

Abstract
Mutations in the common gamma chain (γc, CD132, encoded by the IL2RG gene) can lead to B+T−NK− X-linked severe combined immunodeficiency, as a consequence of unresponsiveness to γc-cytokines such as interleukins-2, -7 and -15. Hypomorphic mutations in CD132 may cause combined immunodeficiencies with a variety of clinical presentations. We analyzed peripheral blood mononuclear cells of a 6-year-old boy with normal lymphocyte counts, who suffered from recurrent pneumonia and disseminated mollusca contagiosa. Since proliferative responses of T cells and NK cells to γc -cytokines were severely impaired, we performed IL2RG gene analysis, showing a heterozygous mutation in the presence of a single X-chromosome. Interestingly, an IL2RG reversion to normal predominated in both naïve and antigen-primed CD8+ T cells and increased over time. Only the revertant CD8+ T cells showed normal expression of CD132 and the various CD8+ T cell populations had a different T-cell receptor repertoire. Finally, a fraction of γδ+ T cells and differentiated CD4+CD27− effector-memory T cells carried the reversion, whereas NK or B cells were repeatedly negative. In conclusion, in a patient with a novel IL2RG mutation, gene-reverted CD8+ T cells accumulated over time. Our data indicate that selective outgrowth of particular T-cell subsets may occur following reversion at the level of committed T progenitor cells.
Introduction
Somatic mosaicism is defined as the presence of genetically distinct populations of somatic cells in a given organism.1 Although somatic mosaicism is assumed to be frequently masked, it can also result in major phenotypic changes and reveal the expression of otherwise lethal genetic mutations. Mosaicism can be caused by DNA mutations, epigenetic alterations of DNA, chromosomal abnormalities and the spontaneous reversion of inherited mutations. Like somatic mosaicism due to de novo mutations during embryogenesis, mosaicism due to reversions to normal of an inherited mutation have been discovered because of milder than expected clinical courses and/or the presence of both phenotypically normal and abnormal cells in vivo and in vitro.2 In the case of spontaneous reversion, the mutation that has reverted to normal in some cells has been inherited from a parent.
Establishment of significant somatic mosaicism following reversion to normal could modify disorders in which revertant cells have a selective advantage in vivo. The rate of cell turnover may be a factor that contributes to the chance of spontaneous reversion as such. Most spontaneous reversions have been found to occur in severe skin disease and hematologic or immunological diseases.2–6 The growth advantage of revertant cells has been the central dogma for the success of gene therapy in severe combined immunodeficiency (SCID).7,8
SCID is a collection of various gene defects that result in the absence of T lymphocytes with or without the involvement of B cells and/or NK cells.9 To date, spontaneous gene reversions have been reported in several patients suffering from ADA-SCID.10–14 This form of SCID that causes a metabolic disease is due to defective adenine deaminase (ADA) activity in all cells. It has been found that lymphocytes are particularly sensitive to the toxic effects of the accumulating purine metabolites in ADA deficiency. Reversed mutations have also been found in patients with Wiskott-Aldrich syndrome15–17 and other primary T-cell immunodeficiencies such as those caused by mutations in CD3zeta, RAG-1, LAD and NEMO.5,18–21 The most common type of SCID in humans shows X-linked inheritance and is caused by mutations in the IL2RG gene.22,23 This gene encodes an essential subunit, the common γ-chain (γc, CD132), of a cytokine receptor subfamily that is essential for lymphocyte development, T-cell homeostasis, and peripheral immune responses.24 To date, only three examples of gene reversion of an inherited mutation have been reported in X-linked SCID and in all three cases were in the common T-cell precursors, hence resulting in reverted CD4+ and CD8+ T cells of both the TcRαβ and TcRγδ signature.25–27
CD132 is a type I transmembrane glycoprotein that serves as a subunit for the receptors (R) of interleukin (IL)-2, IL-4, IL-7, IL-9, IL-15 and IL-21. The IL-2R and IL-15R are heterotrimers consisting of unique α-chains and shared IL-2Rβ (CD122) and the CD132 subunit, whereas the IL-4R, IL-7R, IL-9R, and IL-21R are heterodimers of unique α-subunits and CD132. For each of the receptors CD132 contributes to ligand binding affinity, but also to signal transduction through the direct association of Jak-3 to its cytoplasmic tail.22–24,28 Mutations in CD132 abolish the function in each of these cytokine receptors, resulting in the absence or diminished numbers of T and NK cells, while B-cell development is normal. X-SCID is diagnosed early in life, but some exceptional cases and families have been reported in which CD132 mutations resulted in an immunological phenotype distinct from classical X-SCID.29–34 These so-called X-CID (combined immune deficiency) patients may have impaired rather than absent function of CD132. Mosaicism due to somatic gene reversion has been observed in the immune system as well as in various other disease entities.1–6,17,35–37
Here we report a novel X-(S)CID family with a unique mutation in the extracellular part of CD132.
Design and Methods
Additional details on the design and methods of this study are provided in the Online Supplement.
