necessary for AR transcriptional activity. We found that EPI analogs covalently bound the NTD to block transcriptional activity of AR and its splice variants and reduced the growth of CRPC xenografts. These findings suggest that the development of small-molecule inhibitors that bind covalently to intrinsically disordered proteins is a promising strategy for development of specific and effective anticancer agents.
Introduction
Intrinsically disordered proteins (IDPs) are prevalent in eukaryotes and are associated with cancer, diabetes, and neurodegenerative and cardiovascular disorders. The lack of structure may be throughout the entire protein, or the protein may contain substantial regions of disorder. These proteins are involved in signaling and gene regulation, with protein-protein interactions being central to their mechanism. IDPs have flexibility, thereby providing the plasticity to enable interactions with multiple partners where high-specificity and low-affinity interactions are critical for reversible binding (1). IDPs such as c-myc, p53, EWS-Fli1, and androgen receptor (AR) N-terminal domain (NTD) play central roles in cancer, thereby making them ideal targets of anticancer therapies. To our knowledge, no drug targeting an IDP has reached clinical testing, nor has the binding of any small-molecule inhibitor to an NTD of a steroid receptor ever been described.
Prostate cancer recurs in 20%–40% of patients with high-grade disease after primary treatment. For these patients, androgen ablation therapy is employed, using approaches that target the AR ligand-binding domain (LBD), including antiandrogens that all directly bind LBD, or that reduce levels of circulating and tissue androgens with LHRH/GnRH analogs and CYP17 inhibitors (2). Although these therapies are initially effective in 90% of patients, the disease will inevitably recur as lethal castration-resistant prostate cancer (CRPC). In spite of castrate levels of androgen, development of CRPC is considered to be causally related to continued transactivation of AR by mechanisms that may include amplification or overexpression of AR (3, 4), gain-of-function mutations that allow AR to be activated by steroids or antiandrogens (5, 6), ligand-independent activation of the AR NTD by interleukin-6 or kinases (7–10), overexpression of AR coactivators (11–14), intracrine signaling by increased intratumoral androgens (15), and expression of constitutively active splice variants of AR that lack the C-terminal LBD and are correlated with poor prognosis (16–19). Patients succumb to metastatic CRPC usually within 2 years of onset. In vivo proof-of-principle demonstration of therapeutic response by targeting the AR NTD in CRPC was first shown with decoy proteins (20), and then with EPI-001, a small molecule that inhibits transactivation of AR NTD (21).
AR is a member of the steroid receptor family of transcription factors that share structurally conserved domains consisting of a DNA-binding domain (DBD), LBD, NTD, and a hinge region that contains a nuclear localization sequence. Unlike the intrinsically disordered NTD, the DBD and LBD of AR are intrinsically ordered with resolved crystal structures. Consistent with the properties of IDPs, AR interacts with more than 160 proteins (22), and protein-protein interactions with the activation function-1 (AF1) region in the NTD are essential for AR transcriptional activity (23–26). AR NTD has less than 15% homology with other steroid receptors that also have predominantly intrinsically disordered NTDs (27–31). Malleability of intrinsically disordered NTDs of these transcription factors is crucial for their function that requires interactions with many binding partners. Since AR NTD lacks enzymatic activity or rigid binding clefts for receptor-ligand interaction, small-molecule inhibitors would work by disruption of essential protein-protein interactions from active transcriptional complexes. The AR transcriptional complex is composed of many proteins, including CBP and RAP74 (26, 32). Our previous investigation showed that EPI-001 inhibits these protein-protein interactions by attenuation of AR transcriptional activity, increased apoptosis, and decreased proliferation, all of which are essential for CRPC tumor maintenance (21). Small-molecule inhibitors of the AR NTD may overcome the shortcomings of current therapies targeting the AR LBD for CRPC and represent the first in a new class of antitumor therapies against IDPs being clinically developed.
Targeting IDPs by small molecules to block protein-protein interactions is a rapidly evolving field, as the importance of these proteins in disease becomes established. The plasticity of IDPs with labile regions that can be shaped by their environment and interactions provides potential for small-molecule binding (33). However, the general property of reversible, low-affinity binding of IDPs to many interacting partners to facilitate the exchange of binding partners may forecast a requirement of irreversible binding for any small-molecule inhibitor to have a sustained therapeutic effect. On the basis of these observations, the mechanism of targeting the AR NTD by EPI-001 and its analogs may provide precedent in drug development against other IDPs. Here, we showed that EPI (a) bound covalently to AF1 in the intrinsically disordered AR NTD and did not bind to denatured AF1; (b) had no stereospecificity for covalent binding to AR in living cells; (c) inhibited constitutively active AR splice variants lacking LBD that are suspected in resistant mechanisms to current therapies; (d) was unique from antiandrogens, in that EPI did not cause AR nuclear translocation and its efficacy was not compromised by elevated levels of androgen; and (e) had excellent pharmacokinetic properties. These findings suggest that EPI compounds are promising small molecules to develop therapeutics for CRPC.
Results
EPI has a unique mechanism of action.
EPI-001 is an effective and specific inhibitor of AR transcriptional activity (21). EPI-001 has 2 chiral centers and is a mixture of 4 stereoisomers, EPI-002 (2R, 20S), EPI-003 (2S, 20R), EPI-004 (2R, 20R), and EPI-005 (2S, 20S) (Figure 1A). Consistent with EPI compounds targeting the NTD, inhibition of AR activity could not be competed away with increasing concentrations of androgen, as shown with endogenous AR in LNCaP human prostate cancer cells transfected with the AR-driven PSA(6.1kb)-luciferase reporter, which is induced by the synthetic androgen R1881 (Figure 1B). Antiandrogens, such as bicalutamide and MDV3100, bound to the AR LBD to act as competitive inhibitors of androgen. As expected, when the concentrations of R1881 were increased, the ability of bicalutamide to inhibit AR activity was significantly reduced. At R1881 concentrations of 1–5 nM, bicalutamide (10 μM) completely blocked AR activity, measured as PSA-luciferase activity. However, at 50 nM R1881, this same concentration of bicalutamide was a poor inhibitor, at only approximately 30% inhibition. EPI-001 (25 μM) inhibited AR activity consistently, regardless of increasing levels of androgen, and at 50 nM R1881, EPI-001 still inhibited AR activity by approximately 80%. Elevated androgen level also reverses the inhibitory effects of MDV3100 on androgen-dependent proliferation of VCaP cells (34). This general property of antiandrogens competing with androgen for the LBD may forecast their potential failure when androgen becomes elevated in CRPC and also with resistance to abiraterone (15, 35). These data support that EPI does not bind to the AR LBD, consistent with data from the fluorescent polarization assay showing no competition with the fluoromone for the AR LBD (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI66398DS1), as previously reported (21). Moreover, no binding to LBD was detected with related steroid hormone receptors, as expected, based on previous studies confirming specificity of EPI-002 for blocking transcriptional activity of AR at concentrations that had no effect on the transcriptional activities of related steroid hormone receptors (21).
