

Abstract
The intrauterine environment is a major contributor to increased rates of metabolic disease in adults. Intrahepatic cholestasis of pregnancy (ICP) is a liver disease of pregnancy that affects 0.5%–2% of pregnant women and is characterized by increased bile acid levels in the maternal serum. The influence of ICP on the metabolic health of offspring is unknown. We analyzed the Northern Finland birth cohort 1985–1986 database and found that 16-year-old children of mothers with ICP had altered lipid profiles. Males had increased BMI, and females exhibited increased waist and hip girth compared with the offspring of uncomplicated pregnancies. We further investigated the effect of maternal cholestasis on the metabolism of adult offspring in the mouse. Females from cholestatic mothers developed a severe obese, diabetic phenotype with hepatosteatosis following a Western diet, whereas matched mice not exposed to cholestasis in utero did not. Female littermates were susceptible to metabolic disease before dietary challenge. Human and mouse studies showed an accumulation of lipids in the fetoplacental unit and increased transplacental cholesterol transport in cholestatic pregnancy. We believe this is the first report showing that cholestatic pregnancy in the absence of altered maternal BMI or diabetes can program metabolic disease in the offspring.
Introduction
Over the last 2 decades, the prevalence of obesity and type 2 diabetes has grown, resulting in a worldwide epidemic. Sedentary lifestyle, unbalanced dietary habits, and genetic predisposition are major risk factors for cardiovascular-metabolic disease (1). However, these factors alone cannot explain the epidemic levels of metabolic disease. Human studies have shown that adult disease can originate in utero and during infancy. This led to the hypothesis that the environment to which the parents are exposed can confer susceptibility to disease in the offspring in later life (2). For example, type 2 diabetes is more prevalent among people whose mothers experienced malnutrition during the Dutch famine (3), particularly in adults who were exposed to famine in utero after the first half of pregnancy, with the highest rates related to exposure in the third trimester of pregnancy (4). Moreover, maternal obesity and gestational diabetes are associated with a high risk of metabolic syndrome in childhood (5, 6). Animal studies have shown that parental metabolic disorders can predispose the offspring to cardiovascular and metabolic disease in later life (7–9).
Intrahepatic cholestasis of pregnancy (ICP) is a metabolic disease of pregnancy with a complex etiology (10–12). Affected women have genetic variation in the biliary transporters (ABCB4 and ABCB11) and nuclear receptors that influence bile acid (BA) homeostasis (10, 11, 13–16). The phenotype of ICP is likely to be caused by pathologically elevated levels of reproductive hormone metabolites in genetically predisposed women (12, 17–19). Women with ICP present with pruritus and hepatobiliary injury with hypercholanemia (elevated serum BA levels) and dyslipidemia (20). Increased maternal BA levels in ICP are associated with adverse fetal outcomes, including spontaneous preterm labor, fetal hypoxia, and meconium-stained amniotic fluid (21). Fetal BA levels are elevated in cord blood in ICP pregnancies (22). We hypothesized that exposure of the fetus to elevated BA levels in utero in ICP increases susceptibility of the offspring to metabolic disorders in adulthood.
Results
ICP affects the metabolic profile of teenage offspring.
To address whether ICP affects the metabolic health of the offspring, we analyzed the Northern Finland birth cohort 1985–1986 (NFBC 1986) database. We identified 45 ICP cases (27 male and 18 female offspring) that did not have any other known maternal liver/metabolic disease or other complications. No significant differences were observed in maternal BMI, placental size, or birth weight between normal and ICP cases. In both male and female ICP cases, the proportion of premature births (less than 37 weeks of gestational age at birth) (23) was comparable to that of the control pregnancies. Anthropometric and lipid analysis of the 16-year-old adolescents showed that males had significantly increased BMI and fasting insulin compared with the offspring of normal pregnancies. Females of the same age had significantly increased hip girth and waist girth as well as decreased fasting HDL cholesterol relative to females from normal pregnancies (Table 1). These data demonstrate that ICP impacts the subsequent metabolic health of the adolescent offspring.