Samples
Heparinized peripheral blood samples were collected from the patient longitudinally. Peripheral blood mononuclear cells (PBMC) were isolated using standard density gradient centrifugation techniques and in some instances cryopreserved until use. The patient’s samples were analyzed simultaneously with PBMC obtained from healthy (age-matched) control donors. All participants provided written consent after being fully informed about the study. The study was approved by the Medical Ethical Committee of the Academic Medical Center in Amsterdam which acts according to the principles of the Declaration of Helsinki (version Seoul 2008).
Immunofluorescent staining and flow cytometry
Absolute numbers of T cells, B cells and NK cells were determined in whole blood with Multitest six-color reagents (BD Biosciences, Erembodegem, Belgium). For analysis of isolated PBMC, cells were resuspended in phosphate buffered saline (PBS), containing bovine serum albumin - sodium azide and incubated with fluorescently labeled conjugated monoclonal antibodies.
Proliferation assays
PBMC were resuspended in PBS, labeled with of 5,6-carboxy-fluorescein diacetate succinimidyl ester (CFSE) in PBS, washed and resuspended in culture medium and subsequently cultured for 6 days. T cells were stimulated with anti-CD3 with or without anti-CD28 monoclonal antibodies and/or with specific cytokines at an optimal dose for control cells. For antigen-specific CD4+ T-cell stimulation tetanus toxoid, cytomegalovirus (CMV), varicella-zoster virus (VZV) and Candida albicans antigens were used. For B-cell proliferation, cells were stimulated with different combinations of anti-IgM, anti-CD40, IL-21, CpG ODN 2006 and IL-2. Supernatants were tested for secreted IgM and IgG with an in-house enzyme-linked immunosorbent assay.38
IL2RG gene sequence analysis
Genomic DNA was isolated from total peripheral blood leukocytes and sorted subsets of lymphocytes.39 The coding exons of the IL2RG gene (NCBI NM_000206) were amplified by polymerase chain reaction and sequenced using the BigDye Terminator v1.1 cycle sequencing kit and a 3130 genetic analyzer (Applied Biosystems). Short tandem repeats were analyzed with the PowerPlex16 kit (Promega, Madison, WI, USA).40
T-cell receptor repertoire analysis by high throughput sequencing
The T-cell receptor repertoire of sorted cell populations was analyzed as previously described.41,42 The complementarity determining region (CDR)-3 of the TCR-β-chain was used as a clonal tag to identify individual clones. Briefly, mRNA was isolated and cDNA was synthesized. In the first step of linear amplification cDNA was amplified using a modified version of the Vβ primerset described by Dongen et al.43 All V-primers were tailed with the primerB sequence needed for sequencing according to the 454 Titanium protocol for Amplicon sequencing (Roche Diagnostics, Mannheim, Germany). In this article we use the HUGO nomenclature according to Folch and Lefranc.44 After linear amplification a generic polymerase chain reaction was performed to prepare the samples for sequencing. Over 40,000 (bead-bound) TCR sequences were analyzed.
Results
These studies were performed to diagnose and further elucidate the molecular mechanisms of disease of a male patient, the off-spring of non-consanguineous parents, who was repeatedly admitted to the pediatric pulmonology department because of infectious diseases. The initial immunological screening did not point in the direction of any immunological diagnosis and the humoral immune response to vaccination was positive.
When the patient was brought to our attention at the age of 6 years, two notable findings were suspicious. First, the mother’s twin brother had died at the age of 14 years from an unexplained disease with liver failure and progressive bronchiectasis. He had been diagnosed at that time with acrodermatitis eczematicum in the presence of a low serum zinc and/or a less penetrant form of cystic fibrosis, which could not be diagnosed at the DNA level back then. Secondly, the index case was covered with molluscum contagiosum virus lesions, which had not resolved after 3 years.
We repeated serological tests and found a complete absence of antibodies against the standard immunizations that the patient had received in the past [including measles-mumps-rubella (MMR), diphtheria toxoid-tetanus toxoid-inactivated pertussis polio virus vaccines]. Epstein-Barr virus (EBV) and CMV titers were screened as well as VZV serology since the boy had suffered from mild chickenpox in the past. All serological tests were negative in the presence of normal total values of IgG (12.6 g/L), IgA (0.58 g/L) and IgM (0.89 g/L). Upon repeating the vaccinations against pneumococcal polysaccharides (PPS; 23-valent Pneumovax), tetanus toxoid and MMR, we observed normal induction of antibodies. However, upon follow-up the antibodies had completely disappeared within 2–4 months to undetectable levels again.
High resolution computed tomography scanning showed the presence of bronchiectasis. Cystic fibrosis was excluded at the DNA level, zinc levels were normal, other liver/lung diseases were excluded, including α1-antitrypsin deficiency and primary ciliary disease. The patient was put on monthly infusions of intravenous immunoglobulins and co-trimoxazole prophylaxis. His skin was enucleated in a 3-hour course of “peeling”, but molluscum contagiosum lesions returned and disseminated again. During follow-up the clinical course improved considerably. Over the last 7 years he has suffered only once from an atypical pneumonia caused by Mycoplasma pneumoniae, which responded favorably to macrolide antibiotics and after several years his molluscum contagiosum started to disappear spontaneously.
