

Abstract
Sim1 haploinsufficiency in mice induces hyperphagic obesity and developmental abnormalities of the brain. In humans, abnormalities in chromosome 6q16, a region that includes SIM1, were reported in obese children with a Prader-Willi–like syndrome; however, SIM1 involvement in obesity has never been conclusively demonstrated. Here, SIM1 was sequenced in 44 children with Prader-Willi–like syndrome features, 198 children with severe early-onset obesity, 568 morbidly obese adults, and 383 controls. We identified 4 rare variants (p.I128T, p.Q152E, p.R581G, and p.T714A) in 4 children with Prader-Willi–like syndrome features (including severe obesity) and 4 other rare variants (p.T46R, p.E62K, p.H323Y, and p.D740H) in 7 morbidly obese adults. By assessing the carriers’ relatives, we found a significant contribution of SIM1 rare variants to intra-family risk for obesity. We then assessed functional effects of the 8 substitutions on SIM1 transcriptional activities in stable cell lines using luciferase gene reporter assays. Three mutations showed strong loss-of-function effects (p.T46R, p.H323Y, and p.T714A) and were associated with high intra-family risk for obesity, while the variants with mild or no effects on SIM1 activity were not associated with obesity within families. Our genetic and functional studies demonstrate a firm link between SIM1 loss of function and severe obesity associated with, or independent of, Prader-Willi–like features.
Introduction
The basic helix-loop-helix–PER-ARNT-SIM (bHLH-PAS) transcription factor single-minded 1 (SIM1) plays a key role in neuronal differentiation within the paraventricular nucleus of the hypothalamus, which is critical for food intake regulation (1). Sim1-haploinsufficient mice display hypodevelopment of the paraventricular nucleus of the hypothalamus and are hyperphagic, obese, and highly sensitive to diet-induced obesity, whereas Sim1 overexpression in mice leads to a decrease in food intake (2–4). In humans, several deletions in the chromosome 6q16, the region that includes SIM1, have been identified in obese patients presenting with a Prader-Willi–like (PWL-like) syndrome (5), which is characterized in early infancy by global developmental delay, hypotonia, and feeding difficulties and later in life by hyperphagia, obesity, and facial dysmorphisms (6). The 4.1-Mb critical region for PWL syndrome includes SIM1 but also 11 other genes or gene predictions, and the specific role of SIM1 haploinsufficiency in the development of PWL syndrome has not definitively been established (6). Nonetheless, a de novo balanced translocation disrupting SIM1 has been described in a patient presenting with severe obesity but no other clinical features suggestive of a PWL syndrome (7). Another study reported 6 rare non-synonymous SIM1 mutations in extremely obese patients only, but no functional analyses were performed (8).
In the present study, we aimed to assess the contribution of rare mutations in SIM1 to monogenic and syndromic forms of obesity. Coding regions of SIM1 were sequenced in 44 children presenting with PWL syndrome features, 198 children with severe early-onset obesity, 568 morbidly obese adults, and 383 normal-weight controls. Subsequently, we functionally characterized the effects of the mutations on SIM1 activity and analyzed phenotypes in SIM1-deficient patients.
Results and Discussion
By focusing on rare coding variants only (minor allele frequency [MAF] <1%), we identified 4 substitutions (p.I128T, p.Q152E, p.R581G, and p.T714A; Figure 1) in 4 children presenting with clinical features of PWL syndrome, including severe obesity that started between 1 and 2 years of age (Table 1 and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI68035DS1), and 4 other substitutions (p.T46R, p.E62K, p.H323Y, and p.D740H; Figure 1) in 7 morbidly obese adults (Table 1 and Supplemental Table 2).
Figure 1. Location of the 8 SIM1 rare substitutions.

At the top are shown the substitution locations in exons. In the middle are shown the substitution locations in protein domains (only substitutions located in specific domains are reported). At the bottom are shown the substitution locations in the SIM1 766-amino-acid protein.
Table 1.
