

Abstract
Background. Many cancers, including melanoma, exclusively express constitutive proteasomes (cPs) and are unable to express immunoproteasomes (iPs). In contrast, mature DCs used for immunotherapy exclusively express iPs. Since proteasomes generate peptides presented by HLA class I molecules, we hypothesized that mature melanoma antigen–loaded DCs engineered to process antigens through cPs would be superior inducers of antimelanoma immunity in vivo.
Methods. Subjects with metastatic melanoma were vaccinated with mature DCs transfected with RNAs encoding melanoma antigens MART1, MAGE-3, gp100, and tyrosinase. These DCs were derived from monocytes that were untransfected (Arm A; n = 4), transfected with control siRNA (Arm B; n = 3), or transfected with siRNAs targeting the 3 inducible iP subunits (Arm C; n = 5).
Results. Vaccination stimulated antigen-specific T cell responses in all subjects, which peaked after 3–4 vaccinations, but remained elevated in Arm C subjects. Also in Arm C, circulating melanoma cell levels (as detected by quantitative PCR) fell, and T cell lytic activity against autologous melanoma was induced. In HLA-A2+ subjects, CD8+ T cells that bound tetramers loaded with cP-derived melanoma antigenic peptides were found in the peripheral blood only in Arm C subjects. Of 2 subjects with active disease (both in Arm C), one had a partial clinical response, while the other, who exhibited diffuse dermal and soft tissue metastases, had a complete response.
Conclusion. These results suggest that the efficacy of melanoma DC–based immunotherapy is enhanced when tumor antigen–loaded DCs used for vaccination express cPs.
Trial registration. Clinicaltrials.gov NCT00672542.
Funding. Duke Clinical Research Institute/Duke Translational Medicine Institute, Duke Melanoma Consortium, and Duke University Department of Surgery.
Introduction
The proteasome is a multisubunit complex, located within the cytoplasm of every cell, that degrades ubiquitinated proteins into peptides that are then presented on the surface of the cell in the context of HLA class I molecules (1). When normal cells are exposed to inflammatory mediators, such as IFN-γ, the composition of the intracellular proteasomes changes from the constitutive proteasome (cP) to the immunoproteasome (iP), such that the 3 proteolytic subunits, β1, β5, and β7, are replaced by β1i, β5i, and β7i, respectively, altering the peptides generated by cleavage of a given intracellular protein (2). In contrast to normal cells, various cancers, including melanoma, fail to express the iP when exposed to inflammatory mediators (3).
As the most potent antigen-presenting cells known, DCs continue to hold promise for cancer immunotherapy (4). Since vaccination with antigen-loaded immature DCs has been shown to induce immune tolerance (5), DC-based cancer immunotherapy protocols routinely use tumor-associated antigen–loaded (TAA-loaded) DCs that have been matured, a process that results in DCs exclusively expressing the iP (6). Consequently, there is a mismatch between proteasomes expressed by melanoma cells (cPs) versus those expressed by mature DCs (iPs). Because of this mismatch, we reasoned that TAA-specific immune responses induced by mature DCs may not lead to optimal recognition of tumor cells presenting TAA-derived peptides generated by the cP (Figure 1).
Figure 1. Rationale for the clinical trial.

When TAA-loaded DCs undergo maturation, cPs are no longer expressed; therefore, the TAA-derived peptides presented in the context of HLA class I on the surface of the immunostimulatory mature DCs are exclusively generated by the iP (triangles). CD8+ T cells induced by these DCs will then specifically recognize only iP-generated TAA-derived peptides. In contrast, melanoma cells (as well as other cancer cells) express the cP and not the iP, even when exposed to inflammatory mediators. Therefore, TAA-derived peptides presented in the context of HLA class I on the surface of melanoma cells will be exclusively generated by the cP (squares). T cells induced by the mature TAA–loaded DCs and recognizing iP-generated peptides will then not recognize melanoma cells expressing cP-derived peptides from the same TAA, resulting in a misdirected and suboptimal antimelanoma immune response. By modulating the proteasome of the mature DC from the iP to the cP though transfection of the monocytes from which the DCs were derived with iP siRNA, mature immunostimulatory TAA–loaded DCs then present TAA-derived peptides generated by the cP, stimulating a T cell response that will be appropriately directed against melanoma cells expressing the cP and presenting TAA-derived peptides generated by the cP.
We previously reported that transfection with siRNA targeting the 3 inducible iP subunits (referred to herein as iP siRNA) altered the proteasome content of mature DCs from the iP to the cP (7). When these mature DCs, expressing the cP and the iP, were then transfected with RNA encoding melanoma TAAs, these DCs stimulated superior antimelanoma immunity in vitro. Based on those results, we initiated a phase I clinical trial in which subjects with metastatic melanoma were vaccinated with mature autologous DCs transfected with RNAs encoding the melanoma TAAs MART1, MAGE3, gp100, and TYR (Figure 2), according to the schedule shown in Figure 3. DCs were generated from untransfected monocytes (Arm A; n = 4), control siRNA–transfected monocytes (Arm B; n = 3), or monocytes transfected with iP siRNA (Arm C; n = 5). During the course of vaccination, which was well tolerated by all subjects, no dose-limiting toxicities or adverse events were observed, and TAA-specific immune responses were assessed.
Figure 2. CONSORT diagram for this phase I nonrandomized clinical trial.