Lymphocyte counts and subsets
As indicated by the absolute lymphocyte counts, all subsets were in the low-normal range and remained stable apart from the absolute numbers of CD8+ T cells which increased over time and reached very high numbers resulting in an inverted CD4/CD8 ratio (Figure 1A). T-cell subset analysis showed decreased percentages of naïve (CD45RA+CD27+) CD4+ and CD8+ T cells for age concomitant with increased percentages of memory T cells (CD45RA−CD27+), especially in the CD8+ T-cell subset (Figure 1B). This disturbed CD8+ T-cell differentiation progressed over the years (Online Supplementary Figure S1), whereas little change was seen in CD4+ T-cell differentiation. TREC levels in CD3-sorted T cells were within the normal range (data not shown). In the circulating B-cell fraction, there was a notable lack of switched-memory B cells with the IgD−CD27+ phenotype (<2% over time; normal age-matched range, 6–15%) (Figure 1B).
Figure 1.

Increased numbers and disturbed differentiation of CD8+ T cells. (A) Absolute numbers of total and CD4+ and CD8+ T cells, CD4/CD8 ratio, CD3−CD56+ NK cells and CD19+ B cells. Dotted lines indicate upper and lower limits of normal for corresponding age (changing at age 10 years), for CD4/CD8 ratio only the lower limit of normal is shown. (B) Distribution of naïve, memory and effector-memory T cells in both CD3+CD4+ and CD3+CD8+ T cells (i.e. CD45RA+CD27+, CD45RA−CD27+, CD45RA+or−CD27−, respectively), at 11 years of age compared to values in a 10-year old healthy control. Lower panels: B-cell differentiation (naïve, non-switched or class-switched memory, i.e. sIgD+CD27−sIgD+CD27+ or sIgD−CD27+).
Lymphocyte proliferation tests and cytokine receptor expression
We tested the patient’s CD4+ and CD8+ T cells for αCD3/αCD28-mediated proliferation using CFSE dilution and this was completely normal; almost all T cells underwent several rounds of division (Figure 2A). The antigen-specific memory responses were defined by assessment of T-cell responsiveness against CMV, VZV, tetanus toxoid and Candida albicans. Although the boy had been frequently boosted with tetanus toxoid, had been infected by chickenpox in the past, and must have been colonized by Candida albicans before, no appropriate reaction to these stimuli could be detected (Figure 2B).
Figure 2.

T-cell proliferation in response to recall antigens and cytokines is diminished. Analysis of proliferation of healthy control and patient CFSE-labeled cells after 6 days of culture with the indicated stimuli. (A) Proliferation of CD4+ and CD8+ T cells activated with αCD3/αCD28 monoclonal antibodies. (B) Proliferation of antigen-specific CD4+ T cells to recall antigens. The number indicate percentages of divided cells minus the background in unstimulated samples. (C) Proliferation of T cells in response to CD132 cytokines IL-2, IL-15 and IL-7. The number indicate percentages of undivided cells. No stim = unstimulated condition.
Finally, we tested the proliferative responses of T and NK cells toward the CD132 cytokines IL-2, IL-15 and IL-7 (Figure 2C and data not shown). Proliferation of NK and T cells, especially CD4+ T cells, was diminished compared to that of control cells. Cytokine titration curves also showed that CD4+ T-cell responses were more severely affected than CD8+ T-cell proliferation at different concentrations (Online Supplementary Figure S2).
The proliferative capacity of the boy’s B cells and simultaneous differentiation into CD20lowCD38high plasma cells was analyzed using BCR-independent B-cell stimulation with CpG. B-cell proliferation was normal but CD132-dependent IL-2 signaling had no additional effect as observed in cells from control donors (Online Supplementary Figure S3). A similar defect was found when B cells were activated via the BCR by a combination of αIgM and αCD40 monoclonal antibodies in the absence or presence of IL-21, again the patient’s cells did not respond to the extra cytokine signal. Interestingly, the patient’s B cells did not show any proliferation in response to indirect activation via the agonistic αCD3/αCD28 monoclonal antibody combination for T-cell stimulation (Online Supplementary Figure S3, lower panels), as demonstrated before in another X-linked CID case34. Together these data show that the patient’s B cells are unresponsive to common γ chain cytokines. The culture supernatants were analyzed for in vitro IgM and IgG synthesis, which was absent and hence indicated defective maturation into Ig-secreting cells, irrespective of BCR-dependent or BCR-independent activation (data not shown).