Table 1.
Anthropometric and fasting lipid values in the 16-year-old offspring of ICP pregnancies (NFBC 1986 database)

Maternal hypercholanemia in mouse pregnancy results in metabolic disease of the offspring.
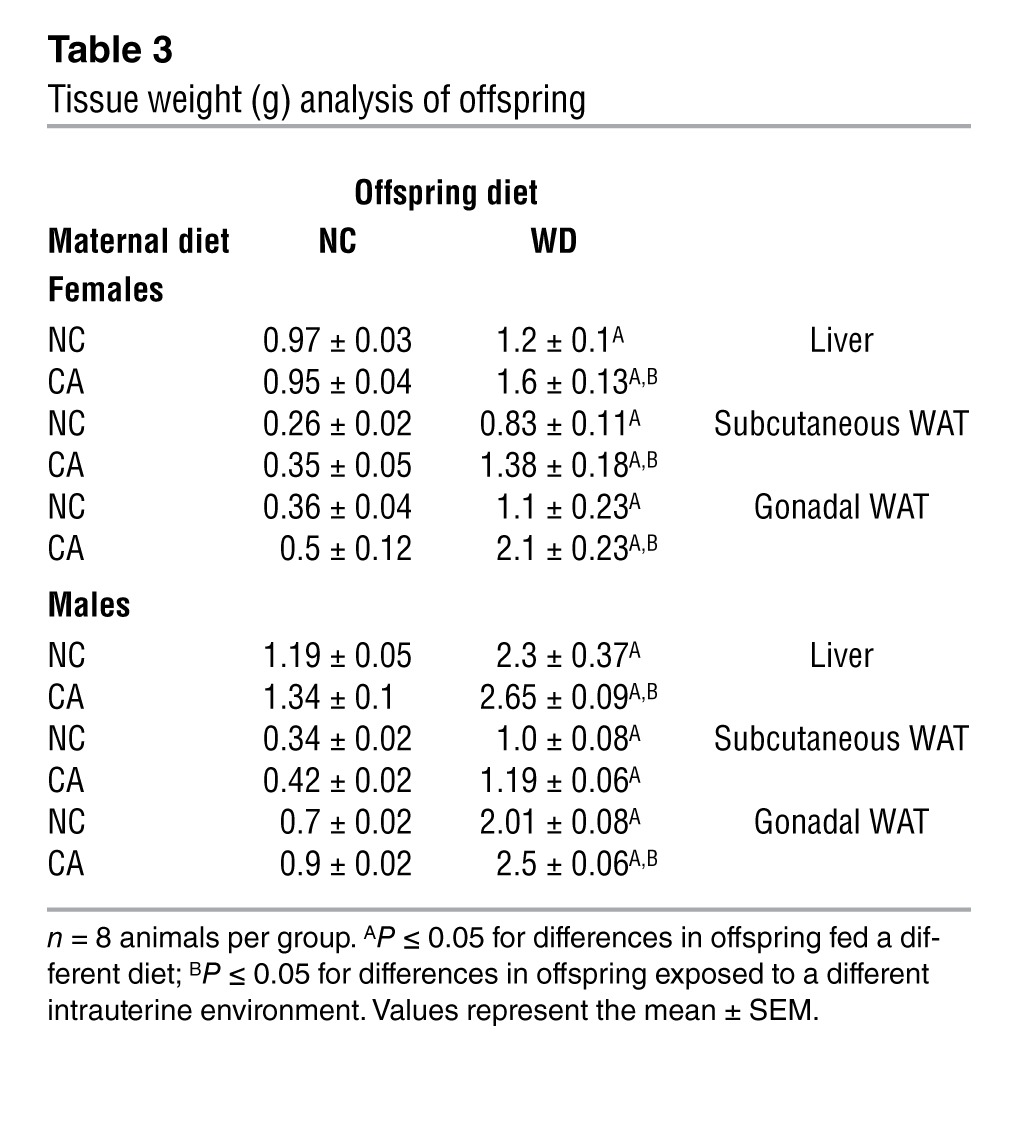
To further investigate the influence of cholestatic pregnancy on subsequent metabolic disease of the offspring, we developed a murine model of maternal hypercholanemia by supplementing the diet of wild-type female mice with 0.5% cholic acid (CA). These mice had increased serum (24) and hepatic (17) BA levels. No differences in glucose tolerance or maternal body weight were observed relative to normal chow–fed (NC-fed) mothers (Supplemental Figure 1, A and B; supplemental material available online with this article; doi: 10.1172/JCI68927DS1). Moreover, there was no significant difference in litter size (albeit a small trend toward a reduced litter size in the CA-fed mice) or fetal body weight between NC-fed and CA-fed mothers (Supplemental Table 1). However, the CA-fed mice developed dyslipidemia, as indicated by increased lipid levels in the serum and liver (Supplemental Figure 1, C and D). We assessed the metabolic phenotype of 18-week-old male and female offspring fed a lifelong NC or Western diet (WD) for 6 weeks. No alterations in body weight or glucose tolerance were observed among the offspring of NC-fed and CA-fed mothers until challenged with a WD. WD-fed female offspring of CA-fed mothers had an accelerated body weight gain after 2 weeks on the diet and developed impaired glucose tolerance as assessed 6 weeks after WD challenge (Figure 1, A and B). Additionally, these females were more dyslipidemic than the WD-fed females of NC-fed mothers, with increased serum cholesterol and LDL cholesterol, elevated levels of hepatic cholesterol, and FFAs (Table 2). Accordingly, increased vacuolization and lipid droplets (Figure 1C), as well as reduced AMPK-α and increased mTOR phosphorylation (Figure 1D) were observed in their livers, consistent with a hepatosteatotic phenotype (25, 26). These females did not respond to insulin stimulation (Figure 1E), had enlarged pancreatic islets (Figure 1F), and had reduced hepatic insulin receptor-β (IR-β) expression (Figure 1G). Moreover, the WD-fed females from CA-fed mothers had significantly increased liver and white adipose tissue (WAT) weight (Table 3), increased serum leptin levels and elevated inflammatory and metabolic markers (Supplemental Figure 2, A and B). Collectively, the WD-fed female offspring of CA-fed mothers developed a more severe obesogenic and diabetic phenotype than the WD-fed female offspring of NC-fed mothers. The hypercholanemic intrauterine environment also affected the metabolic phenotype of male offspring, but the effects were not as profound as in the female offspring. The male offspring of CA-fed mothers had significantly increased liver and WAT weight (Table 3) and showed a trend toward elevated hepatic cholesterol and triglycerides (TG) (Table 2).
Figure 1. Metabolic phenotype of 18-week-old female offspring.

(A) Body weight from the day of weaning to 18 weeks of age; n = 8 animals per group. Dashed line: time of initiation of the WD. (B) Glucose tolerance test; n = 8 animals per group. (C) Representative image of PAS- (upper panel) and toluidine blue–stained (lower panel) livers; n = 6 animals per group. Original magnification, ×20 and ×5 (insets). (D) Immunoblotting against hepatic AMPK and mTOR in pooled samples from 6 animals per group. (E) Insulin tolerance test and serum insulin measurements; n = 6 animals per group. (F) Representative pancreatic H&E-stained sections; n = 6 animals per group. Original magnification, ×20 and ×5 (insets). (G) Immunoblotting for hepatic IR-β in pooled protein samples from 6 animals per group. *P ≤ 0.05 for differences in offspring fed a different diet; #P ≤ 0.05 for differences in offspring exposed to a different intrauterine environment. NC NC, NC-fed offspring from NC-fed mothers; NC WD, WD-fed offspring from NC-fed mothers; CA NC, NC-fed offspring from CA-fed mothers; CA WD, WD-fed offspring from CA-fed mothers.
Table 2.