14 eligible subjects underwent leukapheresis to generate the DC vaccine. 12 subjects were immunized with their autologous vaccine, while 2 subjects who developed CNS metastases (an exclusion criterion) were not vaccinated, and their participation in the trial was discontinued. All 12 vaccinated subjects underwent complete follow-up and analysis.
Figure 3. Clinical timeline for vaccine generation and administration and immunologic testing.

Subjects were screened to determine eligibility (week –6), and then underwent surgery or a biopsy of their metastatic melanoma (week –5). Leukapheresis was then performed (week –4) for collection of PBMCs. The monocytes were then isolated from the PBMCs, electroporated with the appropriate siRNAs, and differentiated into immature DCs over a period of 5 days. After induction of maturation for 2 days, DCs were electroporated with TAA RNAs, then cryopreserved. Over the next 3 weeks, the vaccine was test thawed, and both sterility testing and DC phenotypic analysis was performed. Subjects were then vaccinated weekly (weeks 0–5) by intradermal injection, as indicated. Blood was collected at the time of each vaccination, and repeat leukapheresis was performed 2 weeks after the final vaccination (week 7) for isolation of T cells and monocytes, which were used to assess antitumor immune responses.
Results
Subject characteristics.
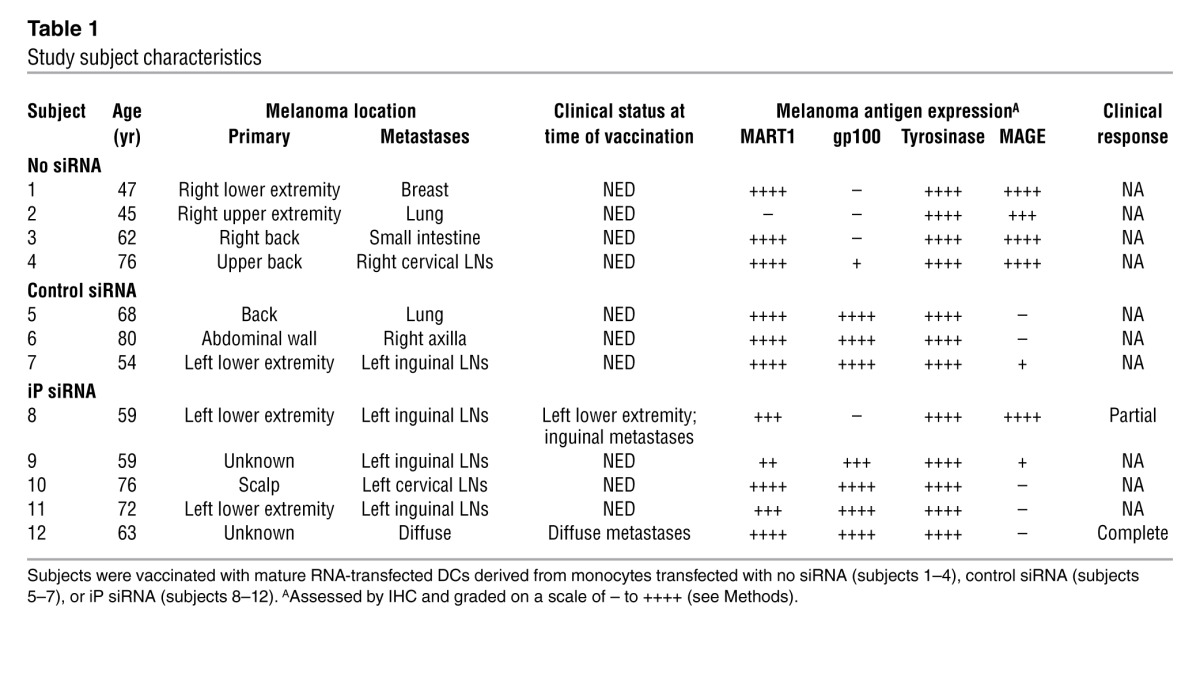
The clinical characteristics of the subjects enrolled in this phase I clinical trial are listed in Table 1, and the CONSORT diagram for this nonrandomized trial is depicted in Figure 2. At the time of vaccination, 2 subjects had active metastatic disease: subjects 8 and 12, both in Arm C. All other subjects were vaccinated after surgical resection of metastatic disease that had rendered them clinically with no evidence of disease (NED). 1.0 × 107 RNA-transfected DCs were administered by intradermal injection weekly, for a total of 6 doses.
Table 1.
Study subject characteristics
DC vaccine characteristics.
DCs for each subject were evaluated for TAA expression, the levels of which were variable and did not correlate with the stimulation of TAA-specific immune responses (data not shown). While the cP and iP content of the DCs for each vaccine was not evaluated in this study, we did confirm that our DC vaccine manufacturing approach, in which monocytes were electroporated with iP siRNA and then differentiated into mature DCs, resulted in cP expression (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI67544DS1).
Induction of antigen-specific T cell responses.
Specific immune responses against each of the 4 melanoma TAAs used for vaccination in this study were measured by IFN-γ ELISPOT using serially collected PBMCs. As shown in Figure 4, both CD4+ and CD8+ anti-TAA immune responses were stimulated by vaccination in all subjects. These maximal TAA-specific IFN-γ ELISPOT responses, which occurred in all subjects after 3–4 vaccinations, were highest in those subjects vaccinated with mature TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes. In addition, levels of TAA-specific T cells, as detected by this IFN-γ ELISPOT assay, were sustained in subjects vaccinated with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes, but returned toward baseline in those subjects vaccinated with DCs derived from untransfected or control siRNA–transfected monocytes (Figure 5 and Supplemental Figures 2 and 3). In none of the study arms did vaccination stimulate IFN-γ ELISPOT responses against control RNA–transfected monocytes.