Family history, X-chromosome inactivation and IL2RG gene analysis
The family history indicated an X-linked pattern of inheritance (Figure 3A). The X-linked nature of this immunodeficiency was further supported by studies in the obligate carriers [mother (II:1) and grandmother (I:2)], demonstrating 100% skewing in X-inactivation in memory CD4+ and CD8+ T cells (data not shown). The mother’s sister (II:4) delivered an affected male during follow-up. He had normal numbers and percentages of CD4+ and CD8+ T cells, B cells and NK cells, normal CD4/CD8 ratio (1:1) and almost all T cells had a naïve phenotype as expected in a neonate (CD4+ T cells 92%, CD8+ T cells 81% naïve). However, γc-cytokine induced proliferation of T and NK cells was strongly diminished.
Figure 3.
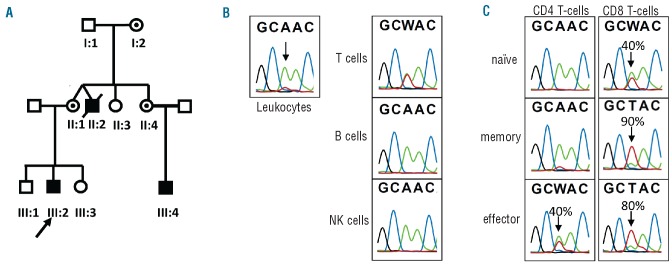
X-linked inheritance of the IL2RG gene locus and mutation analysis. (A) Pedigree of the family indicates the X-linked inheritance by the index (III-2) who is alive and well, and the affected brother (II:2) who died from X-CID. The patient’s mother (II:1), one of her sisters (II:4), and the grandmother (I:2) are carriers of the mutated IL2RG gene (circles with dots). (B) Gene sequence analysis is shown for the patient in leukocytes, and sorted total T, B and NK cell subsets. Fifty percent of T cells had a reversion mutation (back mutation, indicated in red). (C) Percentage reversion in T-cell subsets at t=2 years; (naïve CD45RA+CD27+, memory CD45RA−CD27+, effector CD45RA+/−CD27−). See also Online Supplementary Table S1.
Mutation analysis of the IL2RG gene was performed on DNA isolated from total leukocytes of the index patient and showed a single nucleotide substitution (c.655A>T) representing a missense mutation resulting in a change at amino acid position 219 from a tyrosine into an asparagine residue (p.Tyr219Asn). In addition to the mutated allele, a wild-type allele was detected, although at a lower frequency.
The boy had no stigmata of Klinefelter disease (XXY) which could explain the presence of two alleles. An alternative explanation for this genetic finding was the presence of circulating maternal T cells. However, this was formally excluded by HLA-genotyping and short tandem repeat marker analysis. We, therefore, concluded that the wild-type allele originated from somatic reversion. To determine in which lymphocyte subpopulation the reversed wild-type allele was present, patient’s CD19+ B cells, CD3−CD16/56+ NK cells and CD3+ T cells were sorted from peripheral blood followed by sequence analysis. In T cells the mutated and the reversed wild-type IL2RG allele were detected with a similar frequency, whereas in B and NK cells only the mutated allele was present (Figure 3B). In summary, the patient had a missense mutation in the ILR2G gene that had reverted to a wild-type allele in approximately half of the T cells.
IL2RG mutation analysis in sorted T-cell subsets
Over the years the number of CD8+ T cells increased and this increase was observed in the memory fraction (Online Supplementary Figure S1). To determine in which T-cell subset the reverted allele was present and whether this influenced the observed changes over time in dominance of the CD8+ T-cell subset, CD4+ and CD8+ naïve (CD45RA+CD27+), memory (CD45RA−CD27+), and effector (CD45RA−CD27−) T cells were sorted and the frequency of reversion was determined at two time points (t=0 and t=2 years). In naïve CD4+ T cells only the non-reverted, mutated allele was detected at both time points, in memory CD4+ T cells only a small percentage was reverted (Online Supplementary Table S1). At the second time point 40% of the usually small fraction of CD4+CD27−effector T cells was found to contain the reverted allele (Figure 3C). In contrast, the reverted allele was found in 5% of the naïve CD8+ T cells and in 25% of memory CD8+ T cells, and over a period of 2 years, these values rose to 40% of the naïve CD8+ T cells and 90% of memory CD8+ T cells. The effector CD8+ T cells had a reversion frequency of 80% (t=2 years). Finally, in two additional sorts from different time points, TCRγδ+ T cells were isolated and in this T-cell population the reversion frequency was stable at 20% (Online Supplementary Table S1). These results show that the reverted allele was present in different T-cell lineages, indicating a progenitor T cell as the common source of the circulating revertant T cells.
Properties of the CD8+ T-cell population
When the findings on partial gene reversion became apparent, we tested for the expression of CD132 and clonal composition in both the naïve and memory CD4+ and CD8+ compartments. CD132 expression was tested after activation of the PBMC for 3 days with αCD3/αCD28 monoclonal antibodies. In contrast to the very low CD132 expression on activated CD4+ T cells, CD132 expression was clearly present on CD8+ T cells after stimulation (Figure 4). The highest expression of CD132 was detected on the CD27− memory-effector CD8+ T cells. CD132 was not expressed on the patient’s B and NK cells. Expression of CD25 and CD122 (the other chains of the IL-2 receptor) was strongly up-regulated as normal on both activated CD4+ and CD8+ T cells (data not shown).