Serum and hepatic lipid profile of adult offspring
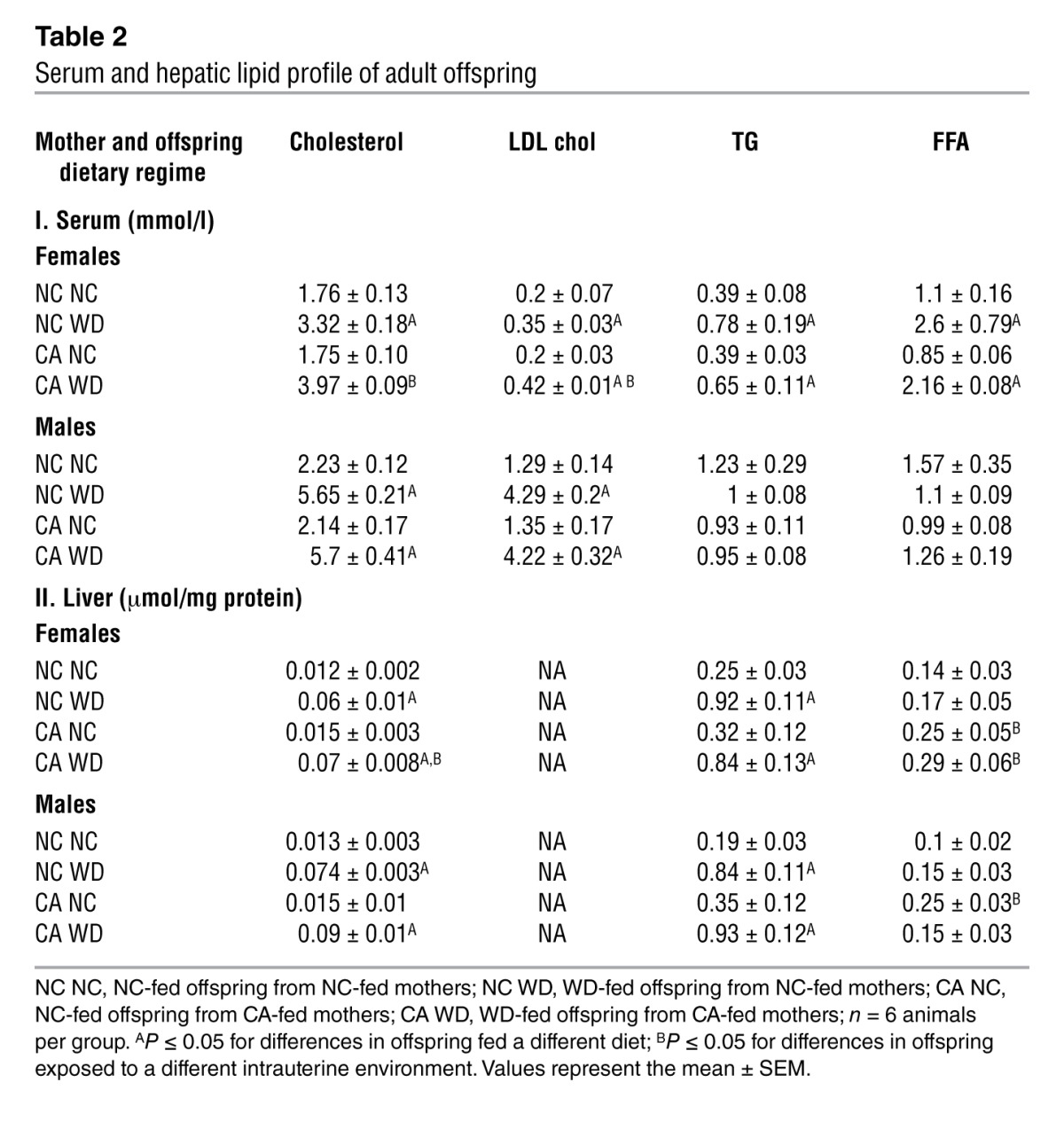
Table 3.
Tissue weight (g) analysis of offspring
Mouse cholestatic pregnancy results in an altered gene expression profile of liver and WAT in the WD-fed female offspring.
To further examine the obesogenic and diabetic phenotype of the WD-fed female offspring of CA-fed mothers, we performed a gene expression microarray analysis of their livers and WAT. Transcriptomic analysis of the livers of WD-fed female offspring from hypercholanemic mothers revealed 531 altered genes (146 downregulated and 385 upregulated), whereas WAT analysis revealed changes in 1,460 genes (308 downregulated and 1,152 upregulated) compared with the liver and WAT of WD-fed female offspring from normal mothers (data not shown). Ingenuity pathway analysis (IPA) of the transcriptomic changes predicted an upregulation of pathways involved in increased fatty acid (FA) synthesis and cleavage, neutrophil infiltration, cell movement and migration, and inflammatory response (Supplemental Tables 2 and 3). Moreover, a comparison of gene expression profiles of WAT-cultured primary adipocytes and snap-frozen WAT, both isolated from the offspring of CA-fed mothers, showed a similar gene expression profile. This provides in vitro evidence for an adipocyte-programming effect (Supplemental Figure 2C).
Offspring from CA-fed mothers are predisposed to metabolic disease.
The data presented here support the hypothesis that offspring exposed to hypercholanemia in utero are predisposed to metabolic disease. Therefore, we investigated the phenotype of NC-fed offspring that were exposed to increased BA in utero. In both NC-fed male and female offspring, there was a trend toward elevated hepatic TG levels (Table 2) as well increased adiposity (Table 3) when exposed to hypercholanemia in utero. In both NC-fed female and male offspring, hepatic FFAs were significantly increased when born from CA-fed mothers (Table 2). Light microscopy revealed mild hepatosteatosis in the NC-fed female offspring of CA-fed mothers, as demonstrated by an increase in glycogen stores and small lipid droplets compared with the female offspring of NC-fed mothers (Figure 2A). Serum proinflammatory cytokines, such as CRP and TNF-α, were increased, and antiinflammatory cytokines, such as IL-11, were decreased in the NC-fed female offspring of CA-fed mothers. The latter also had increased levels of adipocytokines that positively correlate with insulin resistance (Figure 2B). Moreover, an unsupervised transcriptomic assessment of livers from NC-fed female offspring of CA-fed mothers demonstrated differential alterations in 985 genes. Of these changes, 668 were downregulated and 317 were upregulated compared with livers from NC-fed mothers (Figure 2C). IPA predicted an increased occurrence of inflammatory, immunological, cardiovascular, and metabolic disorders (Figure 2D), with upregulation of gene networks associated with NFKB and VEGF signaling (Supplemental Figure 3). Collectively, these data illustrate increased inflammation in females that were exposed to a hypercholanemic intrauterine environment, thereby predisposing them to metabolic disease that is unmasked when challenged with a WD.
Figure 2. Female offspring from a cholestatic pregnancy are predisposed to metabolic disease.

(A) Representative images of PAS- (upper panel) and toluidine blue–stained (lower panel) livers in NC-fed female offspring; n = 6 animals per group. Original magnification, ×20 and ×5 (insets). (B) Alterations in serum adipocytokines in CA NC female offspring as assessed in 6 serum samples per animal group. (C) Unsupervised hierarchical clustering of differentially expressed hepatic genes in CA NC compared with NC NC female offspring; n = 4 animals per group. (D) IPA of differentially expressed genes from C. Threshold denotes cutoff for statistical significance. NC NC and CA NC are as defined in Figure 1.
Cholestatic pregnancy is associated with altered metabolic homeostasis in the fetoplacental unit.