Genetic and functional description of the 8 SIM1 rare substitutions
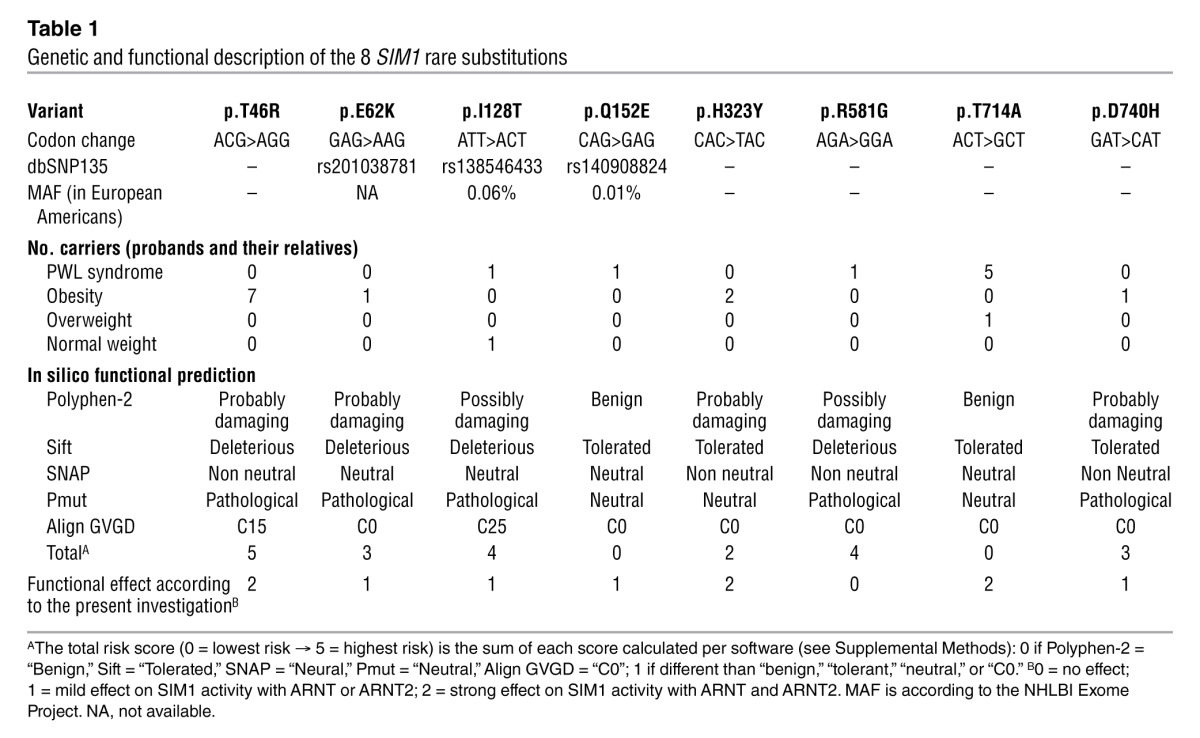
With regard to the obese child SIM1 mutant carriers presenting with PWL syndrome (Supplemental Table 1 and Supplemental Figure 1), the girl carrying the p.I128T variant received the substitution from her normal-weight father. The mother of the girl carrying the p.Q152E variant did not carry this substitution; the father’s DNA and phenotypes were not available. The girl carrying the p.R581G variant was an adopted child. In contrast, we were able to investigate the family of the boy carrying the p.T714A variant. His 3 siblings, his mother, and a maternal aunt also carried this substitution. The mother and her 4 children were severely obese and presented at least one other clinical feature associated with PWL syndrome, while the aunt was only overweight and without clinical abnormalities (Supplemental Table 3).
Among the morbidly obese adult SIM1 mutant carriers (Supplemental Table 2 and Supplemental Figure 2), 4 unrelated patients carried the same p.T46R variant. In the first mutated participant, the p.T46R variant had arisen de novo. In the second mutated participant, the p.T46R variant had been inherited from his obese mother. Furthermore, this participant had an obese sister who also carried the p.T46R variant. Although parental DNA samples were not available for the third p.T46R-carrying participant, her obese son carried the substitution, while her normal-weight sister and normal-weight daughter did not. The family of the fourth p.T46R-carrying participant was not available. The patient carrying the p.E62K variant had a normal-weight mother and 2 normal-weight daughters, none of whom carried the substitution. His severely obese father has died, and no DNA was available. The carrier of the p.H323Y variant had a morbidly obese sister who was also a p.H323Y carrier, while both parents had died. No DNA was available from relatives of the p.D740H carrier.