Figure 4.

Decreased expression of CD132 on lymphocytes of the X-CID patient. CD132 expression on lymphocyte subsets upon 3-day stimulation using antiCD3/antiCD28 monoclonal antibodies. Filled histograms: unstimulated cells, black line: activated cells.
The patient’s TCR repertoire was suggested to be intact by the normal variety found by flow cytometric analysis using 24 Vβ monoclonal antibodies covering about 80% of Vβ antigens on the patient’s T cells (data not shown). Subsequent analysis of the sorted subsets of CD8+ T cells from the patient was performed, showing a broad Vβ repertoire by monoclonal antibody staining in both the sorted naïve (CD45RA+CD27+) as well as memory CD4+ and CD8+ T-cell subsets.
Using subsequent high-throughput TCR-β sequencing, we tested whether the expanded revertants would dominate the memory repertoire by an oligoclonal signature. Data from previous studies on clonality in the naïve and memory compartments were used as a reference.41 By sequencing >7000 individual TCR in the patient’s CD8+ T cells, we found that the clonal distributions in the CD8+CD45RA− (memory) subset and the CD8+CD45RA+CD27+ (naïve) T-cell subset were normal when compared to those of healthy individuals, showing no signs of oligoclonal signatures (Figure 5A). This was further confirmed by the diverse use of the (variable) genes of the TCR-β-chain (Figure 5B). This broad repertoire indicates that normal VDJ-rearrangement had taken place in the naïve T-cell subset, which is similar to that of healthy individuals. The non-revertant component may have contributed to the fully diversified repertoire of the memory CD8 T+ cell subset but as this is a relatively small fraction of the total cell population it seems unlikely that the full diversity can be attributed to these cells. Thus, the revertant populations were highly diverse in their TCR repertoire and not oligoclonal in nature.
Figure 5.

TCR-β analysis in reverted cells shows a diverse repertoire. (A) CD8+CD45RA− (memory) subset and the CD8+CD45RA+CD27+ (naïve) were sorted using FACS (>95% purity). Both samples were prepared for high throughput sequencing using a primer set that covers the complete TCR-β repertoire. From both samples approximately 10,000 TCR-β sequences were sequenced using high throughput sequencing. Each clone is depicted as a dot showing its relative frequency to the complete subset (percentage of total reads). As a comparison we used control samples. (B) Vβ-usage of the same samples used in (A) was determined. Use of V-genes (and J-genes, data not shown) was comparable to that of healthy controls (data not shown).
Discussion
In this study we describe a novel family with non-classical X-(S)CID and a spontaneous reversion of the genetic defect that was identified in some of the CD8+ T cells. The patient was admitted with recurrent pulmonary infections and widely spread molluscum contagiosa virus lesions on his skin. The X-linked pattern of inheritance combined with a defect in cytokine-mediated T-cell proliferation led to the identification of a missense mutation in the gene coding for CD132 (IL2RG) (c.655A>T).
This mutation has not been described before (http://research.nhgri.nih.gov/scid/) and results in replacement of tyrosine 219 by asparagine (p.Tyr219Asn) in the extracellular domain. The amino acids of the linker region are highly conserved in the mammalian CD132 protein, suggesting a role in ligand binding as based on theoretical grounds and epitope mapping studies with anti-human CD132 antibodies. Outside the linker region the residues p.Leu208 to p.Ser211 affect cytokine binding by conformation constraints on the two extracellular loops of CD132.45 Although not proven, the nearby p.Tyr219Asn substitution may similarly affect the quaternary binding interaction of a cytokine with its respective αβγ or αγ receptor complex. The IL2RG mutation most likely acts as a hypomorphic mutation because the non-reverted T and NK cells carrying this mutation were normally detectable in peripheral blood, thus not causing any lymphopenia in the patient. In fact, the patient’s younger cousin who was born some years later carried the same mutation in the absence of any reversion, having normal cord blood lymphocyte counts. However, CD132 expression was strongly reduced on his peripheral blood lymphocytes and the cytokine-induced proliferative responses of his T and NK cells were diminished. This also implies that initial immunological screening is not sufficient for the diagnosis of these types of patients; only analysis of CD132 up-regulation together with cytokine responsiveness can reveal the defect.
Despite the relatively normal serology upon immunization with various vaccines, the specific antibody titers became undetectable again within the following weeks. We observed a reduced number of CD27+ memory B cells that persisted over the years. The patient had lost specific antibodies against VZV and CMV, and virus-specific memory CD4+ T cell reactivity was hardly detected, emphasizing the need for γc signals for maintenance of immunological memory. There was no lymphopenia but a clear and progressive inversion of the CD4/CD8 ratio was observed. Whereas CD132 expression by activated CD4+ T cells was diminished, on CD8+ T cells this was clearly detectable upon cell activation. It is within this population of CD8+ T cells that we had identified the reversion to predominate, which may have contributed to the proportionally increased cell number in the blood counts.