Figure 4. Peripheral blood levels of TAA-specific T cells following vaccination as detected by IFN-γ ELISPOT.

CD4+ and CD8+ T cells were isolated from PBMCs collected serially from all study subjects, then incubated with autologous DCs that had been transfected with RNAs encoding the indicated melanoma TAA in a standard IFN-γ ELISPOT assay. The number of IFN-γ–secreting cells per 105 cells was determined. The maximal response over the time course of vaccination for each subject was graphed. Data represent mean ± SEM of duplicate measurements. *P < 0.05 vs. no siRNA and control siRNA, Student’s t test.
Figure 5. Kinetics of T cell responses to vaccination, as detected by IFN-γ ELISPOT.

The number of TAA-specific IFN-γ–secreting CD8+ and CD4+ T cells for each of the serially collected PBMCs samples of a representative subject from each study arm is graphed over time. Similar trends were noted for all 4 subjects in Arm A (DC), for the 3 subjects in Arm B (DC+CosiRNA), and for the 5 subjects in Arm C (DC+IPsiRNA) (see Supplemental Figures 2 and 3). Data represent mean ± SEM of duplicate measurements.
Detection of circulating melanoma cells.
For each subject, we extracted RNA from the mononuclear cell fraction of whole blood collected serially over the course of vaccination. Using cDNA generated from these RNA samples and TAA-specific primers and probes, quantitative PCR (qPCR) was then performed to assess levels of melanoma TAAs as an indirect indicator of circulating melanoma cells. Expression levels of the TAAs MCAM and MAGE3 in peripheral blood, normalized to levels of the GAPDH housekeeping gene, were highly variable, with subject 5 having no detectable MAGE3 in the blood (Figure 6A), and MAGE3 expression in peripheral blood by qPCR not corresponding with expression of MAGE by each subject’s melanoma (Table 1).
Figure 6. Detection of melanoma cells in peripheral blood by real-time qPCR.

cDNA, generated from RNA extracted from serially collected whole blood mononuclear cells for each study subject, was used as a template for qPCR using primers and probes specific for MAGE3 (a TAA used for vaccination) and MCAM (a melanoma TAA not included in the vaccine). (A) Baseline expression levels of MAGE3 and MCAM for each study subject, expressed relative to housekeeping gene GAPDH. (B) qPCR values for each individual subject and the indicated markers, normalized to baseline at time 0 (day of first vaccination). Data represent mean ± SEM of duplicate measurements.
As shown in Figure 6B, changes in circulating levels of MAGE3 and MCAM were minimal for patients vaccinated with TAA RNA–transfected DCs derived from either untransfected or control siRNA–transfected monocytes, with the exception of MCAM in subject 5. In contrast, in subjects vaccinated with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes, circulating tumor cell levels fell over the course of vaccination and were not detectable 1 month after completion of the full course of vaccination. MCAM was included because it was not one of the antigens used for vaccination. Despite similar assay sensitivity, as assessed using cloned DNA for each TAA gene, circulating levels of TYR, MART1, and gp100 mRNA were each barely detectable in a few patients, and these low levels dropped to undetectable after 1–2 vaccinations (data not shown).
Induction of cytotoxic T lymphocyte (CTL) activity versus autologous melanoma.
Using excised melanoma tissue and a biopsy of normal skin, we were able to establish melanoma cell lines and matching normal fibroblast cell lines, respectively, from 2 subjects in each study arm. We then used these cell lines as targets to evaluate autologous melanoma-specific CTL activity in peripheral blood. We could only assess a single effector/target ratio because of the very limited availability of T cells after other assays had been performed. As shown in Figure 7, only T cells from subjects vaccinated with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes demonstrated increased specific lytic activity against autologous melanoma cells.
Figure 7. Pre- and postvaccination CTL responses against autologous melanoma.

Autologous melanoma cell lines, as well as autologous skin fibroblast cell lines, were established from tissue samples collected from 2 subjects vaccinated in each of the 3 study arms. These cell lines were then used as targets in a europium release lytic assay to assess CTL activity of autologous T cells collected on the day of the first vaccination (i.e., baseline) and approximately 1 month after the final vaccine dose (pre and post, respectively). Percent specific lytic activity is expressed for both autologous fibroblasts (negative control) and autologous melanoma cells. E:T, effector/target ratio. Data represent mean ± SEM of triplicate measurements.
Induction of CD8+ T cells specific for cP-derived peptides.
HLA typing of each study subject revealed that 7 subjects were HLA-A2+, and sufficient cells were available from 6 of these to perform HLA-A2 tetramer analysis. The 2 HLA-A2 tetramers used for this analysis were loaded with peptides generated by the cP rather than the iP: the MART1-derived peptide ELAGIGILTV126–135 and the gp100-derived peptide ITDQVPFSV209–217 (8, 9). As shown in Figure 8, among peripheral blood lymphocytes, the highest percentage of cP-derived peptide CD8+tetramer+ T cells was noted after vaccination with TAA-transfected mature DCs derived from iP siRNA–transfected monocytes. In contrast, in those subjects vaccinated with TAA-transfected DCs derived from monocytes that were not transfected with iP siRNA, the percentage of CD8+ T cells that bound these 2 HLA-A2 tetramers loaded with peptides generated by the cP was reduced after vaccination. Of note, melanomas from all subjects expressed MART1, as detected by IHC staining, with the exception of that from subject 2. Melanoma expression of gp100 was strong for subjects 7, 10, 11, and 12; weak for subject 4; and absent for subject 2. However, the number of CD8+tetramer+ T cells before and after vaccination did not appear to correlate with expression of either of these 2 TAAs by the subject’s melanoma, and all subjects were vaccinated with DCs transfected with MART1 and gp100 as well as MAGE3 and TYR RNAs.