Metabolic disease in the offspring can be programmed in utero through alterations in the structure or function of organs at the fetomaternal interface, e.g., the placenta (27). To test the possibility of programming effects due to changes in fetomaternal tissue structure and function, we investigated the metabolic phenotype on gestational day 18. We showed that fetuses from CA-fed mothers had a cholestatic profile as well as increased hepatic cholesterol and TG (Supplemental Table 4). Moreover, placentas from cholestatic mothers had elevated cholesterol and cholesteryl esters, increased mRNA levels of lipid storage–related genes, such as Adrp (Plin2) and Acat-2 (Figure 3, A and B), and profound steatotic vacuolization in the cytotrophoblasts (Figure 3C). These data suggest that maternal cholestasis affects lipid biosynthesis and transport in the fetoplacental unit. In support of this, we found increased de novo hepatic FAs and cholesterol biosynthesis in the fetuses of CA-fed mothers (Supplemental Table 4). Injection of pregnant mice with 14C-cholesterol between days 12 and 17 of pregnancy, when active cholesterol transport from the mother to the fetus occurs (28), established increased 14C-cholesterol in the placenta and fetal serum of hypercholanemic mice (Figure 3, D and E). These data are consistent with our observations in human pregnancies. We screened 8 ICP placentas and demonstrated increased cholesterol and FFA levels (Figure 3F). Moreover, we performed apolipoprotein A1–dependent (APOA1-dependent) cholesterol efflux assays in primary human cytotrophoblasts and found increased cholesterol efflux in response to 100 μM of taurocholic acid (TCA) (Figure 3G). This concurred with the elevated cholesterol and LDL cholesterol levels as well an increased cholesterol/HDL ratio in the umbilical cord serum of ICP patients (Figure 3H). Overall, these findings directly establish that increased levels of BA in cholestatic pregnancy potentiate excessive lipid transport and lipogenesis in the fetoplacental unit.
Figure 3. Metabolic phenotype of the fetoplacental unit in cholestatic pregnancy.

(A) Lipid measurements of mouse placenta; n = 6 animals per group. (B) Gene expression levels of Adrp and Acat2 mRNA in mouse placenta; n = 6 animals per group. (C) Representative murine toluidine blue–stained placental sections; n = 6 animals per group. Original magnification, ×20 and ×5 (insets). (D and E) Distribution of 14C-cholesterol in placenta and fetal serum/tissues; n = 8 for NC-fed mothers and n = 5 for CA-fed mothers. *P ≤ 0.05. (F) Lipid measurements of human placenta; n = 8 animals per group. (G) APOA1-mediated cholesterol efflux in human cytotrophoblasts. Representative graph of 1 experiment performed in triplicate. Error bars represent SD. *P < 0.05 versus control; #P < 0.05 versus taurocholic acid (TCA) and control. (H) Lipid measurements in umbilical cord serum. ICP cases, n = 19; control cases, n = 9. *P < 0.05 versus control. Chol, cholesterol; LDL-CHOL, LDL cholesterol; HDL-CHOL, HDL cholesterol (as defined in Table 1).
Hypercholanemia in mouse pregnancy is associated with alterations in the epigenome of the offspring.
We used the agouti viable yellow (Avy) mouse (29) to investigate whether increased BA levels during gestation affect the epigenome of the offspring. BA feeding of female mice (a/a) during pregnancy affected the coat color of both Avy/a female and male offspring (Supplemental Figure 4, A and B). Precisely, females shifted toward the pseudoagouti brown (hypomethylation of the Avy cryptic promoter), and males to the agouti yellow phenotype (hypermethylation of the Avy cryptic promoter). To confirm that coat color grouping was consistent with the methylation status of the Avy cryptic promoter, we pyrosequenced at least 4 samples per phenotype and ascertained that coat color is correlated with methylation status (Supplemental Figure 4C and refs. 30, 31).
Discussion
ICP is the most common liver-specific disease of pregnancy that affects up to 2% of pregnant women in different populations and is associated with maternal hypercholanemia and dyslipidemia (20, 22). It can have adverse outcomes for both the mother and the fetus. The majority of ICP research has focused on the etiology of the condition (12, 18, 22) and the causes of adverse pregnancy outcomes (32, 33), whereas there have been no reports of studies undertaken to explore the long-term effects of ICP on the health of the offspring. In the present study, we investigated the possibility of metabolic disease programming in offspring that were exposed to increased levels of BA in utero. To the best of our knowledge, we show for the first time, supported by several lines of evidence, that cholestatic pregnancy can program metabolic disease in the offspring. We performed both human and animal studies to establish, in vitro and in vivo, that maternal hypercholanemia affects the subsequent health of the offspring. Furthermore, we show that metabolic programming of the offspring originates in utero due to altered lipid homeosteotatic pathways in the fetoplacental unit, as a consequence of increased BA levels in the maternal bloodstream and the associated dyslipidemia.
Our initial investigation sought to determine whether ICP is likely to have an impact on human offspring. We interrogated the NFBC 1986 database and found that 16-year-old children of ICP mothers had altered lipid profiles. Also, males had increased BMI, and females had increased waist and hip girth. This phenotype is likely due to the direct result of ICP on the fetus, as pregnant women with diabetes and preeclampsia were excluded, and the data were adjusted to correct for the effects of maternal BMI, parity, and gestational age at birth. Thus, we aimed to avoid the confounding effects of disorders known to program metabolic disease in the offspring (34–36). We also found levels of lipids in umbilical cord serum from ICP cases to be increased at term. We are not aware of any medical interventions before or at delivery that could result in a dyslipidemic phenotype, either in the offspring of ICP cases from the NFBC 1986 database, or in the ICP pregnancies from which umbilical cord serum was collected. In cases of labor induction, the most common methods used were prostaglandins or oxytocin administration. Collectively, these data suggest that no factors other than maternal hypercholanemia in ICP affected the metabolic phenotype of term and adolescent offspring.
To further investigate metabolic disease in the offspring from cholestatic pregnancy, we developed a murine metabolic programming model of maternal hypercholanemia in which the offspring had increased susceptibility to metabolic syndrome. We established that maternal hypercholanemia affects the metabolic phenotype of the male and female offspring, with the female offspring developing a more severe obesogenic and diabetic phenotype that was further unmasked by WD feeding. The female offspring from CA-fed mothers were characterized by increased body weight, impaired glucose tolerance, reduced insulin sensitivity, hepatosteatosis, and an increased inflammatory response in liver and WAT. Although WD-fed male offspring had a less severe phenotype, they did have substantially increased white fat deposition and some features of dyslipidemia. There are several potential explanations for the gender discrepancies between female and male mouse and human offspring in response to a hypercholanemic intrauterine environment, including the relatively short duration of WD feeding, the adolescent nature of the NFBC 1986 offspring, and species-specific differences (37). However, the metabolic profiles of both males and females exposed to increased BA in utero, in both the mouse model and in humans with ICP, were associated with a preobese and prediabetic phenotype as well as altered morphometric and biochemical profiles. This demonstrates an increased susceptibility of the offspring to metabolic disease. We therefore conclude that maternal hypercholanemia in mice and humans has long-term implications for the health of their male and female offspring.