To date, only three cases of IL2RG gene reversion have been reported at the T-cell precursor stage.25–27 A reversion of a specific deleterious point mutation is statistically highly unlikely. However, in vivo selection could allow such a rare event, because any reversion of the inherited IL2RG mutation in T cells would confer a distinct advantage, in terms of growth and differentiation, over cells without functional CD132 chains. In our case, only partial gene reversion was observed to be present in the T cells. This could be explained by the hypomorphic nature of the IL2RG mutation that still allowed T and NK cell development to occur, which would fit with the delayed clinical presentation.
All reverted T cells in the patients reported thus far arose from a single revertant T-cell precursor, as was confirmed by the polyclonal T-cell repertoire.46 Also in our patient the T cell repertoire was polyclonal. In addition, analysis of TCRγδ+ T-cells showed that gene reversion was present in part of the cells. The split in TCRγδ+ and TCRαβ+ T-cell differentiation occurs between the CD4−CD8− double-negative and CD4+CD8+ double-positive thymocyte stage of T-cell differentiation in the thymus. This implies that the genetic reversion occurred in the thymus before or during the double-negative stage, which reflects a stage of T-cell differentiation that does not depend on any CD132 expression.24
A common theme is the selective survival advantage of the revertant immune cells over mutant cells, which has been amply discussed in terms of its implications for gene therapy. Our data confirm the developmental potential of a single T-cell precursor in humans with broadly diverse T-cell repertoire.25,26 The mutations in these two cases also predicted amino acid substitutions (p.Cys115Arg and p.Leu151Pro) in the extracellular part of the protein,25,26 but had – in contrast to our case – a more global T-cell pattern of gene reversion. Importantly, although the reversion must have occurred in a common T-cell progenitor, accumulation of reverted cells was not found in either naïve or memory CD4+ cells nor in the NK or B cells. This suggests that for these populations the reversion offered no selective advantage, or at least a very minor one compared to CD8+ T cells. In this respect, it is of interest to note that the mutated receptor is not completely unable to signal dead as CD4 T+ cells showed a low but detectable response to cytokines such as IL7. The difference in the amount of reversion between the CD4+ and CD8+ T-cell pools suggests that in this patient, who has a partially active common-gamma chain, the dependence on signaling through this receptor differs between the two T-cell populations. The small fraction of effector CD4+ T cells (<2% of total CD4+ T cells), also carried the reversion in a large part of the cells, which suggests that this T-cell subset requires a fully functional CD132 to be generated or persist in vivo. Whether these effector CD4+ T cells largely reside in the tissues or have a shorter life span is unknown, but the reversion did not result in an accumulation in the circulation. Of course, the possibility remains that more revertant CD4+ T cells will emerge over time in the case the kinetics are just much slower than for CD8+ T cells. This could be, for example, because of preferential antigen-driven expansion of CD8+ T cells caused by chronic infection. Another interesting finding was the high percentage of naïve CD8+ T cells carrying the reversion (40%). This implies the persistence of reverted progenitor T cells that continue to supply new revertant naive T cells or a very long half-life of these naïve CD8+ T cells.
In our patient the widely disseminated molluscum contagiosum virus infection of the skin slowly but completely cleared after many years, indicating improved cellular immunity. Although the developmental stage of gene reversion was not investigated, a similar effect of infection-driven proliferative advantage due to gene reversion was recently suggested in XLP patients showing selective expansion of reverted CD8+ T cells only, following EBV infection.47
The preferential expansion of CD8+ reverted T cells makes our X-SCID case unique and suggests a more important role for CD132-signaling in proliferation (homeostatic or antigen-driven) of CD8+ T cells as compared to CD4+ T cells.48 The IL2RG gene reversion in the polyclonal CD8+ T-cell outgrowth did not result in complete recovery of all clinical symptoms demonstrating that rescue of CD8+ T cells alone is not sufficient for full restoration of immunity.
Acknowledgments
The authors would like to thank the technicians of the Laboratory for Medical Immunology, Berend Hooibrink and Ester Remmerswaal for expert technical assistance. We would also like to thank Drs. Hans Zaaijer and Janke Schinkel for their micro-biological expertise and the bioinformatics laboratory (Barbera van Schaik and Antoine van Kampen) for technical support during TCR repertoire analysis.