Figure 8. Peripheral blood levels of CD8+ T cells binding HLA-A2 tetramers loaded with melanoma TAA–derived peptides generated by the cP.

For 6 of the 7 subjects in this trial that were HLA-A2+, sufficient numbers of collected peripheral blood T cells were available for tetramer binding analysis. Using blood samples collected before and approximately 1 month after completion of the course of vaccination, PBMCs were isolated and then stained with an anti-CD8 FITC-conjugated mAb and PE-conjugated HLA-A2 tetramers loaded with cP-generated peptides derived from MART1 or gp100. The pre- and postvaccination percentages of CD8+tetramer+ T cells are graphed for subjects 10, 11, and 12 (each vaccinated with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes), and for subjects 2, 4, and 7, whose TAA RNA–transfected DC vaccine was derived from monocytes that were either untransfected (subjects 2 and 4) or transfected with control siRNA (subject 7). Representative FACS profiles are shown for pre- and postassessment of MART1 CD8+tetramer+ cells for subjects 2 and 12.
Clinical response.
In this heterogeneous group of subjects with metastatic melanoma vaccinated with TAA RNA–transfected DCs, 11 of 12 (92%) subjects were alive after a median follow-up of 35 months. Of these 11 subjects, 6 continued to be NED.
Among all study subjects, 2 subjects in Arm C that were vaccinated with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes had active metastatic melanoma at the time of vaccination. Subject 8 presented with bulky left inguinal adenopathy and multiple left lower extremity darkly pigmented firm dermal metastatic lesions. During the course of vaccination, these dermal metastatic lesions liquefied and changed to white in color. Unfortunately, this response was transient: within 4 weeks of the last vaccination, these lesions were once again firm and dark in character, and the subject died of metastatic disease 7 months after the last vaccination. We thus concluded that vaccination with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes induced a partial clinical response in this subject.
In subject 12, with diffusely distributed multiple dermal as well as soft tissue metastatic melanoma lesions, postvaccination PET/CT scan showed complete resolution of PET-avid lesions (Figure 9). A subsequent PET/CT performed 9 months later continued to show NED. We concluded that vaccination with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes stimulated a complete clinical response in this subject, who remained free of active disease 20 months after the end of the course of 6 vaccinations with TAA RNA–transfected DCs derived from iP siRNA–transfected monocytes.
Figure 9. Resolution of PET-avid metastatic melanoma after DC vaccination.

PET/CT scanning was performed both prior to and 6 weeks after completion of the sixth vaccine dose for subject 12. PET-avid metastatic lesions (circles) were no longer detectable after vaccination.
Discussion
DCs are the most potent antigen-presenting cells of the immune system and possess the unique ability to induce naive T lymphocytes to differentiate into effector T cells with specific cytotoxic activity against a variety of antigens, including those expressed by tumor cells (10). DCs can be cultured from monocytes found in peripheral blood, thereby making available large numbers of these cells for active immunotherapy (11).
Our laboratory has chosen RNA transfection for loading DCs with TAAs because of the many advantages of this approach over other methods of antigen loading that have routinely been used both experimentally and in DC-based clinic immunotherapy trials in subjects with cancer (12–18). This method relies on the DC translating the transfected RNAs into the TAA proteins, which are then degraded into peptides in the cytoplasm by the proteasome. These proteasome-generated peptides are then presented on the cell surface in the context of HLA class I molecules, where they can be recognized by and stimulate TAA peptide–specific CD8+ T cells (1).
DC-based cancer immunotherapy has held promise for many years, with a variety of murine studies demonstrating the efficacy of this approach. In cancer patients, significant immunologic responses have been observed in a number of DC-based cancer vaccination studies, but clinical responses to DC-based vaccines, including those using RNA transfection for DC TAA loading, have been variable (4). Several immunologic factors certainly contribute to this variability, but we hypothesized that the differing proteasome composition of mature monocyte-derived DCs versus melanoma cells might offer one explanation for the variable clinical responses after DC-based cancer immunotherapy.
Because immature antigen-loaded DCs can stimulate immune tolerance, almost all clinical DC-based immunotherapy trials have used DCs that have been induced to maturity. With maturation, DCs express increased levels of costimulatory molecules and secrete immunostimulatory cytokines. As stated above, mature DCs also exclusively express the iP, in contrast to immature DCs that express cPs (as well as high levels of iPs) (6). However, cancer cells, including melanoma, often do not express the iP, even when exposed to inflammatory mediators (3). As such, mature TAA-loaded DCs used in all previous DC-based cancer immunotherapy trials would have predominantly presented TAA-derived peptides generated by the iP. T cells stimulated by these DCs might not optimally recognize tumor cells presenting TAA-derived peptides generated by the cP (Figure 1).
Based on our previously published in vitro studies (7), we expected that iP modulation would generate superior anti-TAA CD8+ T cell responses after vaccination in this clinical trial. Because the proteasome generates peptides for presentation in the context of HLA class I, but not HLA class II, our findings that iP-modulated DCs stimulated superior CD4+ T cell responses against all 4 TAAs used in this study was somewhat unexpected. The proteasome certainly has multiple intracellular functions in addition to generating peptides presented in the context of HLA class I, and our approach for altering the proteasome may have additional (immunostimulatory) effects on the DCs used for vaccination.