The phenotype of offspring exposed to CA in utero was consistent with the metaflammation hypothesis, a proinflammatory state that is directly correlated with obesity and contributes to the development of insulin resistance and metabolic dysfunction (38–40). In support of this, female offspring from CA-fed mothers had a proinflammatory, predisease phenotype that had features of metabolic syndrome after a WD challenge. The accelerated weight gain was not associated with hyperphagia, as littermates consumed equal amounts of food irrespective of the dietary regime they were exposed to prenatally and postnatally (data not shown). Moreover, hyperleptinemia was not present prior to dietary stress. It is plausible that the accelerated weight gain contributed to the impaired glucose tolerance, as this was noted 6 weeks after commencement of the WD. These data reveal that a hypercholanemic in utero environment can increase the sensitivity of the offspring to the aberrant effects of a WD challenge.
Unlike other metabolic diseases of pregnancy (41–43), no differences were observed in body birth weight or placental weight in ICP, indicating that neither of these factors is likely to be responsible for metabolic disease programming in the offspring. However, given that maternal dyslipidemia has been reported in ICP (20) and lipid parameters are raised in our cholestatic mouse model, it was plausible to hypothesize that hypercholanemia in pregnancy affects maternal-fetal nutrient/lipid availability and transfer. We showed, in vivo and in vitro, that hypercholanemia potentiates excessive cholesterol storage and transport in the fetoplacental unit, thereby altering fetal energy homeostasis. This is likely to continue postnatally, resulting in an altered metabolic phenotype in adult life, which can be unmasked when challenged by an environmental insult such as a WD, giving rise to metabolic disease. This is consistent with the thrifty phenotype hypothesis, in which fetal adaptations to survive a poor intrauterine environment are preserved throughout life (44).
Although the precise mechanisms that cause the BA-mediated programming effect in the offspring are not known, we were able to use the Avy mouse model (29–31) to demonstrate that BA feeding has an impact on the epigenome of the offspring. This is an important result, as it has been shown that in utero epigenetic alterations in the placenta and fetal blood can program metabolic disease in the offspring (45, 46). However, more detailed studies are required to investigate whether epigenetic mechanisms affect the lipidemic profile observed in the fetoplacental unit in response to a hypercholanemic environment. It is likely that the programming phenotype reported here shares components of other models of metabolic programming, specifically those precipitated by maternal metabolic disease, such as maternal obesity (47, 48).
These models of metabolic programming can be used to evaluate therapeutic or lifestyle interventions to prevent metabolic syndrome and hepatosteatosis in young adults. Ursodeoxycholic acid (UDCA) is used to treat ICP, as it has been clinically shown to ameliorate the symptoms and improve the BA profiles of affected women and neonates (49–51). In addition to treating the maternal symptoms of ICP, UDCA administration to an affected mother with ICP may improve the future metabolic health of her children. The maternal and fetal serum BA levels achieved with CA feeding of mice (24) are similar to the levels reported in ICP (22). Given that ICP affects up to 2% of women, it is feasible that cholestasis-related development of subsequent metabolic disease is a relatively common occurrence in human pregnancy. Furthermore, asymptomatic hypercholanemia of pregnancy (serum BA levels above the reference range, but no additional clinical features to suggest a diagnosis of ICP) is approximately 3 times more prevalent than ICP (52), implying that metabolic disease of the offspring due to intrauterine exposure to increased BA is a frequent phenomenon.
To our knowledge, this is the first direct evidence showing that cholestatic pregnancy can program metabolic disease in the offspring. We show altered metabolic phenotypes in 16-year-old teenagers from ICP mothers who are not diabetic or lean/obese. Determining the metabolic profiles of these offspring at an older age is warranted, and it will give further insight into the predisposition to cardiometabolic disease in the offspring as a result of maternal cholestasis. Moreover, we show, using a mouse model and an in vitro human model, that maternal hypercholanemia can alter the fetoplacental phenotype, which confers susceptibility of the offspring to metabolic disease.
Methods
Human population study
The NFBC 1986 (53) human database was analyzed for ICP cases. We evaluated mothers and 16-year-old children from 7,808 control (4,034 male and 3,774 female) pregnancies and 45 ICP (27 male and 18 female) pregnancies. UDCA was not used to treat these women, and no medical delivery interventions were reported. In cases of labor induction, oxytocin or prostaglandins were the main methods used. We excluded pregnancies in which disease or a complication other than ICP was diagnosed (e.g., viral hepatitis, obesity, diabetes, or preeclampsia). All parameters analyzed were adjusted for maternal BMI, gestational age at birth, and parity.
Human samples
Placental tissue samples from 8 control pregnancies and 8 ICP pregnancies (serum BA levels ≥40 μM) (21) were collected and snap frozen in liquid nitrogen. Placental extracts were prepared in 0.125 M potassium phosphate buffer (pH 7.4) containing 1 mM EDTA and 0.1% Triton X-100 (all from Sigma-Aldrich) and were subjected to basic lipid measurements (cholesterol, FFAs, and TG) using an LX20 multiplex autoanalyzer (Beckman Coulter). All values were normalized to total protein content measured with a BCA kit (Thermo Scientific). Human umbilical cord serum samples from term ICPs (serum BA levels ≥40 μM) and control pregnancies (18 and 10 cases, respectively) were collected for basic lipid measurements (cholesterol, LDL cholesterol, HDL cholesterol, TG).
Animal handling
Female C57BL6 mice between 10 and 12 weeks of age (Harlan, UK) that had already delivered their first litter were used. Mice were fed an NC diet (RM3 control diet) or an RM3 diet supplemented with 0.5% CA (Lillico) for 1 week before mating and throughout gestation. Pregnant mothers were either sacrificed on day 18 of pregnancy (day 1 of pregnancy was considered the day of plug identification) after a 4-hour fast, following which fetal and maternal tissues and serum were collected, or they were allowed to deliver. Mothers were switched to an NC diet upon delivery, and the litter size was reduced to 4 pups (when possible, 2 males and 2 females). No differences in the female/male ratio were observed in the offspring of NC-fed or CA-fed mice (data not shown). The offspring were weaned after 3 weeks’ lactation. When they reached 12 weeks of age, 1 female and 1 male offspring from each litter were fed a WD (Lillico) for 6 weeks, whereas their littermates were kept on an NC diet. At 18 weeks of age, the offspring were sacrificed for further analysis after a 4-hour fast. Body weight and food intake (in grams) were monitored weekly from weaning to termination. Fasting consistently took place from 8:30 am to 12:30 pm. Two independent cohorts of animals were set up (8 litters from NC-fed and 8 litters from CA-fed mothers, with different mothers for each cohort). Data from 1 animal cohort are presented for simplicity, but both gave consistent results.