Figure 1. Unique mechanism of action of EPI compared with antiandrogens.

(A) Structures of EPI-001 mixture and stereoisomers. (B) AR transcriptional activity, measured in LNCaP cells transiently transfected with the PSA(6.1kb)-luciferase reporter and treated with vehicle (DMSO), 10 μM bicalutamide (BIC), or 25 μM EPI-001 for 1 hour followed by increasing concentrations of R1881 for 48 hours. (C) Effect of bicalutamide (0.1–3.5 μM) and EPI-001 (1–35 μM), alone or in combination (1:10 ratio), on androgen-induced AR transactivation in LNCaP cells transfected with the PSA(6.1kb)-luciferase reporter. (D) Nuclear translocation of AR in LNCaP cells transfected with AR-YFP in serum-free conditions for 24 hours prior to treatment with 1 nM R1881, 10 μM bicalutamide, 10 μM MDV3100, 25 μM EPI-001, or 25 μM EPI-002 for 4 hours. DAPI staining shows the location of the nucleus. Scale bars: 10 μm. Data are mean ± SEM. *P < 0.05; **P < 0.01; #P < 0.001.
Androgen and antiandrogens bind the structured LBD that is accessible for binding ligands due to interaction with chaperones. The AR NTD, an IDP, interacts with many proteins, which suggests that the binding site of EPI may not be continuously accessible throughout the cell cycle. Since the AR LBD and the AR NTD represent completely different drug targets, one being a structured binding pocket and the other being an IDP, this would suggest that drug combinations may yield additive or synergistic responses. Consistent with this theory, a drug combination study using a fixed ratio (1:10) of EPI-001 (1–35 μM) with a suboptimal concentration of bicalutamide (0.1–3.5 μM) significantly improved inhibition of androgen-induced AR activity compared with inhibition by the individual inhibitors (Figure 1C). A suboptimal concentration of bicalutamide (0.5 μM) inhibited androgen-induced AR activity by approximately 27%, similar to the 22% inhibition achieved with 5 μM EPI-001. A cocktail of 0.5 μM bicalutamide and 5 μM EPI-001 significantly reduced AR activity by 70%, thereby supporting combination therapy as a potential strategy for targeting both LBD and NTD.
Another mechanism potentially underlying clinical failure of antiandrogens may involve nuclear translocation of AR. In the absence of androgen, AR is predominantly cytosolic. Antiandrogens, including MDV3100 and ARN-509, induce nuclear translocation of AR (34, 36, 37). As expected, R1881, bicalutamide, and MDV3100 all induced AR nuclear translocation, whereas EPI-001 and EPI-002 did not, with AR remaining in the cytosol (Figure 1D). These data support that EPI-001 has a different mechanism of action compared with antiandrogens and highlight aspects of antiandrogens that may contribute to their clinical failure.
Optimal chirality of EPI for inhibition of AR transcriptional activity.
Drug enantiomers and/or stereoisomers are considered different chemical compounds that may vary considerably in potency, pharmacological activities, off-targets, and pharmacokinetics. In fact, with a mixture of 4 stereoisomers, as much as 75% of the mixture could be considered contaminants, with potentially only 1 stereoisomer possessing the desirable qualities necessary for efficacy. Due to the potential differences in biological activity among stereoisomers, the FDA requires that each stereoisomer be evaluated when developing chiral drugs. Therefore, dose response curves using PSA-luciferase reporter were used to calculate IC50 values for EPI-001 and each stereoisomer (Supplemental Figure 1B). EPI-001 had an IC50 of 12.63 ± 4.33 μM, whereas the value for EPI-002 was 7.40 ± 1.46 μM (Supplemental Table 1). Significant differences between stereoisomers were only observed between EPI-002 and EPI-003 and between EPI-002 and EPI-004. Reporter specificity was investigated using 3 well-characterized AR-driven reporter gene constructs that included PSA-, probasin- (PB-), and ARR3-luciferase reporters. All stereoisomers inhibited the transcriptional activity of AR, as measured using these reporters (Figure 2A). Significant differences compared with EPI-001 were shown for EPI-002 with PB-luciferase, EPI-005 with PB-luciferase, and EPI-005 with ARR3-luciferase. EPI-002 and EPI-005 decreased AR activity to approximately 24% for PSA(6.1kb)-luciferase and 40% for PB-luciferase; for ARR3-luciferase, EPI-002 inhibited AR activity to 61%, whereas EPI-005 inhibited AR activity to 38% (Supplemental Table 1). All stereoisomers inhibited transactivation of the AR NTD induced by forskolin (Figure 2B).
Figure 2. Stereospecificity of EPI-001 on AR transcriptional activity.

(A) LNCaP cells were transfected with luciferase reporters and treated with 1 nM R1881 for 48 hours. Data represent percent of control (DMSO). **P < 0.01, #P < 0.001 vs. DMSO; §P < 0.05 vs. EPI-001. (B) AR NTD transactivation assay in LNCaP cells treated with indicated concentrations of EPI-001 stereoisomers prior to treatment with 50 μM forskolin or DMSO. (C) Inhibition of androgen-induced DNA synthesis in LNCaP cells by stereoisomers of EPI-001 (25 μM) or bicalutamide (10 μM) treated with 0.1 nM R1881 for 48 hours. Data represent percent S-phase cells staining positive for BrdU incorporation (bivariate flow cytometric) from a representative experiment. (D) Effects of EPI-002 on androgen-dependent proliferation of LNCaP cells treated with R1881 compared with PC3 cell viability. (E) Decrease of CRPC LNCaP tumor volume in castrated mice administered EPI-001 mixture and stereoisomers (i.v. 50 mg/kg body weight) every other day for a total of 7 doses. Bicalutamide (10 mg/kg body weight) was administered daily by oral gavage. (F) Comparison of tumor volume from treatment with single stereoisomers. (G) Percent change of tumor volume of individual animals treated with stereoisomers or bicalutamide. (H) Body weight change at day 14 versus day 0. (I) mRNA levels of full-length AR and androgen-regulated genes measured from the LNCaP xenografts. Intact, noncastrated control group (n = 3). Values were normalized to housekeeping gene RPL13A. Data are mean ± SD (A and B) or mean ± SEM (D–F, H, and I). *P < 0.05; **P < 0.01; #P < 0.001.
EPI analogs decrease proliferation and S-phase.
Cell cycle analysis was performed on androgen-dependent growth of LNCaP cells in response to EPI. In the absence of EPI analogs (i.e., DMSO vehicle), approximately 21% of cells were in S-phase in response to androgen (Supplemental Table 2). BrdU uptake in S-phase cells was decreased about 2-fold or more after exposure to each stereoisomer, with a concomitant increase of cells in G1-phase (Figure 2C and Supplemental Table 2). There were no statistical differences among the individual stereoisomers, with each inhibiting androgen-dependent DNA synthesis associated with proliferation. The specificity of EPI-001 for blocking AR-dependent growth, while having no effect on the proliferation of cells that do not depend on AR for growth and survival, was previously reported (21). Here, EPI-002 also had no effect on the viability of PC3 human prostate cancer cells that do not express functional AR, at concentrations that reduced AR-dependent proliferation of LNCaP cells (Figure 2D).