In summary, we identified 21 (adults and children) carriers of a rare heterozygous SIM1 variant (11 probands and 10 of their relatives) and 16 non-carrier relatives, and we found a strong contribution of SIM1 rare variants to intra-family risk for obesity (odds ratio[95% confidence interval]: OR = 20.9[3.5–126.5]; P = 9.3 × 10–4).
Of note, 3 of the 8 rare variants have been listed in the Single Nucleotide Polymorphism database (dbSNP 137) — p.E62K/rs201038781, p.I128T/rs138546433, and p.Q152E/rs140908824 — but are very rare in the general population, according to the NHLBI Exome Project and the 1000 Genomes Project (MAF <0.1%). When we sequenced 383 normal-weight controls, we found a single carrier of the p.I128T variant (in the heterozygous state). A previous study reported that p.I128T did not cosegregate with either obesity or overweight phenotype in a family of European descent (9). Therefore, this variant is unlikely to have a significant effect on obesity or PWL-related clinical features. We then genotyped 2,896 normal-weight individuals but did not find any carriers of the substitutions p.T46R, p.E62K, p.Q152E, p.H323Y, p.R581G, p.T714A, or p.D740H.
Subsequently, we aimed to functionally assess the 8 rare substitutions. Using five types of in silico prediction software, we found that the mutation with the highest risk score of damaging effect was p.T46R, while the substitutions with the lowest risk score were p.Q152E and p.T714A (Table 1). One of the aryl hydrocarbon receptor nuclear translocators, either ARNT or ARNT2, is required as a dimerization partner for SIM1 to function as a transcription factor, with mouse knockout studies suggesting that ARNT2 is the in vivo partner in the hypothalamus (10, 11). We constructed a homology model to estimate structural features of the SIM1:ARNT2 dimer at the bHLH and PASA domains, which was based upon the crystal structure for the N-terminal half of the CLOCK:BMAL1 dimer (12). We found that the N-terminal amino acids (T46, E62, I128, and Q152) likely lie on the surface of the protein (Figure 2A). E62, I128, and Q152 amino acids were located in unstructured loops (Figure 2, B and C); thus, their possible contributions to perturbations of SIM1 structure could not be estimated. However, T46 lay in the middle of a helix that was in close proximity to a helix of ARNT2 (Figure 2B); thus, the larger arginine residue may critically perturb the dimerization interface.
Figure 2. Homology model of the SIM1:ARNT2 heterodimer.

SIM1 is shown in yellow and ARNT2 in green. (A) Surface representation of SIM1, with amino acids that harbor variants shown in red. (B and C) Ribbons diagram of the SIM1:ARNT2 heterodimer, with amino acids that harbor variants depicted in pink.
We then assessed the functional effects of the 8 substitutions on the transcriptional properties of the SIM1:ARNT and SIM1:ARNT2 dimers in vitro using luciferase gene reporter assays in human 293 Flp-In T-Rex stable cell lines expressing the WT protein or one of the 8 SIM1 mutants (Figure 3, A and B). Importantly, with this system, only 1 copy of the mutated (or WT) SIM1 is integrated at a specific and predefined insertion site in the cell genome. This renders activity levels independent of the number of inserted copies and the insertion site, and allowed an accurate comparison of the mutants’ activities. We found a strong loss-of-function effect of p.T46R, p.H323Y, and p.T714A mutations on SIM1 transcriptional activity with both ARNT and ARNT2 (P < 0.001; Figure 3C). These mutations were located in the bHLH, PASB, and C-terminal domains, respectively (Figure 1). The 3 mutations were identified in morbidly obese adults (n = 9 carriers of p.T46R or p.H323Y), in 1 overweight adult (carrier of p.T714A), and in severely obese subjects with PWL syndrome (n = 4 carriers of p.T714A). Together, these loss-of-function mutations were associated with a strong intra-family risk for obesity (OR = 28.0[2.7–295.7]; P = 5.6 × 10–3). The p.I128T and p.Q152E variants, which were identified either in normal-weight participants (n = 2 carriers of p.I128T) or in severely obese subjects with PWL syndrome (n = 2 carriers of p.I128T or p.Q152E), had a milder loss-of-function effect on SIM1 activity with ARNT or ARNT2 (Figure 3C). Two substitutions (p.E62K and p.D740H) which were identified in 2 morbidly obese participants, had a gain-of-function effect on SIM1 activity with ARNT only (Figure 3C). We did not detect any effect of p.R581G on SIM1 activity (Figure 3C), which was identified in a severely obese participant with PWL syndrome. Together, the rare variants with mild or no effects on SIM1 activity were not significantly associated with obesity within families (OR = 7.5[0.5–122.7]; P = 0.158). When we compared our functional data with the in silico predictions, only 62.5% concordance was found.