For the epigenomic studies, the Avy mouse strain was used. This was a gift from Mark Christian (Institute of Reproductive and Developmental Biology, Imperial College London, London, United Kingdom). Females (10 to 12 weeks of age, a/a) were fed with an NC or CA diet for 1 week before mating with an Avy/a male (30). CA feeding continued throughout gestation and switched to an NC diet upon delivery. Seventy Avy/a offspring per maternal dietary regime were collected and separated into males and females. Male and female offspring were grouped according to their coat color as previously described (30). Liver, kidney, and brain, representing all the embryonic germ layers, were collected (31).
Glucose and insulin tolerance tests
Glucose and insulin tolerance tests (GTT and ITT, respectively) were performed after a 6-hour fast (from 8:30 am to 2:30 pm). For pregnancy experiments, mothers were assessed on day 18 of pregnancy, and for offspring studies, mice fed an NC diet or a WD were assessed 48 hours before euthanasia. Different littermates were assessed for GTT and ITT. Glucose (1 g/kg of body weight; Sigma-Aldrich) or insulin (humalin; 0.75 U/kg of body weight; Eli Lilly) was injected intraperitoneally. Blood glucose levels were monitored for 2 hours (from 2:30 to 4:30 pm) at the indicated time points after injection with a Freestyle Lite glucose sensor (Abbott Diabetes Care). Tail blood was also collected for insulin measurements.
Biochemical analysis
Serum and tissue biochemical parameters (cholesterol, LDL cholesterol, HDL cholesterol, TG, and FFAs) were measured using an LX20 autoanalyzer (Beckman Coulter). Liver and placenta were extracted in a 0.125 M potassium phosphate buffer and normalized to the total protein content as described above. Free cholesterol in tissue extracts and insulin and leptin in serum were measured using commercially available kits (Alpha Laboratories, for free cholesterol; Mercodia, for insulin; and Millipore, for leptin). Adipocytokines were assessed in pooled serum samples from 5 animals per group with a commercial array kit (R&D Systems), and several of these adipokines (e.g., leptin and adiponectin) were reassessed with ELISA kits (Millipore) to validate the data.
Histologic and morphometric analysis
Upon euthanasia, tissues were weighed before snap freezing or fixation. PAS and/or H&E staining were performed in liver, placenta, and pancreas (≥6 per dietary regime). Placentas (6 from NC-fed mothers and 6 from CA-fed mothers) and livers from female offspring (6 per dietary regime) were osmicated with tetroxide, followed by toluidine blue staining. A histopathologist blinded to the group’s identity assessed the samples.
Western immunoblotting
Equal concentrations of protein samples from the same group of animals (6 per group) were pooled, and 30 μg of protein was size fractionated with SDS-PAGE and transferred onto PVDF membranes. Membranes were probed for phospho-AMPK-α (T172; Cell Signaling Technology, 2535), AMPK-α (Cell Signaling Technology, 2532), phospho-mTOR (Ser2448; Cell Signaling Technology, 2971), mTOR (Cell Signaling Technology, 2972), and insulin receptor-β (sc-711; Santa Cruz Biotechnology). GAPDH (MAB374; Millipore) and α-tubulin (T9026; Sigma-Aldrich) were used as loading controls.
Quantitative real-time PCR
Total RNA was extracted from livers of at least 6 animals per group with the RNeasy mini kit (QIAGEN). RNA (1 μg) was reverse transcribed into cDNA with the Superscript II kit (Invitrogen). Real-Time PCR was performed with the SYBR green (Sigma-Aldrich) reagent. The primer sequences (Sigma-Aldrich) are given in Supplemental Table 5.
Transcriptomic analysis
Total RNA (4 samples per group) was extracted, and 100 ng RNA with RNA integrity number (RIN) 7.5 was reverse transcribed with a transplex whole transcriptome amplification kit (Sigma-Aldrich). Transcriptomic analysis was performed using Roche NimbleGen mouse gene expression arrays, and data were analyzed with Partek and IPA software.
Bisulfite conversion and pyrosequencing
Identification of DNA methylation levels at individual CpG sites within the Avy cryptic promoter (IAP LTR retrotransposon) was performed (30, 31). Genomic DNA from liver, brain, and kidney of 3-week-old Avy/a male and female offspring from NC- or CA-fed mothers was extracted (QIAGEN), and bisulfite was modified using the EZ DNA Methylation-Gold kit (Zymo Research). Target DNA was amplified with Invitrogen Platinum Taq polymerase, followed by pyrosequencing (54) using previously published primers (31). At least 4 samples per phenotype were pyrosequenced. No differences in CpG methylation were observed in the 3 different tissue extracts from the same animal (data not shown).
Primary adipocyte culture and differentiation
Primary adipocytes were isolated and differentiated as described (55). Pooled fat tissue dissected from 4 females fed a WD (4 from NC-fed mothers, 4 from CA-fed mothers) was used. Equal numbers of cells were seeded in duplicate and were grown to confluence. RNA was extracted, in parallel, from undifferentiated cells and from cells that were differentiated for 1 week in DMEM/10% FBS plus insulin, dexamethasone, transferrin, and T3 (Sigma-Aldrich). Quantitative PCR (qPCR) was performed for Ap2 to confirm differentiation. Differentiated cells were then assessed for genes of interest (Ctgf, Col4a1, Nos). Target gene expression levels were normalized to Ap2 mRNA levels. The primers used are listed in Supplemental Table 5.