Stereoisomers of EPI-001 inhibit CRPC.
In vitro, EPI-002 and EPI-005 were the most potent stereoisomers in their ability to block AR transcriptional activity depending upon the reporter. To determine whether these in vitro responses could predict superior antitumor activity in vivo, the LNCaP CRPC xenograft model was used. All EPI analogs significantly inhibited CRPC tumor growth compared with DMSO control (Figure 2E). Consistent with in vitro responses, EPI-002 as well as EPI-005 had better antitumor activity compared with EPI-003 and EPI-004 (Figure 2F). EPI-002 and EPI-005 both have the S configuration for the chlorohydrin, whereas EPI-003 and EPI-004 have the R configuration. Tumor regression was attained in 60% of animals treated with EPI-002 and EPI-005, although EPI-002 caused greater regression and was superior to that achieved with bicalutamide (Figure 2G). The 10-mg/kg daily oral dose of bicalutamide has been previously shown to be effective in LNCaP xenografts (37). No significant loss of body weight was measured in animals treated with EPI-002, in contrast to EPI-005 and the other stereoisomers (Figure 2H). This difference in effect on body weight was the criteria for focusing on EPI-002 rather than EPI-005 in subsequent studies.
In vitro, EPI-001 blocks transcription of androgen-regulated genes in response to R1881 (21). Levels of expression of these genes were examined using xenografts from castrated hosts treated for 14 days with EPI-001, EPI-002, and bicalutamide. Under castrated conditions, no significant changes in levels of full-length AR, PSA (also known as KLK3), KLK2, and FKBP5 transcripts were observed in xenografts treated with EPI-001 and EPI-002 compared with DMSO in castrated hosts (Figure 2I and Supplemental Figure 2). However, levels of NKX3.1 and TMPRSS2 transcripts were significantly decreased with EPI-001 and EPI-002 (Figure 2I). RHOU, SLC41A1, GOLPH3, and PAK1IP1 were all significantly decreased with both bicalutamide and EPI-002, but not with EPI-001 (Supplemental Figure 2).
EPI chlorohydrin analogs covalently and specifically bind AR in living cells.
EPI compounds that have a chlorohydrin group are active while those analogs that lack the chlorohydrin such as BADGE.2H2O are inactive (21). The chlorohydrin group of EPI compounds may be required for activity to block AR transcriptional activity, and its chemical structure suggests a possible mechanism of covalent binding. To elucidate the mechanism of binding of EPI compounds to the AR and potentially other cellular proteins, cells were incubated with modified EPI probes containing an alkyne group to allow for Click-chemistry to add biotin to the EPI probe, followed by SDS-PAGE and Western blot analysis (Figure 3A). Whereas modified EPI probes containing the chlorohydrin group (Figure 3B) were active in cells and inhibited AR activity, EPI compounds lacking the chlorohydrin had relatively poor activity (Supplemental Figure 3 and Supplemental Table 3). LNCaP cells were exposed to EPI probes for 24 hours before lysing and Click-chemistry. Nonchlorinated EPI-051 and EPI-063 were negative controls, by analogy with the inactive analog BADGE.2H2O (21). SDS-PAGE disrupts noncovalent interactions and is used to determine covalent binding. Western blot analysis using an antibody against biotin revealed a band corresponding to AR that was specific to EPI-046– and EPI-047–treated samples and was not present in the whole cell lysates of cells treated with DMSO or the inactive analog EPI-051 that lacks the chlorohydrin (Figure 3C, left, red outline). Lack of biotin bands detected in DMSO-treated and EPI-051–treated cells were not due to nondetectable levels of AR, as shown when the membrane was reprobed with an antibody against AR (Figure 3C, middle). EPI probes with the chlorohydrin, such as EPI-054 — closest to the structure of EPI-002 — did not bind an abundance of other cellular proteins (Figure 3D, top). Only 3 bands between 200 and 75 kDa were detected using an antibody to biotin that were unique to EPI-054 treatment compared with DMSO. Confirmation that the protein band at 110 kDa corresponded to AR was shown by detection of AR pulled down from streptavidin beads only in lanes treated with EPI-054, not from DMSO-treated cells (Figure 3D, bottom). Together, these data support the notion that the biotinylated band detected at 110 kDa with EPI-054 treatment corresponded to AR. All EPI probes with a chlorohydrin, regardless of chirality, bound covalently to full-length AR (FL-AR), while the nonchlorinated analogs did not (Figure 3E). Confirmation that chlorinated EPI probes interacted with the NTD was obtained using cells transfected with FLAG-tagged chimera of AR NTD (Figure 3F). Together, these data support that EPI analogs containing a chlorohydrin covalently bind to AR NTD in cells.
Figure 3. Covalent binding of EPI-001 probes to AR in cells.

(A) Click-chemistry experiment. (B) EPI probes used for Click-chemistry. (C) LNCaP cells treated with EPI-046, EPI-047, and EPI-051 were lysed prior to Click-chemistry. Biotin-labeled probes covalently bound to cellular proteins were detected using an antibody against biotin (left), and AR protein was detected using an antibody against AR (middle). β-actin is a loading control (right). Dotted red outlines denote AR and β-actin bands, as indicated. (D) Western blot analysis of biotin-labeled probes covalently bound to cellular proteins from LNCaP cells treated with EPI-054 before (right) or after Click-chemistry and streptavidin enrichment (left). Gels were probed using anti-biotin or anti-AR antibodies, as indicated. Arrows denote bands corresponding to AR (110 kDa). (E) LNCaP cells treated with EPI chiral probes were lysed prior to Click-chemistry, enriched using streptavidin, and probed using anti-biotin antibody (top). AR levels prior to Click-chemistry were detected using anti-AR441 antibody (input; bottom). (F) Cells transfected with FLAG-AR NTD or vector were treated with EPI analogs. Proteins were detected using anti-biotin, anti-AR NTD, or anti-FLAG antibodies following Click-chemistry and streptavidin enrichment (top) or cell lysates before Click-chemistry (input; middle and bottom). Asterisks denote proteins that were also detected with anti-biotin antibody in DMSO samples not treated with EPI-probes. Lanes in D–F were run on same gel but were noncontiguous (black lines).
Chemical mechanism of EPI binding to AR AF1.