We considered the possibility that our DC-based vaccine might be inducing Tregs, and that the use of DCs with downmodulated iP expression (and increased cP expression) might be inducing fewer of these immunoregulatory cells. Given the fact that CD8+ T cells are HLA class I restricted, and proteasome modulation affects the peptides presented in the context of HLA class I, it stands to reason that if there is any effect with respect to Tregs, then it is mediated by CD8+, rather than CD4+, Tregs. At least some CD8+ Tregs might specifically recognize peptides generated by the iP and help to prevent the induction of autoimmunity by immature DCs that present both iP- and cP-generated peptides. IL-6, secreted by the mature DCs used for vaccination, might help relieve such CD8+ Treg–mediated suppression.
We used promoter methylation analysis to assess the percentage of FoxP3-expressing Tregs among peripheral blood CD3+ T cells in PBMC samples collected over the course of vaccination, and did not see any consistent pattern of change in peripheral blood Treg levels (or Th17 levels; ref. 19 and data not shown). It is certainly possible that Treg levels were altered in the LNs to which our intradermally injected DCs migrated, in other secondary lymphoid organs, or in the bone marrow, but Treg levels in these compartments were not assessed in this study. Assessment of Treg levels using this FoxP3 promoter methylation approach, or most other approaches, would also have been unable to detect any changes in the levels of TAA-specific Tregs.
Since a majority of subjects in this study were clinically NED, we used changes in circulating melanoma cell levels as a surrogate for clinical antimelanoma responses. Several methods have been used to detect circulating tumors cells in patients with a variety of cancers, including melanoma (20), and the clearance of circulating tumor cells from the blood is being used clinically to provide an early indicator of the response to chemotherapy in patients with metastatic breast cancer (21).
For melanoma, Koyanagi et al. have shown using a whole blood qPCR-based approach that circulating levels of melanoma antigens can predict outcomes in response to therapy of metastatic melanoma (22). Using a similar method, we found that vaccination with DCs derived from iP siRNA–transfected monocytes consistently led to the elimination of circulating melanoma cells over the course of vaccination (Figure 6B). While our qPCR-based approach for measuring circulating TAA levels must be validated as a surrogate marker for clinical outcomes in larger-scale trials, our results suggest the utility of this assay for monitoring response to immunotherapy in the setting of minimal residual disease. Of note, MAGE-3 message was detectable in the peripheral blood of all study subjects prior to vaccination. Currently, a mAb specific for MAGE-3 is not commercially available (ref. 23 and A. Jungbluth, personal communication), and therefore we have not yet been able to specifically assess MAGE-3 expression in each subject’s melanoma by IHC. It is also possible that because of the high degree of homology among the multiple members of the MAGE-A family, our qPCR test for MAGE-3 may also be detecting message for other MAGE-A family members expressed by the melanomas of these patients, especially MAGE-6 (24). Finally, metastatic lesions derived from MAGE-negative primary tumors can be MAGE-positive (25).
Using melanoma cell lines and autologous control fibroblast cell lines established from study subjects, we found that increased CTL activity against autologous melanoma cells was only stimulated in subjects vaccinated with DCs derived from iP siRNA–transfected monocytes (Figure 7). These findings were again consistent with our hypothesis that vaccination with TAA-loaded DCs expressing the cP, through downmodulation of the iP, are superior at inducing relevant immune responses that target the autologous melanoma. While we did not directly assess the proteasome content of our vaccine DCs, our HLA-A2+ tetramer analysis of peripheral blood T cells strongly suggested that mature DCs generated from iP siRNA–transfected monocytes do express the cP, generate cP-derived peptides, and stimulate T cell responses against cP-derived peptides (Figure 8).
As this was a phase I study for evaluating safety and toxicity, the effect of vaccination on overall survival cannot be meaningfully assessed. However, the clinical responses observed in 2 patients with measurable disease, both in Arm C, strongly support the notion that TAA-pulsed DCs modified with iP siRNA are capable of eliciting clinically relevant and biologically active immunologic responses in patients with metastatic cancer. Furthermore, the study subjects’ >90% survival with median follow-up of 35 months is encouraging for patients with stage III and IV melanoma.
The present results suggest that our TAA RNA–transfected DC vaccine consistently stimulates anti-TAA immune responses. Furthermore, DC iP downmodulation, with concomitant expression of the cP, enhanced the potency of our TAA RNA–transfected DC–based vaccine. Most importantly, no adverse side effects were noted, which suggests that this approach of downmodulating the iP in the DCs used for vaccination is also safe. It is also possible that our approach of transfecting monocytes with iP siRNA may not only have downmodulated the iP in DCs generated from these monocytes, but also altered additional DC functions. The use of DCs from a recently developed iP-deficient mouse line (26) for immunotherapy in a murine melanoma model may further confirm that DCs expressing the cP, rather than the iP, are superior inducers of antimelanoma immunity.
Based on our results, the new iP inhibitory drug ONX 0914 (27), developed by Onyx Pharmaceuticals, might provide an additional approach for specifically downmodulating the iP in DCs used for vaccination and enhancing DC vaccine immunogenicity. Rather than using iP siRNA transfection for iP downmodulation, we envision generating cP-expressing DCs from monocytes by culturing these cells in medium supplemented with IL-4, GM-CSF, and this specific iP inhibitor.