Footnotes
The online version of this article has a Supplementary Appendix
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3(10):748–58 [DOI] [PubMed] [Google Scholar]
- 2.Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40(10):721–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregory JJ, Jr, Wagner JE, Verlander PC, Levran O, Batish SD, Eide CR, et al. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA. 2001;98(5):2532–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T, Schurman SH, Otsu M, Garabedian EK, Ochs HD, Nelson DL, Candotti F. Somatic mosaicism in Wiskott-Aldrich syndrome suggests in vivo reversion by a DNA slippage mechanism. Proc Natl Acad Sci USA. 2001;98(15):8697–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uzel G, Tng E, Rosenzweig SD, Hsu AP, Shaw JM, Horwitz ME, Linton GF, et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1). Blood. 2008;111(1):209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasmooij AM, Pas HH, Deviaene FC, Nijenhuis M, Jonkman MF. Multiple correcting COL17A1 mutations in patients with revertant mosaicism of epidermolysis bullosa. Am J Hum Genet. 2005;77(5):727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavazzana-Calvo M, Hacein-Bey-Abina S, Fischer A. Gene therapy of X-linked severe combined immunodeficiency. Curr Opin Allergy Clin Immunol. 2002;2(6):507–9 [DOI] [PubMed] [Google Scholar]
- 8.Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–58 [DOI] [PubMed] [Google Scholar]
- 9.Buckley RH. The multiple causes of human SCID. J Clin Invest. 2004;114(10):1409–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschhorn R, Israni A, Yang DR, Israni A, Huie ML, Ownby DR. Somatic mosaicism for a newly identified splice site mutation in a patient with adenosine deaminase deficient immunodeficency (ADA-SCID) and spontaneous clinical recovery. Am J Hum Genet. 1994;55(1):59–68 [PMC free article] [PubMed] [Google Scholar]
- 11.Hirschhorn R, Yang DR, Puck JM, Huie ML, Jiang CK, Kurlandsky LE. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet. 1996;13(3):290–5 [DOI] [PubMed] [Google Scholar]
- 12.Ariga T, Oda N, Yamaguchi K, Kawamura N, Kikuta H, Taniuchi S, et al. T cell lines from 2 patients with adenosine deaminase (ADA) deficiency showed the restoration of ADA activity resulted from the reversion of an inherited mutation. Blood. 2001;97 (9):2896–9 [DOI] [PubMed] [Google Scholar]
- 13.Arredondo-Vega FX, Santisteban I, Richard E, Bali P, Koleilat M, Loubser M, et al. Adenosine deaminase deficiency with mosaicism for a ‘second-site suppressor’ of a splicing mutation: decline in revertant T lymphocytes during enzyme replacement therapy. Blood. 2002;99(3):1005–13 [DOI] [PubMed] [Google Scholar]
- 14.Moncada-Vélez M, Vélez-Ortega A, Orrego J, Santisteban I, Jagadeesh J, Olivares M, et al. Somatic mosaicism caused by monoallelic reversion of a mutation in T cells of a patient with ADA-SCID and the effects of enzyme replacement therapy on the revertant phenotype. Scand J Immunol. 2011;74(5):471–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis BR, Yan Q, Bui JH, Felix K, Moratto D, Muul LM, et al. Somatic mosaicism in the Wiskott-Aldrich syndrome: molecular and functional characterization of genotypic revertants. Clin Immunol. 2010;135(1): 72–83 [DOI] [PubMed] [Google Scholar]
- 16.Wada T, Konno A, Schurman SH, Garabedian EK, Anderson SM, Kirby M, et al. Second-site mutation in the Wiskott-Aldrich syndrome (WAS) protein gene causes somatic mosaicism in two WAS siblings. J Clin Invest. 2003;111(9):1389–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lutskiy MI, Beardsley DS, Rosen FS, Remold-O’Donnell E. Mosaicism of NK cells in a patient with Wiskott-Aldrich syndrome. Blood. 2005;106(8):2815–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rieux-Laucat F, Hivroz C, Lim A, Mateo V, Pellier I, Selz F, et al. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. N Engl J Med. 2006;354(18): 1913–21 [DOI] [PubMed] [Google Scholar]
- 19.Wada T, Toma T, Okamoto H, Kasahara Y, Koizumi S, Agematsu K, et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood. 2005;106(6):2099–101 [DOI] [PubMed] [Google Scholar]
- 20.Tone Y, Wada T, Shibata F, Toma T, Hashida Y, Kasahara Y, et al. Somatic revertant mosaicism in a patient with leukocyte adhesion deficiency type 1. Blood. 2007; 109(3):1182–4 [DOI] [PubMed] [Google Scholar]
- 21.Nishikomori R, Akutagawa H, Maruyama K, Nakata-Hizume M, Ohmori K, Mizuno K, et al. X-linked ectodermal dysplasia and immunodeficiency caused by reversion mosaicism of NEMO reveals a critical role for NEMO in human T-cell development and/or survival. Blood. 2004;103(12):4565–72 [DOI] [PubMed] [Google Scholar]
- 22.Conley ME. Molecular approaches to analysis of X-linked immunodeficiencies. Annu Rev Immunol. 1992;10:215–38 [DOI] [PubMed] [Google Scholar]
- 23.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73(1):147–57 [DOI] [PubMed] [Google Scholar]