As demonstrated above, EPI analogs with a chlorohydrin covalently bound to AR in cells. To further elucidate the chemical mechanism of binding, EPI-054 (chlorohydrin) or inactive EPI-063 (no chlorohydrin) was incubated with purified recombinant AF1 protein under cell-free conditions prior to Click-chemistry to add fluorescein to the EPI probe, followed by SDS-PAGE and detection of the fluorescent band corresponding to AF1 protein. The ratio of AR AF1 protein to EPI analog was examined as well as binding time. After 1 and 20 hours of binding reaction, EPI-054 covalently bound to AF1 in a dose-dependent manner, in contrast to EPI-063 (Figure 4A). Quantification of the fluorescein/AF1 complex normalized to the corresponding coomassie blue band for each lane is also shown graphically from 4 separate experiments. After 20 hours, the binding reaction with EPI-054 was not significantly increased with a 1:10 AF1/EPI-054 ratio compared with the binding achieved with a 1:5 ratio. Even after 20 hours, the amount of covalent binding was relatively low compared with the total amount of AF1 protein available in each lane.
Figure 4. Chemical mechanism of EPI binding to AF1.

(A) AF1 protein was incubated with EPI-054, EPI-063 (inactive), or DMSO (vehicle) at the indicated molar ratios on ice for 1 hour or 20 hours, prior to Click-chemistry for fluorescein labeling, SDS-PAGE, and detection of fluorescein-labeled probe covalently bound to AF1. Quantification of fluorescein band intensity, normalized to Coomassie blue bands, is also shown. The value from each EPI condition was normalized to the value of DMSO for each individual experiment (n = 4 separate experiments). (B) AF1 protein was incubated with DMSO, EPI-054, EPI-063, EPI-056, or EPI-096 (AF1/EPI 1:3 molar ratio) at 25°C for 18 hours, prior to Click-chemistry for fluorescein labeling, SDS-PAGE, and detection of fluorescein-labeled probe covalently bound to AF1. (C) EPI analogs do not alkylate glutathione or mercaptoethanol. A mixture of glutathione (127 μM) and EPI-001 (25 μM), or a mixture of EPI-001 (55 μM) and 2-mercaptoethanol (155 μM), was monitored by proton and carbon NMR over a period of 7 days. There was no evidence for reaction of EPI-001 with either glutathione or mercaptoethanol. (D) Steady-state spectra of 1 μM recombinant wild-type AF1 protein in buffer, buffer plus 2.9 μM EPI-001, buffer plus 6 M urea, or buffer plus 6 M urea and 2.9 μM EPI-001. (E) Prostate weights from mice treated with DMSO (i.v.), EPI-093 or EPI-002 (50 mg/kg body weight; i.v.), or bicalutamide (10 mg/kg body weight; gavage daily) for 14 days. Data are mean ± SEM. There was no significant difference between EPI and bicalutamide. #P < 0.001.
EPI-054, which has the same absolute configuration as EPI-002, contains the chlorohydrin substructure found in EPI-001 and its stereoisomers. EPI-063 is a mixture of stereoisomers that are simply missing the primary chloride that is present in EPI-001. EPI-096 is missing the secondary alcohol component of the chlorohydrin that is present in the EPI-001 stereoisomers, and EPI-056 has the chlorohydrin converted to an epoxide (Figure 3B). Importantly, although EPI-054 bound covalently to the AF1 protein, the reaction was slow and never reached completion during the experiment exposure times, whereas the epoxide containing probe EPI-056 reacted quickly and gave a much higher yield of covalent adduct (Figure 4B). Thus, EPI-054 bound covalently, and neither EPI-063 (which is simply missing the chloride functionality) nor EPI-096 (which is simply missing the secondary alcohol) bound covalently, to the AF1 protein (Figure 4B). These results demonstrate that the entire chlorohydrin substructure in the EPI-001 series was required for covalent binding, while a simple primary chloride, as found in EPI-096, was not sufficient. EPI analogs with chlorohydrins were not random alkylating agents, as shown by the lack of adducts when incubated with glutathione or mercaptoethanol (Figure 4C).
AR AF1 is intrinsically disordered, with approximately only 16% predicted α-helix secondary structure (27). To determine whether EPI can bind to denatured AF1 protein or whether EPI requires the limited secondary structure for binding, the putative helical regions within AF1 were disrupted using urea, and changes in conformation of the AF1 protein were measured by steady-state fluorescence, which could be altered by both reversible and irreversible interaction with EPI-001. The steady-state fluorescence spectrum of AF1 denatured by urea showed a distinct peak for tyrosine and red shift for tryptophan (i.e., 343 nm to 350 nm), indicative of the tryptophan becoming more solvent exposed and the polypeptide being unstructured (28). Consistent with a requirement for some structure in the AF1 protein in order for EPI to bind, EPI-001 failed to bind to denatured AF1 protein to alter the steady-state spectrum. Instead, this spectrum of EPI with denatured AF1 was similar to the AF1 spectrum in urea without EPI-001, with a λmax for tryptophan of 349 nm and a distinct peak for the tyrosine emission (Figure 4D). These results suggest that some secondary structure of AF1 is necessary for EPI-001 to bind. Finally, to provide an indication of whether the chlorohydrin group of EPI analogs may be necessary for in vivo activity, loss of weight of androgen-dependent tissue in mature male mice was examined, since this is the gold standard for on-target activity of drugs targeting the AR. Consistent with the requirement of a chlorohydrin and covalent binding for in vivo activity, only EPI-002 caused a significant reduction in prostate weight compared with DMSO control, similar to the reduction seen with bicalutamide, whereas EPI-093 had no significant effect (Figure 4E).
EPI inhibits constitutively active AR splice variants.
Constitutively active AR splice variants that lack LBD have been shown in clinical samples of CRPC (16–19, 38). Antiandrogens that bind the AR LBD do not inhibit the activity of ARv567es, which lacks LBD and is constitutively both nuclear and active (17). Variant ARv567es is solely expressed in 20% of metastases and coexpressed with FL-AR in approximately 60% of CRPC metastases (17). Expression of ARv567es in COS-1 cells, which lack endogenous AR, resulted in elevated PB-luciferase activity that was not altered by R1881, as previously reported (17). EPI-001 effectively attenuated ARv567es activity (Figure 5A). A mixed population of FL-AR with ARv567es was next examined in LNCaP cells. In the absence of ARv567es, MDV3100 at both 1 and 10 μM inhibited FL-AR induced by androgen, as measured with ARR3-luciferase reporter (Figure 5B, left). However, MDV3100 had no effect in blocking AR activity, either in the presence or absence of androgen, when ARv567es was introduced into LNCaP cells (Figure 5B, right). Consistent with the results obtained with MDV3100 using ARR3-luciferase reporter in the presence of ARV567es, bicalutamide also had no effect on AR activity in LNCaP cells expressing both FL-AR and variant ARv567es, as measured with PSA(6.1kb)-luciferase reporter. EPI-001 showed good activity against FL-AR as well as mixed populations of FL-AR with variant ARv567es (Figure 5C). Thus, unlike the antiandrogens MDV3100 and bicalutamide, EPI inhibited FL-AR, ARv567es, and mixtures of FL-AR and ARv567es. Direct comparison of EPI, bicalutamide, and MDV3100 on solely endogenous FL-AR or endogenous FL-AR combined with ARv567es using the PB-luciferase reporter in LNCaP cells in the presence and absence of androgen additionally confirmed the efficacy of EPI to significantly inhibit AR activity under conditions in which bicalutamide and MDV3100 failed to have any significant effect (Figure 5D). Western blot analysis using an antibody against the AR NTD confirmed the approximate 1:1 ratio of FL-AR to ARv567es in whole cell lysates of LNCaP cells treated with EPI, MDV3100, and bicalutamide (Figure 5E). Thus, EPI analogs are the first reported inhibitors of constitutively active AR splice variants.