Cholesterol transport assays in mice
Pregnant NC-fed (n = 8) or CA-fed mice (n = 5) (Harlan) were injected into the tail vein with 2 μCi 14C-cholesterol per day (specific activity 45 mCi/mmol; PerkinElmer) dissolved in 0.5 ml of saline solution (150 mM NaCl) containing 2% ethanol. 14C-cholesterol was administered for 6 days until day 17 of pregnancy. On day 18 of pregnancy, mice were fasted for 4 hours before collecting serum and tissue samples. Total cholesterol concentrations in maternal serum were determined enzymatically to calculate the corrected specific radioactivity of 14C-cholesterol, which was used to determine cholesterol levels in the maternal and fetal serum and tissues by radioactivity measurement. Tissues were homogenized in saline solution and then chemically digested with soluene and H2O2, mixed with isopropanol and scintillation cocktail, and radioactivity was measured in an LS-1800 Beckman scintillation counter (Beckman Coulter) (56).
Cholesterol efflux studies in primary human cytotrophoblasts
Cholesterol efflux was performed in primary human cytotrophoblasts isolated from term human placentas as previously described (57). Cells were seeded and grown overnight and then treated with 1 μCi/ml of 3H-cholesterol (PerkinElmer) for 24 hours. Efflux was induced with 0.1 μM of TO901317 (LXR agonist; Sigma-Aldrich) in the presence or absence of 100 μM taurocholic acid (TCA; Sigma-Aldrich) for 24 hours followed by treatment with 10 μM of APOA1 (Sigma-Aldrich) for 8 hours. Medium was then collected and cells were washed 3 times in PBS supplemented with 1 mg/ml of BSA and then dissolved in 0.1M of NaOH. Subsequently, 3H-cholesterol in media and cells were counted in liquid scintillation fluid using a Tri-Carb 2100TR Liquid Scintillation Counter (Packard). Cholesterol efflux was calculated with the following equation: cholesterol efflux % = cpm in supernatant / (cpm [cells + supernatant]). Efflux assays were set up in 4 independent cytotrophoblast cultures, and triple replicates were performed within each culture.
Statistics
For the NFBC 1986 database analysis, we used Stata version 11.2 (StataCorp). All measurements analyzed were adjusted for confounding factors such as maternal BMI, gestational age at birth, and parity. For human sample comparisons, nonparametric, 2-tailed Mann-Whitney U testing was used. For animal studies, single comparisons were performed with unpaired 2-tailed t testing, whereas multiple measures of ANOVA and Newman-Keuls post-hoc testing were run for multiple comparisons. The significance cutoff was set at P ≤ 0.05.
Study approval
Human studies.
The NFBC 1986 ( http://kelo.oulu.fi/NFBC/) was approved by the Ethics Committee of the University of Oulu and Northern Osthrobothnia Hospital District. The review board of the University of Bern approved the ethics of the study of human placental samples. Analysis of umbilical cord samples was approved by the Hammersmith Hospital Research Ethics Committee (97/5197 and 08/H0707/21). Written informed consent was obtained from all participants.
Animal studies.
All experimental procedures were approved by the Ethical Committee for Animal Welfare at Imperial College London, and all animal studies were performed in accordance with the UK Animals (Scientific Procedures) Act of 1986 and the guidelines from the Biological Sciences Unit at Imperial College London. Cholesterol transport studies were approved by the University of Salamanca Ethical Committee for the Use of Laboratory Animals.
Supplementary Material
Acknowledgments
This project was funded by the Genesis Research Trust, Imperial College London; the Biomedical Research Centre, Imperial College Healthcare NHS Trust; and the Wellcome Trust. NFBC 1986 is funded by the Academy of Finland, University Hospital Oulu; the European Commission; the Medical Research Council (United Kingdom); and the EU Framework Programme 7. The authors would like to acknowledge Shikta Das, Ville Huikari, and Marika Kaakinen for assistance with NFBC 1986 data accumulation, and Paul T. Seed for advice and guidance in statistical analysis of the NFBC 1986 database. We thank Mark Christian for providing the Avy mouse strain, Ela Begun-Laroy of Hammersmith Hospital, for assessing umbilical cord serum samples, and Kirsty J. Flower for technical support in pyrosequencing. We also appreciate the assistance with microarray data analysis provided by the Partek customer service staff.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2013;123(7):3172–3181. doi:10.1172/JCI68927.
See the related Commentary beginning on page 2786.
References
- 1.Swinburn BA, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378(9793):804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4(2B):611–624. doi: 10.1079/phn2001145. [DOI] [PubMed] [Google Scholar]
- 3.van Abeelen AF, et al. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes. 2012;61(9):2255–2260. doi: 10.2337/db11-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravelli AC, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351(9097):173–177. doi: 10.1016/S0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 5.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 6.Tzoulaki I, et al. Relation of immediate postnatal growth with obesity and related metabolic risk factors in adulthood: the northern Finland birth cohort 1966 study. Am J Epidemiol. 2010;171(9):989–998. doi: 10.1093/aje/kwq027. [DOI] [PubMed] [Google Scholar]
- 7.Samuelsson AM, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51(2):383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 8.Sheau-Fang N, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467(7318):963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- 9.Burns SP, et al. Gluconeogenesis, glucose handling, and structural changes in livers of the adult offspring of rats partially deprived of protein during pregnancy and lactation. J Clin Invest. 1997;100(7):1768–1774. doi: 10.1172/JCI119703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu-Hayyeh S, et al. Inhibition of Na+-taurocholate Co-transporting polypeptide-mediated bile acid transport by cholestatic sulfated progesterone metabolites. . J Biol Chem. 2010;285(22):16504–16512. doi: 10.1074/jbc.M109.072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon PH, et al. Contribution of variant alleles of ABCB11 to susceptibility to intrahepatic cholestasis of pregnancy. Gut. 2009;58(4):537–544. doi: 10.1136/gut.2008.159541. [DOI] [PubMed] [Google Scholar]
- 12.Abu-Hayyeh S, et al. Intrahepatic cholestasis of pregnancy levels of sulfated progesterone metabolites inhibit FXR resulting in a pro-cholestatic phenotype. Hepatology. 2013;57(2):716–726. doi: 10.1002/hep.26055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Mil SW, et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133(2):507–516. doi: 10.1053/j.gastro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Dixon PH, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet. 2000;9(8):1209–1217. doi: 10.1093/hmg/9.8.1209. [DOI] [PubMed] [Google Scholar]