In summary, the results of this phase I clinical trial suggest that our strategy of altering the proteasome of mature TAA RNA–transfected DCs from the iP to the cP enhances vaccine-induced antimelanoma immune responses. We believe that these results support the novelty and potential efficacy of proteasome-modified DCs for use in cancer immunotherapy and that this approach warrants further clinical evaluation in patients with metastatic melanoma, as well as other cancers.
Methods
Subjects
Adults with metastatic melanoma were eligible to participate in this phase I clinical trial, and informed consent was obtained from all subjects. HLA typing was performed for all subjects by the Duke University Medical Center Tissue Typing Laboratory, but study participation was not limited based on a specific haplotype. Each subject’s surgically excised metastatic melanoma tissue was analyzed using immunohistochemical (IHC) staining performed by the Immunopathology Laboratory at Duke University Medical Center for expression of MART1, gp100, and tyrosinase using mAbs A103, HMB-45, and T311, respectively (all purchased from Dako) and for expression of MAGE using mAb 6C1 (Santa Cruz Biotechnology). Expression levels were scored by an experienced dermatopathologist using a scale of – to ++++. Study participation was not based on TAA expression by a given subject’s melanoma. See clinical protocol in the supplemental material.
Establishment of melanoma and fibroblast cell lines
Metastatic melanoma tissue and a sample of normal skin, collected either by biopsy performed in the outpatient clinic or in the operating room at the time of the subject’s melanoma surgery, were placed in sterile medium in a tissue culture dish and mechanically minced. After several days in culture, melanoma cells were isolated using Ab-conjugated magnetic bead separation (anti-melanoma microbeads; Miltenyi Biotec). Melanoma cell lines were established using these isolated cells and were maintained in culture. Autologous fibroblast cell lines were established from the adherent fibroblasts from the cultured minced skin specimens.
Reagents
Serum- and antibiotic-free DC culture medium was purchased from CellGenix. T150 tissue culture flasks were purchased from BD Biosciences. 290-C Teflon culture bags were purchased from American Fluoroseal. Cytokines IL-4, GM-CSF, IL-1β, IL-6, and TNF-α were purchased from CellGenix. PGE2 and DMSO were purchased from Sigma-Aldrich. siRNAs targeting the 3 inducible iP subunits were purchased from Integrated DNA Technologies. Human serum albumin was purchased from Grifols Biologicals. Human AB serum was purchased from Valley Biomedical.
RNA
Cloning of melanoma TAAs MART1, MAGE3, TYR, and gp100.
The full-length coding sequences for each of these TAAs, cloned using PCR, were inserted into a DNA plasmid immediately downstream of a T7 promoter and immediately preceding a gene segment containing 64 adenine nucleotides. An SpeI restriction site was located at the 3′ end of the 64 adenines for linearization of the DNA prior to in vitro RNA transcription. Plasmid DNA was isolated from transformed DH5α E. coli using the endotoxin-free Maxi-Prep kit (Qiagen). The presence of the correct full-length inserted TAA genes within each TAA-containing plasmid was confirmed by DNA sequencing.
In vitro RNA transcription.
Each plasmid was linearized with SpeI (New England Biolabs), then used as a template for in vitro RNA transcription using a commercial kit containing recombinant T7 RNA polymerase (mMessageMachine; Ambion). After treatment with recombinant DNase, RNA was isolated using endotoxin-free spin columns (Qiagen) and then stored at –80°C. An aliquot of each RNA transcription reaction was subjected to denaturing gel electrophoresis to confirm the synthesis of full-length RNA.
RNA and siRNA transfection.
Monocytes and mature DCs were transfected with siRNAs and TAA RNAs, respectively, using electroporation. For subjects 4 and 12, the Maxcyte system was used for electroporation; for all other subjects, cells were resuspended in normal saline and electroporated using 4-mm gap cuvettes and a BTX 830 square wave electroporator (BTX) at 500V for 500 μs.
DC vaccine generation
The timeline of vaccine generation and administration is shown in Figure 3. PBMCs were collected by standard clinical leukapheresis. Monocytes were then isolated from the PBMCs either magnetically, using anti-CD14 mAb–coated beads and the CliniMACS System (Miltenyi; subjects 1–3 and 5–10), or by elutriation, using the Elutra Cell Separation System (Terumo BCT; subjects 4, 11, and 12). The monocytes were then not transfected with siRNA (Arm A; subjects 1–4), transfected with control siRNA (Arm B; subjects 5–7), or transfected with iP siRNA (Arm C; subjects 8–12), then placed into sterile flasks or Teflon tissue culture bags in serum- and antibiotic-free medium supplemented with IL-4 and GM-CSF. After 5 days of incubation at 37°C in 5% CO2, a cytokine maturation cocktail (IL-1β, IL-6, TNF-α, and PGE2) was added to induce DC maturation. 48 hours later, DCs were harvested, a sample of the pooled DCs in the culture medium was submitted for mycoplasma testing (Clongen Laboratories), and the mature DCs were then transfected with TAA RNAs using electroporation, resuspended in either 10% DMSO and 90% human AB serum or Crystor CS10 (BioLife Solutions), and cryopreserved in aliquots.
For all DC preparations, a test thaw was performed, and an aliquot of these DCs was submitted for sterility testing. The test-thawed DCs were also analyzed by flow cytometry to document the generation of phenotypically mature DCs. In addition, DCs were analyzed by flow cytometry after intracellular staining with TAA-specific mAbs to confirm intracellular DC translation of TAA RNAs into TAA proteins within the DCs.