Figure 5. EPI inhibits splice variant ARv567es.

(A) COS-1 cells were transfected with PB-luciferase reporter and the ARv567es variant and treated with DMSO or 25 μM EPI-001 plus 1 nM R1881 for 24 hours. (B) ARR3-luciferase activity in LNCaP cells with endogenous FL-AR (left) or with both FL-AR and ARv567es (right), with or without MDV3100 (1 or 10 μM). (C) PSA(6.1kb)-luciferase activity in LNCaP cells with endogenous FL-AR (left) or with both FL-AR and ARv567es (right). Cells were treated with DMSO, 25 μM EPI-001, or 10 μM bicalutamide with or without 1 nM R1881 for 48 hours. (D) PB-luciferase activity in LNCaP cells with endogenous FL-AR (left) or with both FL-AR and ARv567es (right). Cells were treated with 25 μM EPI-001, 10 μM bicalutamide, and 5 μM MDV3100 for 1 hour prior to treatment with 1 nM R1881 for 24 hours. (E) Protein levels of FL-AR and ARv567es from samples in D, detected using AR-N20 antibody. Data are mean ± SEM (A and B) or mean ± SD (C and D). n = 3 separate experiments. *P < 0.05; **P < 0.01; #P < 0.001.
EPI-002 inhibits the growth of CRPC xenografts that express AR splice variants.
The effect of EPI-002 on CRPC that express AR variants and FL-AR was investigated using VCaP xenografts and oral dosing in castrated hosts. VCaP cells exhibited amplified FL-AR and expressed AR variant within 14 days after castration. Pharmacokinetic studies indicated that EPI-001 had 86% bioavailability, a half-life of approximately 3.3 hours, and a slow clearance rate of 1.75 l/h/kg; moreover, blood levels of 10 μg/ml (25 μM) were achieved, which was the effective concentration in vitro (Supplemental Figure 4 and Supplemental Table 4). VCaP tumor volume in animals treated with EPI-002 was significantly less than the control and bicalutamide-treated groups (Figure 6, A and B). Animals treated daily for 28 days showed no changes in behavior and only minor body weight loss (Figure 6C). Weight loss with EPI-002 may be associated with frequent (twice daily) gavage and/or slight toxicity.
Figure 6. Oral dosing of EPI-002 blocks AR transcriptional program and inhibits growth of VCaP CRPC xenografts that express AR splice variants.

(A) VCaP tumor growth in castrated mice administered EPI-002 (200 mg/kg body weight) or bicalutamide (10 mg/kg body weight) daily by gavage for a total of 28 doses. Tumors were harvested 2 days after the last treatment. (B) Photographs of tumors harvested at day 28 from animals as in A. Scale bars: 10 mm. (C) Body weight change over the duration of the experiment. (D) Transcript levels of FL-AR and AR variants (V7, V567es) normalized to RPL13A using total RNA isolated from VCaP xenografts from castrated hosts treated with bicalutamide (n = 8), EPI-002 (n = 8), or DMSO control (CMC; n = 7) for 28 days. (E) Protein levels of AR and AR variants from harvested xenografts treated with EPI-002 or bicalutamide or vehicle control. Quantification of protein bands (FL-AR and AR variant), normalized to β-actin, is also shown. (F) Transcript levels of UBE2C, AKT1, CDC20, CYCLINA2, PSA, and ERG, normalized to levels of RPL13A. (G) Proliferation (Ki67) and apoptosis (caspase-3) index, measured in harvested VCaP xenografts. Data are mean ± SEM. *P < 0.05; **P < 0.01; #P < 0.001.
Contrary to abiraterone and MDV3100, which increase levels of both FL-AR and variant ARs (35, 38), EPI-002 did not increase either transcript or protein levels of FL-AR and splice variant ARs in harvested tumors (Figure 6, D and E). The transcriptional program associated with expression of AR variant was blocked in vivo by EPI-002, as supported by significantly decreased levels of UBE2C, AKT1, CDC20, and CYCLINA2 transcripts in harvested tumors (Figure 6F). AR selectively upregulates expression of these M-phase cell cycle genes in CRPC (39), and their expression is associated with increased levels of AR variant in CRPC bone metastases (19). Bicalutamide inhibited FL-AR and had no effect on AR splice variants lacking LBD, and thus had no significant effect on the transcript levels of these M-phase genes (Figure 6F). Expression of genes regulated by FL-AR revealed that after castration and 28 days of treatment, neither bicalutamide nor EPI-002 had any significant effects on PSA, TMPRSS2-ERG, or FKPB5 transcripts compared with DMSO (Figure 6F and data not shown). ERG transcript levels were significantly decreased with EPI-002, whereas bicalutamide did not achieve statistical significance (Figure 6F). Consistent with EPI-002 decreasing tumor volume and having an inhibitory effect on expression of M-phase genes associated with variant AR and CRPC, EPI-002 also significantly decreased proliferation and increased apoptosis, whereas bicalutamide had no significant effect, as determined by immunohistochemistry of sections of xenografts stained for Ki67 or caspase-3 (Figure 6G and Supplemental Figure 5).
Discussion
AR NTD is a unique therapeutic target for CRPC and potentially other diseases of the androgen axis. Functional NTD is necessary for AR transcriptional activity (23–25). The small molecule EPI-001 is a mixture of 4 stereoisomers that inhibits protein-protein interactions with CBP and RAP74 (21) that are required for AR transcriptional activity (26, 32). Here, our preclinical study of EPI-001 revealed (a) no stereospecificity in binding of stereoisomers to AR, although both in vitro and in vivo, the single stereoisomer EPI-002 (2R, 20S) had improved properties compared with other stereoisomers; (b) the chlorohydrin was required for covalent binding of EPI analogs to AF1 in the AR NTD; (c) EPI covalent binding was specific for AR; (d) EPI-001 did not bind to denatured AF1; (e) EPI-001 and EPI-002 inhibited a constitutively active AR splice variant that lacks LBD; (f) oral delivery of EPI-002 reduced the growth of CRPC xenografts expressing the AR variant; and (g) AR transcriptional program was blocked in vivo by EPI-002. The lead compound EPI-002 showed that AR NTD could be blocked, with a detrimental effect on CRPC. These findings revealed that small-molecule inhibitors can be developed against IDPs, such as the AR NTD, with excellent in vivo pharmacokinetics, efficacy, and specificity.