- 15.Mullenbach R, et al. ABCB4 gene sequence variation in women with intrahepatic cholestasis of pregnancy. J Med Genet. 2003;40(5):e70. doi: 10.1136/jmg.40.5.e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauli-Magnus C, et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14(2):91–102. doi: 10.1097/00008571-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Milona A, et al. Raised hepatic bile acid concentrations during pregnancy in mice are associated with reduced farnesoid X receptor function. Hepatology. 2010;52(4):1341–1349. doi: 10.1002/hep.23849. [DOI] [PubMed] [Google Scholar]
- 18.Reyes H, Sjovall J. Bile acids and progesterone metabolites in intrahepatic cholestasis of pregnancy. Ann Med. 2000;32(2):94–106. doi: 10.3109/07853890009011758. [DOI] [PubMed] [Google Scholar]
- 19.Papacleovoulou G, Abu-Hayyeh S, Williamson C. Nuclear receptor-driven alterations in bile acid and lipid metabolic pathways during gestation. Biochim Biophys Acta. 2011;1812(8):879–887. doi: 10.1016/j.bbadis.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Dann AT, Kenyon AP, Wierzbicki AS, Seed PT, Shennan AH, Tribe RM. Plasma lipid profiles of women with intrahepatic cholestasis of pregnancy. Obstet Gynecol. 2006;107(1):106–114. doi: 10.1097/01.AOG.0000189096.94874.9c. [DOI] [PubMed] [Google Scholar]
- 21.Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: Relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40(2):467–474. doi: 10.1002/hep.20336. [DOI] [PubMed] [Google Scholar]
- 22.Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049–2066. doi: 10.3748/wjg.15.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milona A, et al. The normal mechanisms of pregnancy-induced liver growth are not maintained in mice lacking the bile acid sensor Fxr. Am J Physiol Gastrointest Liver Physiol. 2010;298(2):G151–G158. doi: 10.1152/ajpgi.00336.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Um SH, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 26.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jansson T, Ekstrand Y, Bjorn C, Wennergren M, Powell TL. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes. 2002;51(7):2214–2219. doi: 10.2337/diabetes.51.7.2214. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida S, Wada Y. Transfer of maternal cholesterol to embryo and fetus in pregnant mice. J Lipid Res. 2005;46(10):2168–2174. doi: 10.1194/jlr.M500096-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolinoy DC, Hu D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolinoy DC, Weinhouse C, Jones TR, Rozek LS, Jirtle RL. Variable histone modifications at the A(vy) metastable epiallele. Epigenetics. 2010;5(7):637–644. doi: 10.4161/epi.5.7.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miragoli M, et al. A protective antiarrhythmic role of ursodeoxycholic acid in an in vitro rat model of the cholestatic fetal heart. Hepatology. 2011;54(4):1282–1292. doi: 10.1002/hep.24492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Germain AM, Kato S, Carvajal JA, Valenzuela GJ, Valdes GL, Glasinovic JC. Bile acids increase response and expression of human myometrial oxytocin receptor. Am J Obstet Gynecol. 2003;189(2):577–582. doi: 10.1067/S0002-9378(03)00545-3. [DOI] [PubMed] [Google Scholar]
- 34.Kim SY, England JL, Sharma JA, Njoroge T. Gestational diabetes mellitus and risk of childhood overweight and obesity in offspring: a systematic review. Exp Diabetes Res. 2011;2011:541308. doi: 10.1155/2011/541308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis EF, et al. Pre-eclampsia and offspring cardiovascular health: mechanistic insights from experimental studies. Clin Sci (Lond). 2012;123(2):53–72. doi: 10.1042/CS20110627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vuguin P, Raab E, Liu B, Barzilai N, Simmons R. Hepatic insulin resistance precedes the development of diabetes in a model of intrauterine growth retardation. Diabetes. 2004;53(10):2617–2622. doi: 10.2337/diabetes.53.10.2617. [DOI] [PubMed] [Google Scholar]
- 37.Aiken CE, Ozanne SE. Sex differences in developmental programming models. Reproduction. 2013;145(1):R1–R13. doi: 10.1530/REP-11-0489. [DOI] [PubMed] [Google Scholar]
- 38.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 39.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140(3):338–348. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36(1):62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 42.Barker DJ, Martyn CN, Osmond C, Hales CN, Fall CH. Growth in utero and serum cholesterol concentrations in adult life. BMJ. 1993;307(6918):1524–1527. doi: 10.1136/bmj.307.6918.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hales CN, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303(6809):1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35(7):595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 45.El Hajj N, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62(4):1320–1328. doi: 10.2337/db12-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Godfrey KM, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60(5):1528–1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mouralidarane A, et al. Hepatology. Maternal obesity programs offspring non-alcoholic fatty liver disease via innate immune dysfunction in mice [published online ahead of print January 12, 2013]. doi: 10.1002/hep.26248 . [DOI] [PubMed] [Google Scholar]
- 48.McCurdy CE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119(2):323–335. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chappell LC, Gurung V, Seed PT, Chambers J, Williamson C, Thornton JG. Ursodeoxycholic acid versus placebo, and early term delivery versus expectant management, in women with intrahepatic cholestasis of pregnancy: semifactorial randomised clinical trial. BMJ. 2012;344:e3799. doi: 10.1136/bmj.e3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bacq Y, et al. Efficacy of ursodeoxycholic acid in treating intrahepatic cholestasis of pregnancy: a meta-analysis. Gastroenterology. 2012;143(6):1492–1501. doi: 10.1053/j.gastro.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis of pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42(6):1399–1405. doi: 10.1002/hep.20952. [DOI] [PubMed] [Google Scholar]
- 52.Pascual MJ, Serrano MA, El-Mir MY, Macias RI, Jimenez F, Marin JJ. Relationship between asymptomatic hypercholanaemia of pregnancy and progesterone metabolism. Clin Sci (Lond). 2002;102(5):587–593. doi: 10.1042/CS20010258. [DOI] [PubMed] [Google Scholar]
- 53.Jarvelin MR, et al. Ecological and individual predictors of birthweight in a northern Finland birth cohort 1986. Paediatr Perinat Epidemiol. 1997;11(3):298–312. doi: 10.1111/j.1365-3016.1997.tb00007.x. [DOI] [PubMed] [Google Scholar]
- 54.Brennan K, et al. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012;72(9):2304–2313. doi: 10.1158/0008-5472.CAN-11-3157. [DOI] [PubMed] [Google Scholar]
- 55.Gupta RK, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15(2):230–239. doi: 10.1016/j.cmet.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vicens M, et al. Inhibition of the intestinal absorption of bile acids using cationic derivatives: mechanism and repercussions. Biochem Pharmacol. 2007;73(3):394–404. doi: 10.1016/j.bcp.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 57.Nikitina L, Wenger F, Baumann M, Surbek D, Korner M, Albrecht C. Expression and localization pattern of ABCA1 in diverse human placental primary cells and tissues. Placenta. 2011;32(6):420–430. doi: 10.1016/j.placenta.2011.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.