- 24.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephan V, Wahn V, Le Deist F, Dirksen U, Broker B, Müller-Fleckenstein I, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335(21):1563–7 [DOI] [PubMed] [Google Scholar]
- 26.Speckmann C, Pannicke U, Wiech E, Schwarz K, Fisch P, Friedrich W, et al. Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency. Blood. 2008;112(10):4090–7 [DOI] [PubMed] [Google Scholar]
- 27.Wada T, Yasui M, Toma T, Nakayama Y, Nishida M, Shimizu M, et al. Detection of T lymphocytes with a second-site mutation in skin lesions of atypical X-linked severe combined immunodeficiency mimicking Omenn syndrome. Blood. 2008;112 (5):1872–5 [DOI] [PubMed] [Google Scholar]
- 28.Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O’Shea JJ. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol Rev. 2005;203:127–42 [DOI] [PubMed] [Google Scholar]
- 29.Brooks EG, Schmalstieg FC, Wirt DP, Rosenblatt HM, Adkins LT, Lookingbill DP, et al. A novel X-linked combined immunodeficiency disease. J Clin Invest. 1990; 86(5):1623–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saint-Basile G, Le Deist F, Caniglia M, Lebranchu Y, Griscelli C, Fischer A. Genetic study of a new X-linked recessive immunodeficiency syndrome. J Clin Invest. 1992;89(3):861–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiSanto JP, Rieux-Laucat F, Dautry-Varsat A, Fischer A, de Saint-Basile G. Defective human interleukin 2 receptor gamma chain in an atypical X chromosome-linked severe combined immunodeficiency with peripheral T cells. Proc Natl Acad Sci USA. 1994;91(20):9466–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morelon E, Dautry-Varsat A, Le Deist F, Hacein-Bay S, Fischer A, de Saint-Basile G. T-lymphocyte differentiation and proliferation in the absence of the cytoplasmic tail of the common cytokine receptor gamma c chain in a severe combined immune deficiency X1 patient. Blood. 1996;88(5):1708–17 [PubMed] [Google Scholar]
- 33.Sharfe N, Shahar M, Roifman CM. An interleukin-2 receptor gamma chain mutation with normal thymus morphology. J Clin Invest. 1997;100(12):3036–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuijpers TW, Baars PA, Aan de Kerk DJ, Jansen MH, Derks IA, Bredius RG, et al. A novel mutation in CD132 causes X-CID with defective T-cell activation and impaired humoral reactivity. J Allergy Clin Immunol. 2011;128(6):1360–3 [DOI] [PubMed] [Google Scholar]
- 35.Tone Y, Wada T, Shibata F, Toma T, Hashida Y, Kasahara Y, et al. Somatic revertant mosaicism in a patient with leukocyte adhesion deficiency type 1. Blood. 2007;109(3):1182–4 [DOI] [PubMed] [Google Scholar]
- 36.Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34(12):1313–20 [DOI] [PubMed] [Google Scholar]
- 37.Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330(6000):94–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120(1):214–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Burg M, Kreyenberg H, Willasch A, Barendregt BH, Preuner S, Watzinger F, et al. Standardization of DNA isolation from low cell numbers for chimerism analysis by PCR of short tandem repeats. Leukemia. 2011;25(9):1467–70 [DOI] [PubMed] [Google Scholar]
- 40.van der Burg M, Weemaes CM, Preijers F, Brons P, Barendregt BH, van Tol MJ, et al. B-cell recovery after stem cell transplantation of Artemis-deficient SCID requires elimination of autologous bone marrow precursor-B-cells. Haematologica. 2006;91(12):1705–9 [PubMed] [Google Scholar]
- 41.Klarenbeek PL, Tak PP, van Schaik BD, Zwinderman AH, Jakobs ME, Zhang Z, et al. Human T-cell memory consists mainly of unexpanded clones. Immunol Lett. 2010;133(1):42–8 [DOI] [PubMed] [Google Scholar]
- 42.van Gisbergen KP, Klarenbeek PL, Kragten NA, Unger PP, Nieuwenhuis MB, Wensveen FM, et al. The costimulatory molecule CD27 maintains clonally diverse CD8(+) T cell responses of low antigen affinity to protect against viral variants. Immunity. 2011;35(1):97–108 [DOI] [PubMed] [Google Scholar]
- 43.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98–3936. Leukemia 2003;17(12):2257–317 [DOI] [PubMed] [Google Scholar]
- 44.Folch G, Lefranc MP. The human T cell beta variable (TRBV) genes. Exp Clin Immunogenet. 2000;17(1):42–54 [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bousso P, Wahn V, Douagi I, Horneff G, Pannetier C, Le Deist F, et al. Diversity, functionality, and stability of the T cell repertoire derived in vivo from a single human T cell precursor. Proc Natl Acad Sci USA. 2000;97(1):274–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palendira U, Low C, Bell AI, Ma CS, Abbott RJ, Phan TG, et al. Expansion of somatically reverted memory CD8+ T cells in patients with X-linked lymphoproliferative disease caused by selective pressure from Epstein-Barr virus. J Exp Med. 2012;209(5): 913–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park JH, Adoro S, Guinter T, Erman B, Alag AS, Catalfamo M, et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat Immunol. 2010;11(3): 257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]