Figure 3. Functional assessment of each rare non-synonymous SIM1 variant.

(A) WT and mutant SIM1 proteins display similar levels of expression in doxycycline-induced 293 Flp-In T-Rex cells. Western blot analysis of Myc-tagged SIM1 overexpression in whole cell extracts from 2 independently derived WT stable cell lines, 8 SIM1 mutant cell lines, and 1 empty vector (EV) cell line. (B) Independently derived WT SIM1 cell lines display similar levels of activity in doxycycline-induced 293 Flp-In T-Rex cells. Two independently derived WT SIM1 293 Flp-In T-Rex stable cell lines and 1 EV cell line were analyzed. Results shown are the mean of at least 3 experiments performed in triplicate ± SEM. (C) Effect of SIM1 variants on the transcriptional activity of SIM1. Two independently derived WT SIM1 293 Flp-In T-Rex stable cell lines, 8 SIM1 mutant cell lines, and 1 EV cell line were analyzed. Results are the mean of at least 3 experiments performed in triplicate ± SEM expressed relative to the 2 WT lines, which have been combined into a single average value and normalized to 100% (*P < 0.05, #P < 0.01, †P < 0.001 vs. WT).
Our genetic and functional studies, together with the findings of Ramachandrappa et al. (13), convincingly demonstrate a link between SIM1 loss of function and severe to morbid obesity that may also be associated with PWL-related clinical features including developmental delay (or intellectual disability) and facial dysmorphism. The observed effects of SIM1 loss-of-function mutations on both development and body weight regulation are in line with the key role of SIM1 in development of the paraventricular nucleus of the hypothalamus (1). Importantly, the necdin protein (NDN), which is believed to be involved in the Prader-Willi syndrome (14), regulates the activity of the SIM1:ARNT2 dimer (15), reinforcing the putative role of SIM1 in the PWL syndrome.
SIM1 non-synonymous mutations were present in numerous patients with severe obesity associated with, or independent of, PWL-related clinical features. Some mutations, such as p.T46R, dramatically reduced SIM1 activity on a reporter gene and were only found in obese individuals, suggesting that loss-of-function SIM1 mutants may underpin a monogenic form of obesity. However, some substitutions with marked loss-of-function effect (e.g., p.T714A) did co-segregate with overweight/obesity but were not always associated with severe obesity or PWL syndrome. Moreover, the p.I128T variant, which caused mild loss of SIM1 function, was unlikely to contribute to obesity. Therefore, mutation of SIM1 is not always responsible for a fully penetrant form of obesity. Similar features have been repeatedly found in MC4R loss-of function mutation carriers, where a permissive role of the environment on disease expression has been suggested (16, 17). The degree of penetrance is expected to be determined by the severity of loss of function of a particular mutant in combination with environment and genetic background. In obese patients presenting with the clinical features associated with Prader-Willi syndrome, if chromosomes 15q11 and 6q16 are found not to contain abnormalities, then SIM1 sequencing and subsequent molecular characterization should be performed, in order to demonstrate the presence of causative loss-of-function mutations.
Methods
Further information is available in Supplemental Methods.
Statistics.
For the genetic study, effects of mutation on obesity risk were calculated through univariate logistic regression models using SPSS 14.0. For the functional analysis, univariate ANOVA was performed on the log values generated for each mutant with reference to WT using SPSS 17.0. Separate analyses were performed for ARNT- and ARNT2-transfected cells. A P value less than 0.05 was considered significant.
Study approval.