In vitro, stereoisomers with 20S chlorohydrin (EPI-002 and EPI-005) were significantly better in blocking AR transcriptional activity, depending on the reporter gene construct, than the 20R stereoisomers. Reporter specificity potentially involves recruitment of different binding partners to AR on androgen response elements (AREs). Since EPI inhibits protein-protein interactions, together, these data indicate that some androgen-regulated genes may have more sensitivity to EPI stereoisomer configuration. In vivo, stereoisomer EPI-002 had superior antitumor activity compared with the other stereoisomers and the EPI-001 mixture, which may reflect potential differences in EPI stereoisomers on the transcriptional program. This notion is supported by the finding that EPI-002 achieved statistical significance for decreasing RHOU, SLC41A1, GOLPH3, and PAK1IP1, whereas the EPI-001 mixture did not, although differences in pharmacokinetic properties may also be involved. AR-regulated gene expression substantially differs between VCaP and LNCaP cells in vitro in response to androgen and AR silencing (40). This may be due to the fact that VCaP cells have 5 extra copies of the AR gene (41) and 11-fold more AR mRNA than LNCaP cells (40); that VCaP cells express AR variants that have unique transcriptomes, while parental LNCaP cells do not express variant, but have a mutated AR and cell-specific differences in coregulators and signaling pathways; or that VCaP cells express the AR-regulated TMPRSS2-ERG fusion (42); or it may be due to differences in cellular/intratumoral levels of androgen. Differences observed here between gene expression profiles in VCaP and LNCaP xenografts from castrated hosts in response to bicalutamide and EPI may also involve the different times of analysis after castration and drug treatment. LNCaP xenografts were harvested from hosts 21 days after castration and 14 days of drug treatment, while VCaP xenografts were harvested 35 days after castration and 28 days of treatment. Levels of PSA were not further decreased after castration by EPI compounds in either xenograft, whereas bicalutamide had an effect in LNCaP, but not VCaP, xenografts. PSA mRNA is not a sensitive marker of AR action (40), nor have PSA mRNA levels proved reliable as a prognostic marker for prostate cancer (43), in spite of serum levels of PSA being one of the best biomarkers used in oncology. Instead, 2 other well-characterized AR-regulated genes, NKX3.1 and TMPRSS2, were significantly decreased by EPI in LNCaP xenografts from castrated hosts. Importantly, EPI-002 decreased transcript expression of the M-phase cell cycle genes UBE2C, AKT1, CDC20, and CYCLINA2, which are increased in CRPC and regulated by AR variant (39), in VCaP xenografts.
It is important to note that all stereoisomers covalently bound to the endogenous AR in cells. To our knowledge, these studies are the first to show binding of the different stereoisomers to an IDP in living cells; others have relied on functional assays or used recombinant proteins. The plasticity of IDPs that permits these proteins to bind multiple partners with an induced fit may result in less dependence on stereospecific properties, compared with structured proteins with rigid clefts and pockets. Thus, the demonstrated lack of stereospecific properties of the EPI analogs for binding to AR may reflect a malleable binding surface or large region for interaction on the AR NTD; alternatively, such lack of stereospecific properties may be a reflection of the potential flexible structure of the EPI compounds. The high-specificity and low-affinity interactions that are essential for reversible binding of multiple proteins to IDPs support that covalent binding of a small molecule may be optimal for sustained binding and therapeutic response. In support of this hypothesis, the noncovalent binding EPI analog EPI-093, which lacks the chlorohydrin, had no in vivo effects on the androgen axis, whereas the covalent binder EPI-002 decreased the weight of androgen-dependent tissue. The EPI compounds were not general alkylating agents, as indicated by the inability of EPI-001 to form adducts with glutathione and mercaptoethanol and from Click-chemistry experiments in living cells showing that EPI probes did not bind an abundance of cellular proteins.
Based on the evidence in Figures 3 and 4, we propose the following model of the chemical mechanism for the selective covalent of EPI-001 analogs to the AR NTD. First, the AR AF1 requires some secondary structure, since EPI compounds did not bind the denatured protein (Figure 4D). Then, the initial binding step possibly involves a fast reversible interaction between EPI-001 and the AR AF1 region (Figure 7). This reversible binding potentially situates the secondary alcohol of the chlorohydrin functionality adjacent to a basic site in AF1. In a slow and essentially irreversible step, the base might remove the proton from the secondary alcohol, leading to formation of an intermediate epoxide. The epoxide could then react with an adjacent nucleophilic site on an amino acid side chain (e.g., -SH [cysteine], -NH2 [lysine, ornithine], phenoxide [tyrosine], or imidazole [histidine]) to form the covalent bond. The selectivity of this covalent binding may come from a combination of a requirement for a strong reversible binding interaction with AR AF1 and the necessity of having a basic functionality located adjacent to the chlorohydrin secondary alcohol in this reversibly bound EPI-001 that can form the reactive epoxide. The slow rate of covalent binding of EPI-054 compared with the epoxide EPI-056 may reflect the slow rate of conversion of the chlorohydrin EPI-054 to the epoxide EPI-056 on the AF1.
Figure 7. Covalent binding reaction of EPI compounds to AR AF1 region.
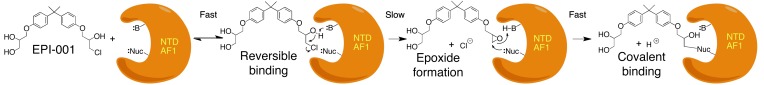
First, there is a fast reversible interaction between EPI-001 and the AR AF1 region that places the secondary alcohol of the chlorohydrin functionality next to a basic site in AF1. Then, in a slow rate-determining step, the base removes the proton from the secondary alcohol to form an intermediate epoxide. The reactive epoxide reacts rapidly and irreversibly with a nucleophilic site on an amino acid side chain to form a covalent bond.
Approximately 40 drugs that are covalent binders have been approved by the FDA, including clopidogrel, lansoprazole, esomeprazole, abiraterone, aspirin, and therapeutics for long-term use (44). However, EPI is the first reported covalent binder to an IDP and is in clinical development for human studies. EPI analogs overcome some of the limitations of current therapies for CRPC, including EPI’s low propensity for developing gain-of-function mutations because of the intrinsic disorder of the NTD and covalent binding. Importantly, EPI analogs are the only known inhibitors of constitutively active AR splice variants that are correlated to CRPC, poor prognosis, and resistance to abiraterone (16–19, 35, 38). This paradigm for drug development could be applied to other IDPs that are associated with cancer and other diseases.
Methods
Cells, plasmids, and reporter assays.
LNCaP, PC3, and VCaP cells as well as PSA(6.1kb)-luciferase, PB-luciferase, ARR3-luciferase, 5xGAl4UAS-TATA–luciferase, AR1–558-Gal4DBD, ARv567es plasmids, and transfection of cells have been described previously (17, 21).
Fluorescence polarization, microscopy, and spectroscopy.