DC vaccine administration
Cryopreserved DCs were thawed, washed, and resuspended in saline. Cell count and viability was determined using Trypan blue exclusion, and an aliquot of DCs was submitted for immediate Gram stain to the Clinical Microbiology Laboratory at Duke University Medical Center. The endotoxin level in the final wash buffer was measured using the LAL assay system. After confirmation that Gram stain was negative, endotoxin level was <5 EU/kg, and DC viability was >70%, a syringe containing the DCs was transported to the clinic. Each study subject was then vaccinated, receiving 4 200-μl intradermal injections of DCs (2.5 × 106 cells per injection site) in the skin of the upper or lower extremity, for a total dose of 1.0 × 107 DCs per vaccination.
Immune monitoring
IFN-γ ELISPOT analysis.
For every subject, the frequency of vaccine-induced antigen-specific CD4+ and CD8+ T cells was assessed using an IFN-γ ELISPOT assay (28). CD8+ and CD4+ T cells were isolated from PBMCs collected from each subject, then stimulated in vitro with autologous TAA RNA– or control RNA–transfected DCs in a standard IFN-γ ELISPOT assay.
Tetramer analysis.
For those subjects who were HLA-A0201 positive on HLA typing, PBMCs were stained with FITC-conjugated anti-CD8 mAb and PE-labeled HLA-A0201 tetramers loaded with the cP-generated peptides ELAGIGILTV126–35 or ITDQVPFSV209–217 (corresponding to MART1 and gp100, respectively; Beckman Coulter). Peptide-specific CD8+ T cells were then enumerated using flow cytometry.
Treg and Th17 cell levels in peripheral blood.
Circulating Treg levels in whole blood over the course of vaccination were assessed using Treg-specific demethylated region (TSDR) methylation analysis (19), and circulating Th17 cell levels were assessed by IL-17A demethylation analysis. For these assays, performed by Epiontis, DNA was extracted from PBMCs collected serially over the course of vaccination.
CTL activity assay.
A europium release lytic assay (28) was used to assess the induction of antitumor CTL activity against the same target cells described for IFN-γ ELISPOT. After a single in vitro restimulation with TAA RNA–transfected DCs, the T cells were incubated with cellular targets that included autologous melanoma cells and fibroblasts (as negative controls) in subjects from whom such stable cell lines could be derived.
qPCR analysis of tumor markers in whole blood.
10-ml blood samples were collected serially for qPCR analysis. After removal of rbcs, RNA was extracted from the mononuclear cells and stored at –80°C. cDNA synthesized from this RNA was then used as a template for qPCR assessment of each of the 4 TAAs used for vaccination as well as the additional melanoma marker MCAM and GAPDH as an internal control. TAA- and GAPDH-specific primers and probes were purchased from Integrated DNA Technologies, and the qPCR reaction was performed using a Stratagene Mx3005P cycler (Agilent Technologies). For each TAA qPCR reaction, copy numbers were normalized with respect to GAPDH levels.
Flow cytometric analysis of proteasome subunits
Proteasome subunit–specific mAbs were provided by S. Ferrone (Roswell Park Cancer Institute, Buffalo, New York, USA) and used as previously described (29). Briefly, DCs were fixed with 2% paraformaldehyde, microwave treated, and permeabilized with 0.1% saponin. Cells were incubated with the indicated Abs under saturating conditions. After additional incubation with FITC-conjugated F(ab)2 fragments of goat anti-mouse Ig (Jackson Laboratories), cells were analyzed by FACS.
Statistics
Data are presented as mean and SEM. Statistical differences between groups were determined using 2-tailed Student’s t test; a P value of 0.05 or less was considered significant.
Study approval
This clinical study was conducted according to Declaration of Helsinki principles under an FDA-approved Investigational New Drug (IND) Application, with Recombinant DNA Advisory Committee (RAC) and Duke University IRB approval, and was registered with Clinicaltrials.gov (NCT00672542). Written informed consent was received from all participants prior to inclusion in the study.
Supplementary Material
Acknowledgments
This study was supported in part by the Duke Clinical Research Institute/Duke Translational Medicine Institute (CTSA grant UL1RR024128). D.S. Tyler is supported by VA Merit Review funding. The authors acknowledge the financial support of Mike Muehr and Golf Pros Beating Cancer. They also thank Joe and Shelby Sherwood, as well as the Duke Melanoma Consortium and the Duke Department of Surgery, for supporting this research. Hilliard Seigler and the Duke Melanoma Research Fund have been an unwavering source of support and input for this study. Robert Schmittling and Carrie Komatas provided excellent technical assistance in vaccine preparation. Virginia Burns supervised the cell manufacturing facility, while Bruce Burnett provided invaluable help with regulatory filings. We also thank Udo Baron and Epiontis for performing the DNA methylation analysis for determining Treg and Th17 cell levels in blood.
Footnotes
Conflict of interest: Smita Nair and David Boczkowski are coinventors on a patent that describes the use of DCs transfected with tumor antigen–encoding RNA that has been licensed by Argos Therapeutics through Duke University. Smita Nair and David Boczkowski have no financial interests in Argos Therapeutics and are not compensated by Argos Therapeutics.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article:J Clin Invest. 2013;123(7):3135–3145. doi:10.1172/JCI67544.
Rebecca Haley’s present address is: Puget Sound Blood Center, Seattle, Washington, USA.
Scott K. Pruitt’s present address is: Merck Research Laboratories, Rahway, New Jersey, USA.