The study protocol was approved by the ethics committees of Lille II University, the University of Antwerp, the Canton of Zurich, and the University of Paris-Sud and the Institutional Review Board of the University of Florida, and study participants signed informed consent agreements. For children younger than 18 years, oral consent was obtained, and parents provided written informed consent.
Supplementary Material
Acknowledgments
We are indebted to all families who participated in this study. We thank the Société Francophone du Diabète, 200 Familles pour Vaincre le Diabète et l’Obésite, the UK Medical Research Council, the Conseil National de la Recherche Scientifique Libanais, the Conseil Régional Nord Pas de Calais/FEDER, the European EurOCHIP Obesity FP7 Consortium (to P. Froguel), and the Australian Research Council (to M.L. Whitelaw) for their financial support. We thank J. Pelletier for the pML-6xCME-Luc vector and V. Negre, J. Thierry, and T. Kuijpers for their diagnoses.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2013;123(7):3037–3041. doi:10.1172/JCI68035.
References
- 1.Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998;12(20):3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michaud JL, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10(14):1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 3.Holder JL, Jr, et al. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab. 2004;287(1):E105–E113. doi: 10.1152/ajpendo.00446.2003. [DOI] [PubMed] [Google Scholar]
- 4.Yang C, et al. Adenoviral-mediated modulation of Sim1 expression in the paraventricular nucleus affects food intake. J Neurosci. 2006;26(26):7116–7120. doi: 10.1523/JNEUROSCI.0672-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilhuis HJ, van Ravenswaaij CM, Hamel BJ, Gabreels FJ. Interstitial 6q deletion with a Prader-Willi-like phenotype: a new case and review of the literature. Eur J Paediatr Neurol. 2000;4(1):39–43. doi: 10.1053/ejpn.1999.0259. [DOI] [PubMed] [Google Scholar]
- 6.Bonaglia MC, et al. Detailed phenotype-genotype study in five patients with chromosome 6q16 deletion: narrowing the critical region for Prader-Willi-like phenotype. Eur J Hum Genet. 2008;16(12):1443–1449. doi: 10.1038/ejhg.2008.119. [DOI] [PubMed] [Google Scholar]
- 7.Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9(1):101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 8.Ahituv N, et al. Medical sequencing at the extremes of human body mass. Am J Hum Genet. 2007;80(4):779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung CC, et al. Studies of the SIM1 gene in relation to human obesity and obesity-related traits. Int J Obes (Lond). 2007;31(3):429–434. doi: 10.1038/sj.ijo.0803443. [DOI] [PubMed] [Google Scholar]
- 10.Michaud JL, DeRossi C, May NR, Holdener BC, Fan CM. ARNT2 acts as the dimerization partner of SIM1 for the development of the hypothalamus. Mech Dev. 2000;90(2):253–261. doi: 10.1016/S0925-4773(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 11.Woods SL, Whitelaw ML. Differential activities of murine single minded 1 (SIM1) and SIM2 on a hypoxic response element. Cross-talk between basic helix-loop-helix/per-Arnt-Sim homology transcription factors. J Biol Chem. 2002;277(12):10236–10243. doi: 10.1074/jbc.M110752200. [DOI] [PubMed] [Google Scholar]
- 12.Huang N, et al. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science. 2012;337(6091):189–194. doi: 10.1126/science.1222804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandrappa S, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. . J Clin Invest. 2013;123(7):3042–3050. doi: 10.1172/JCI68106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jay P, et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat Genet. 1997;17(3):357–361. doi: 10.1038/ng1197-357. [DOI] [PubMed] [Google Scholar]
- 15.Friedman ER, Fan CM. Separate necdin domains bind ARNT2 and HIF1alpha and repress transcription. Biochem Biophys Res Commun. 2007;363(1):113–118. doi: 10.1016/j.bbrc.2007.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stutzmann F, et al. Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: the two facets of a Janus obesity gene. Hum Mol Genet. 2007;16(15):1837–1844. doi: 10.1093/hmg/ddm132. [DOI] [PubMed] [Google Scholar]
- 17.Saeed S, Butt TA, Anwer M, Arslan M, Froguel P. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol Genet Metab. 2012;106(1):121–126. doi: 10.1016/j.ymgme.2012.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.