Androgen, progesterone, and glucocorticoid receptor PolarScreen Competitor Assay kits (Invitrogen) were used according to the manufacturer’s protocol. Serial dilution was done for each small molecule, and solvent was compensated to ensure equal volume of DMSO and ethanol in each sample. Fluorescence polarization at excitation wavelength 470 nm and emission at 530 nm were measured in Greiner 384 black clear-bottomed plates using Infinite M1000 (TECAN).
For microscopy, LNCaP cells were transiently transfected with an expression vector for AR-YFP using serum-free and phenol red–free RPMI media for 24 hours prior to treatment of compounds. 4 hours after treatment, cells were fixed and stained for DAPI and examined using fluorescence microscopy.
Steady-state fluorescence spectroscopy was measured as described previously (21, 28), and on-site competition curve best-fit analysis was performed using GraphPad Prism version 6.01 software.
BrdU cell cycle analysis.
LNCaP cells were treated with inhibitors for 1 hour, followed by addition of 0.1 nM R1881 under serum-free and phenol red–free conditions for 48 hours. Cells were pulse labeled with 10 μM BrdU for 2 hours and fixed in 70% ethanol. BrdU-labeled cells were probed with anti-BrdU–FITC antibody, and DNA was counterstained with DAPI. List mode files were collected using a dual laser Epics Elite-ESP flow cytometer. Bivariate analysis was performed using FlowJo 7 software (Ashland).
Viability and proliferation assays.
PC3 and LNCaP cells were plated in 96-well plates in respective media plus 0.5% FBS. The next day, PC3 cells were treated with vehicle and EPI-002 for 2 days, and LNCaP cells were pretreated with vehicle and EPI-002 for 1 hour before treating with 0.1 nM R1881 for 3 days. Cell viability was measured using alamarBlue Cell Viability Assay (Invitrogen) following the manufacturer’s protocol.
Binding assays.
LNCaP cells were treated for 24 hours with vehicle or with alkyne-containing EPI analogs. To examine binding to the AR NTD, LNCaP cells were transiently transfected with Flag-ARN plasmid or empty vector using lipofectin (Invitrogen) and treated with vehicle or modified EPI-001 analogs for 24 hours. Proteins were extracted from treated cells with lysis buffer containing 50 mM HEPES (pH 8.0), 150 mM NaCl, 1% (v/v) Triton-X100, and EDTA-free protease inhibitors and were subjected to Click-chemistry conditions for 3 hours at 25°C in buffer containing 0.1% SDS, 5% t-butanol, 100 μM tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (Sigma-Aldrich), 1 mM tris-(2-carboxyethyl) phosphine (TCEP), 100 μM biotin-azide reagent, and 1 mM CuSO4. Samples were dialyzed overnight in 50 mM HEPES (pH 8.0), 150 mM NaCl, 0.1% SDS, and 1% Triton-X100 to remove excess biotin-azide reagent. Biotinylated EPI probes covalently bound to proteins were enriched using streptavidin-agarose resin (Thermo Fisher Scientific). Biotin-EPI-proteins were separated by SDS-PAGE and subjected to Western blot analysis using anti-biotin antibody.
The cell-free binding assay was performed with AR AF1 recombinant protein that was expressed and purified as previously described (21, 28), with additional purification by size exclusion chromatography. Recombinant AR AF1 was mixed with alkyne-containing EPI probes, and binding reaction was carried out under the conditions indicated in the figures and legends prior to heating (90°C for 5 minutes). EPI probes were labeled with fluorescein by Click-chemistry reaction at 25°C for 1 hour in buffer containing fluorescein azide (in amounts exceeding those of the EPI probes), 0.1 mM ascorbic acid, and 0.1 mM copper(II)-tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine complex (Lumiprobe). Samples were resolved on 12.5% SDS-PAGE, and fluorescein was visualized using Fujifilm FLA-7000 image analyzer (GE Healthcare). The same gel was stained with Coomassie blue R-250. The intensities of bands for fluorescein or Coomassie blue were quantified using ImageJ.
Alkylation reaction.
Test solutions were prepared by placing 10 μg EPI-001 into a NMR tube in DMSO/HEPES buffer (4:1, v/v, 0.10 M HEPES, pH 7.4), adding thiols (neat liquid or solid form), and diluting the solution with 100 μl TCEP (0.5 M). The NMR spectra experiments were set to be monitored at 25°C at 0, 1, 3, 5, 7, and 24 hours as well as 7 days after the addition of thiols.
For glutathione screening, glutathione (39 mg, 0.127 mmol) and TCEP (100 μl) were added to a solution of EPI-001 (10 μg, 0.025 mmol) in DMSO-d6/HEPES buffer (500 μl). After shaking several times, the reaction mixture was monitored by NMR.
For 2-mercaptoethanol screening, 2-mercaptoethanol (11 μl, 0.155 mmol) was added to a solution of EPI-001 (22 μg, 0.055 mmol) in DMSO-d6. After shaking several times, the reaction mixture was monitored by NMR.
Prostate weight.
Mature male mice were treated every 3 days with EPI-093 (i.v.), EPI-002 (i.v.), DMSO control (i.v.) or daily with bicalutamide (10 mg/kg body weight by gavage). 2 days after the last dose, prostates were dissected and weighed.
Xenografts.
Male NOD-SCID mice bearing subcutaneous tumors were castrated when tumor volume was approximately 100 mm3. Animals bearing LNCaP xenografts were injected i.v. with 50 mg/kg body weight of EPI-001 mixture or stereoisomers every other day or were treated by oral gavage with bicalutamide (10 mg/kg body weight). Animals bearing VCaP xenografts were administered 200 mg/kg body weight of EPI-002 (100-mg/kg dose twice daily), 10 mg/kg body weight of bicalutamide, or vehicle daily by oral gavage. Tumors were excised 2 days after the last dose and prepared for gene expression analysis, Western blot analyses, and immunohistochemistry.
QRT-PCR gene expression analysis.
Total RNA was extracted from harvested xenografts using TRIzol reagent (Invitrogen) and PureLIink RNA Mini Kit (Invitrogen) according to the manufacturer’s protocols specified for On-column PureLink DNase treatment. cDNA was subsequently synthesized using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Diluted cDNA and gene-specific primers were mixed with Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen). The transcripts were measured by ABI PRISM 7900 Sequence Detection System (Invitrogen). For all qRT-PCR experiments, each tumor sample was tested in technical triplicates. Gene expression levels were normalized to the housekeeping gene RPL13A. Primers were as previously described (21, 38).
Immunohistochemistry and Western blot analysis.