References
- 1.Kloetzel PM. Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat Immunol. 2004;5(7):661–669. doi: 10.1038/ni1090. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama K, et al. Replacement of proteasome subunits X and Y by LMP7 and LMP2 induced by interferon-gamma for acquirement of the functional diversity responsible for antigen processing. FEBS Lett. 1994;343(1):85–88. doi: 10.1016/0014-5793(94)80612-8. [DOI] [PubMed] [Google Scholar]
- 3.Heink S, Fricke B, Ludwig D, Kloetzel P-M, Kruger E. Tumor cell lines expressing the proteasome subunit isoform LMP7E1 exhibit immunoproteasome deficiency. Cancer Res. 2006;66(2):649–652. doi: 10.1158/0008-5472.CAN-05-2872. [DOI] [PubMed] [Google Scholar]
- 4.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macagno A, Gilliet M, Sallusto F, Lanzavecchia A, Nestle FO, Groettrup M. Dendritic cells up-regulate immunoproteasomes and the proteasome regulator PA28 during maturation. Eur J Immunol. 1999;29(12):4037–4042. doi: 10.1002/(SICI)1521-4141(199912)29:12<4037::AID-IMMU4037>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 7.Dannull J, et al. Immunoproteasome down-modulation enhances the ability of dendritic cells to stimulate antitumor immunity. Blood. 2007;110(13):4341–4350. doi: 10.1182/blood-2007-04-083188. [DOI] [PubMed] [Google Scholar]
- 8.Morel S, et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity. 2000;12(1):107–117. doi: 10.1016/S1074-7613(00)80163-6. [DOI] [PubMed] [Google Scholar]
- 9.Van den Eynde BJ, Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr Opin Immunol. 2001;13(2):147–153. doi: 10.1016/S0952-7915(00)00197-7. [DOI] [PubMed] [Google Scholar]
- 10.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 11.Romani N, et al. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods. 1996;196(2):137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 12.Su Z, et al. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003;63(9):2127–2133. [PubMed] [Google Scholar]
- 13.Kalady MF, Onaitis MW, Padilla KM, Emani S, Tyler DS, Pruitt SK. Enhanced dendritic cell antigen presentation in RNA-based immunotherapy. J Surg Res. 2002;105(1):17–24. doi: 10.1006/jsre.2002.6435. [DOI] [PubMed] [Google Scholar]
- 14.Heiser A, et al. Induction of polyclonal prostate cancer-specific CTL using dendritic cells transfected with amplified tumor RNA. J Immunol. 2001;166(5):2953–2960. doi: 10.4049/jimmunol.166.5.2953. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Wahab Z, et al. Induction of anti-melanoma CTL response using DC transfected with mutated mRNA encoding full-length Melan-A/MART-1 antigen with an A27L amino acid substitution. Cell Immunol. 2003;224(2):85–96. doi: 10.1016/j.cellimm.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Abdel-Wahab Z, et al. Cotransfection of DC with TLR4 and MART-1 RNA induces MART-1-specific responses. J Surg Res. 2005;124(2):264–273. doi: 10.1016/j.jss.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Boczkowski D, Lee J, Pruitt S, Nair S. Dendritic cells engineered to secrete anti-GITR antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther. 2009;16(12):900–911. doi: 10.1038/cgt.2009.39. [DOI] [PubMed] [Google Scholar]
- 18.Pruitt SK, et al. Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur J Immunol. 2011;41(12):3553–3563. doi: 10.1002/eji.201141383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wieczorek G, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69(2):599–608. doi: 10.1158/0008-5472.CAN-08-2361. [DOI] [PubMed] [Google Scholar]
- 20.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192(3):373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graves H, Czerniecki BJ. Circulating tumor cells in breast cancer patients: an evolving role in patient prognosis and disease progression. Patholog Res Int. 2011;2011:621090. doi: 10.4061/2011/621090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyanagi K, et al. Serial monitoring of circulating melanoma cells during neoadjuvant biochemotherapy for stage III melanoma: outcome prediction in a multicenter trial. J Clin Oncol. 2005;23(31):8057–8064. doi: 10.1200/JCO.2005.02.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rimoldi D, Salvi S, Schultz-Thater E, Spagnoli GC, Cerottini J-C. Anti-MAGE-3 antibody 57b and anti-MAGE-1 antibody 6C1 can be used to study different proteins of the MAGE-A family. Int J Cancer. 2000;86(5):749–751. doi: 10.1002/(SICI)1097-0215(20000601)86:5<749::AID-IJC24>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 24.Jungbluth AA, et al. The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood. 2005;106(1):167–174. doi: 10.1182/blood-2004-12-4931. [DOI] [PubMed] [Google Scholar]
- 25.Russo V, Traversari C, Verrecchia A, Mottolese M, Natali PG, Bordignon C. Expression of the mage gene family in primary and metastatic human breast cancer: Implications for tumor antigen-specific immunotherapy. Int J Cancer. 1995;64(3):216–221. doi: 10.1002/ijc.2910640313. [DOI] [PubMed] [Google Scholar]
- 26.Kincaid EZ, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol. 2012;13(2):129–135. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber EM, et al. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. 2012;148(4):727–738. doi: 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 28.Dannull J, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115(12):3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandoh N, et al. Development and characterization of human constitutive proteasome and immunoproteasome subunit-specific monoclonal antibodies. Tissue Antigens. 2005;66(3):185–194. doi: 10.1111/j.1399-0039.2005.00462.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.