Cells that were positive for Ki67 and caspase-3 staining were counted in sections from 3 xenografts per treatment. At least 2,000 cells per xenograft were counted. The total number of cells counted was as follows: 7,647 (DMSO, Ki67), 7,690 (EPI-002, Ki67), 7,901 (bicalutamide, Ki67), 8,799 (DMSO, caspase-3), 8,075 (EPI-002, caspase-3), and 7,843 (bicalutamide, caspase-3). For analysis of AR protein, concentrations of lysates of homogenized VCaP xenografts were measured by BCA assay after albumin depletion. Proteins (10 μg) were resolved on a NuPAGE 8%–12% Bis Tris gradient gel, transferred to nitrocellulose membrane, and probed for AR species using antibodies to the NTD (AR441; Santa Cruz). Protein levels of FL-AR and AR567 were detected using AR-N20 (Santa Cruz).
Statistics.
Statistical analysis was performed using GraphPad Prism (version 6.01; GraphPad Software). Except where specified, comparisons between groups were performed with 2-tailed Student’s t test, and differences were consider statistically significant at P values less than 0.05.
Study approval.
All experiments involving animals conformed to the relevant regulatory and ethical standards, and the University of British Columbia Animal Care Committee approved the experiments.
Supplementary Material
Acknowledgments
This research was supported by grants from the NCI (2R01 CA105304), the Canadian Institutes of Health Research (MOP79308 and PPP90150), the Canadian Cancer Society (017289), the US Army Medical Research and Materiel Command Prostate Cancer Research Program (W81XWH-11-1-0551), NCI Pacific Northwest Prostate Cancer SPORE (2 P50 CA 097186-06 Project 4), PO1 CA85859, and the Department of Veterans Affairs to S. Plymate. We are grateful to Country Meadows Senior Men’s Golf Charity Classic, the FORE PAR Prostate Awareness Research Charity Golf Classic, and Safeway for their financial support of this research.
Footnotes
Conflict of interest: Carmen A. Banuelos, Javier Garcia Fernandez, Nasrin R. Mawji, Jun Wang, Raymond J. Andersen, and Marianne D. Sadar are coinventors of EPI; however, Raymond J. Andersen and Marianne D. Sadar had to waive all rights to inventor royalties. Stephen Plymate, Raymond J. Andersen, and Marianne D. Sadar are consultants to ESSA Pharma Inc. Carmen A. Banuelos, Javier Garcia Fernandez, Nasrin R. Mawji, Jun Wang, Stephen Plymate, Raymond J. Andersen, and Marianne D. Sadar own ESSA Pharma Inc. stock.
Citation for this article:J Clin Invest. 2013;123(7):2948–2960. doi:10.1172/JCI66398.
References
- 1.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40(3):1623–1634. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 2.Sadar MD. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011;71(4):1208–1213. doi: 10.1158/0008-5472.CAN_10-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visakorpi T, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Fenton MA, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997;3(8):1383–1388. [PubMed] [Google Scholar]
- 6.Yoshida T, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65(21):9611–9616. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 7.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999;274(12):7777–7783. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 8.Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277(9):7076–7085. doi: 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- 9.Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem. 2002;277(41):38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- 10.Blaszczyk N, et al. Osteoblast-derived factors induce androgen-independent proliferation and expression of prostate-specific antigen in human prostate cancer cells. Clin Cancer Res. 2004;10(5):1860–1869. doi: 10.1158/1078-0432.CCR-0974-3. [DOI] [PubMed] [Google Scholar]
- 11.Fujimoto N, Mizokami A, Harada S, Matsumoto T. Different expression of androgen receptor coactivators in human prostate. Urology. 2001;58(2):289–294. doi: 10.1016/S0090-4295(01)01117-7. [DOI] [PubMed] [Google Scholar]
- 12.Agoulnik IU, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66(21):10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 13.Comuzzi B, et al. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J Pathol. 2004;204(2):159–166. doi: 10.1002/path.1609. [DOI] [PubMed] [Google Scholar]
- 14.Debes JD, et al. p300 in prostate cancer proliferation and progression. Cancer Res. 2003;63(22):7638–7640. [PubMed] [Google Scholar]
- 15.Montgomery RB, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68(11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Z, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun S, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu R, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornberg E, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6(4):e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quayle SN, Mawji NR, Wang J, Sadar MD. Androgen receptor decoy molecules block the growth of prostate cancer. Proc Natl Acad Sci U S A. 2007;104(4):1331–1336. doi: 10.1073/pnas.0606718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen RJ, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 22.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28(7):778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 23.Jenster G, et al. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5(10):1396–1404. doi: 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- 24.Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266(1):510–518. [PubMed] [Google Scholar]
- 25.Rundlett SE, Wu XP, Miesfeld RL. Functional characterizations of the androgen receptor confirm that the molecular basis of androgen action is transcriptional regulation. Mol Endocrinol. 1990;4(5):708–714. doi: 10.1210/mend-4-5-708. [DOI] [PubMed] [Google Scholar]
- 26.McEwan IJ, Gustafsson J. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc Natl Acad Sci U S A. 1997;94(16):8485–8490. doi: 10.1073/pnas.94.16.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavery DN, McEwan IJ. Structural characterization of the native NH2-terminal transactivation domain of the human androgen receptor: a collapsed disordered conformation underlies structural plasticity and protein-induced folding. Biochemistry. 2008;47(11):3360–3369. doi: 10.1021/bi702221e. [DOI] [PubMed] [Google Scholar]
- 28.Reid J, Kelly SM, Watt K, Price NC, McEwan IJ. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. Influence of structure-stabilizing solutes and protein-protein interactions. J Biol Chem. 2002;277(22):20079–20086. doi: 10.1074/jbc.M201003200. [DOI] [PubMed] [Google Scholar]
- 29.Fischer K, Kelly SM, Watt K, Price NC, McEwan IJ. Conformation of the mineralocorticoid receptor N-terminal domain: evidence for induced and stable structure. Mol Endocrinol. 2010;24(10):1935–1948. doi: 10.1210/me.2010-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R, Litwack G. Structural and functional relationships of the steroid hormone receptors’ N-terminal transactivation domain. Steroids. 2009;74(12):877–883. doi: 10.1016/j.steroids.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R, Thompson EB. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol. 2003;17(1):1–10. doi: 10.1210/me.2002-0258. [DOI] [PubMed] [Google Scholar]
- 32.Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein in androgen receptor-mediated signaling. Proc Natl Acad Sci U S A. 1998;95(5):2122–2127. doi: 10.1073/pnas.95.5.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhalla J, Storchan GB, MacCarthy CM, Uversky VN, Tcherkasskaya O. Local flexibility in molecular function paradigm. Mol Cell Proteomics. 2006;5(7):1212–1223. doi: 10.1074/mcp.M500315-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mostaghel EA, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17(18):5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002;277(29):26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 37.Clegg NJ, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72(6):1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6(11):e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makkonen H, Kauhanen M, Jaaskelainen T, Palvimo JJ. Androgen receptor amplification is reflected in the transcriptional responses of Vertebral-Cancer of the prostate cells. Mol Cell Endocrinol. 2011;331(1):57–65. doi: 10.1016/j.mce.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Liu W, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008;10(8):897–907. doi: 10.1593/neo.08428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomlins SA, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448(7153):595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 43.Mostaghel EA, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67(10):5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 44.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.