

Abstract
ATM abnormalities are frequent in chronic lymphocytic leukemia and represent an important prognostic factor. Sole 11q deletion does not result in ATM inactivation by contrast to biallelic defects involving mutations. Therefore, the analysis of ATM mutations and their functional impact is crucial. In this study, we analyzed ATM mutations in predominantly high-risk patients using: i) resequencing microarray and direct sequencing; ii) Western blot for total ATM level; iii) functional test based on p21 gene induction after parallel treatment of leukemic cells with fludarabine and doxorubicin. ATM dysfunction leads to impaired p21 induction after doxorubicin exposure. We detected ATM mutation in 16% (22 of 140) of patients, and all mutated samples manifested demonstrable ATM defect (impaired p21 upregulation after doxorubicin and/or null protein level). Loss of ATM function in mutated samples was also evidenced through defective p53 pathway activation after ionizing radiation exposure. ATM mutation frequency was 34% in patients with 11q deletion, 4% in the TP53-defected group, and 8% in wild-type patients. Our functional test, convenient for routine use, showed high sensitivity (80%) and specificity (97%) for ATM mutations prediction. Only cells with ATM mutation, but not those with sole 11q deletion, were resistant to doxorubicin. As far as fludarabine is concerned, this difference was not observed. Interestingly, patients from both these groups experienced nearly identical time to first treatment. In conclusion, ATM mutations either alone or in combination with 11q deletion uniformly led to demonstrable ATM dysfunction in patients with chronic lymphocytic leukemia and mutation presence can be predicted by the functional test using doxorubicin.
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by a poor curability and distinctively variable clinical course.1 The mutation status of the immunoglobulin heavy-chain variable region (IGHV)2 and the presence of recurrent cytogenetic aberrations3 represent two major prognostic factors with clear clinical impact. The worst prognosis is associated with the occurrence of 17p deletion (17p-) and/or TP53 mutation.3–5 The heterozygous deletion at the locus 11q22-23 (11q-) affecting the Ataxia Telangiectasia-Mutated (ATM) gene may also significantly contribute to inferior patient outcome, especially in younger patients.3,6,7 The pathogenic role of the ATM gene has been unambiguously proven by demonstrating the presence of somatic ATM mutations in a proportion of patients.8–10ATM abnormalities (deletions and mutations) are typically associated with extensive lymphadenopathy in CLL patients11 and have recently been identified as the most common negative genetic defect at CLL diagnosis.12 Nevertheless, the functional impact of ATM defects and their relevance in CLL still remains controversial13–15 and further studies are needed.
Patients with both ATM alleles affected (deletion and mutation or two mutations) lack ATM activity, while patients with monoallelic mutation may have preserved ATM function.16,17 Therefore, it is important to monitor status of both alleles, because ATM mutations only overlap poorly with 11q-.12,17 In addition, various types of ATM mutations may have a different impact on the resulting ATM activity. In autosomal recessive disorder Ataxia-Telangiectasia (AT), characterized by progressive neurodegeneration, immunodeficiency, and predisposition to lymphoid malignancies,18 the clinical heterogeneity can be attributed to different types of inherited ATM mutations. While truncating mutations are associated with full AT phenotype, splicing and missense mutations lead to milder clinical appearance due to partially preserved ATM expression and function.19,20
ATM plays a pivotal role in the DNA-damage response (DDR) pathway after DNA double-strand break (DSBs) recognition by Mre11/Rad50/Nbs1 (MRN) complex21 through phosphorylation of many different targets, including p53 protein.22 In CLL, Stankovic et al.23 demonstrated that transcriptional response to DSBs is entirely dependent on ATM, where only part of this activity is transferred to the p53 pathway, leading to pro-apoptotic signaling. Monitoring of ATM function may be a feasible tool for disclosing ATM mutations. Several slightly modified tests have been suggested, based on monitoring p53 and p21 accumulation after cell exposure to ionizing radiation (IR).17,24–26 An alternative approach utilizing etoposide and nutlin-3a was also used which enabled efficient differentiation of TP53 and ATM defects.27
Despite undeniable progress, ATM mutation identification in CLL remains challenging due to: i) an extreme gene size (62 coding exons) with lack of well-characterized (hot-spot) mutations; ii) the difficult interpretation of polymorphisms and pathogenic mutations resulting from only vague information about their functional consequences. Therefore, relevant information is lacking about the clinical impact of ATM mutations, including the response of affected patients to chemoimmunotherapy.
In our CLL patient cohort, we found that all ATM defects involving mutation(s) resulted in disruption of ATM activity towards p53 pathway activation. In addition, we present a novel functional test based on monitoring p21 induction after parallel treatment of CLL cells with doxorubicin and fludarabine that have different DNA damage mechanisms. This test proved to be an effective means to search for ATM mutations, which had been selected in a dominant proportion of leukemic cells.
Design and Methods
Patients’ samples
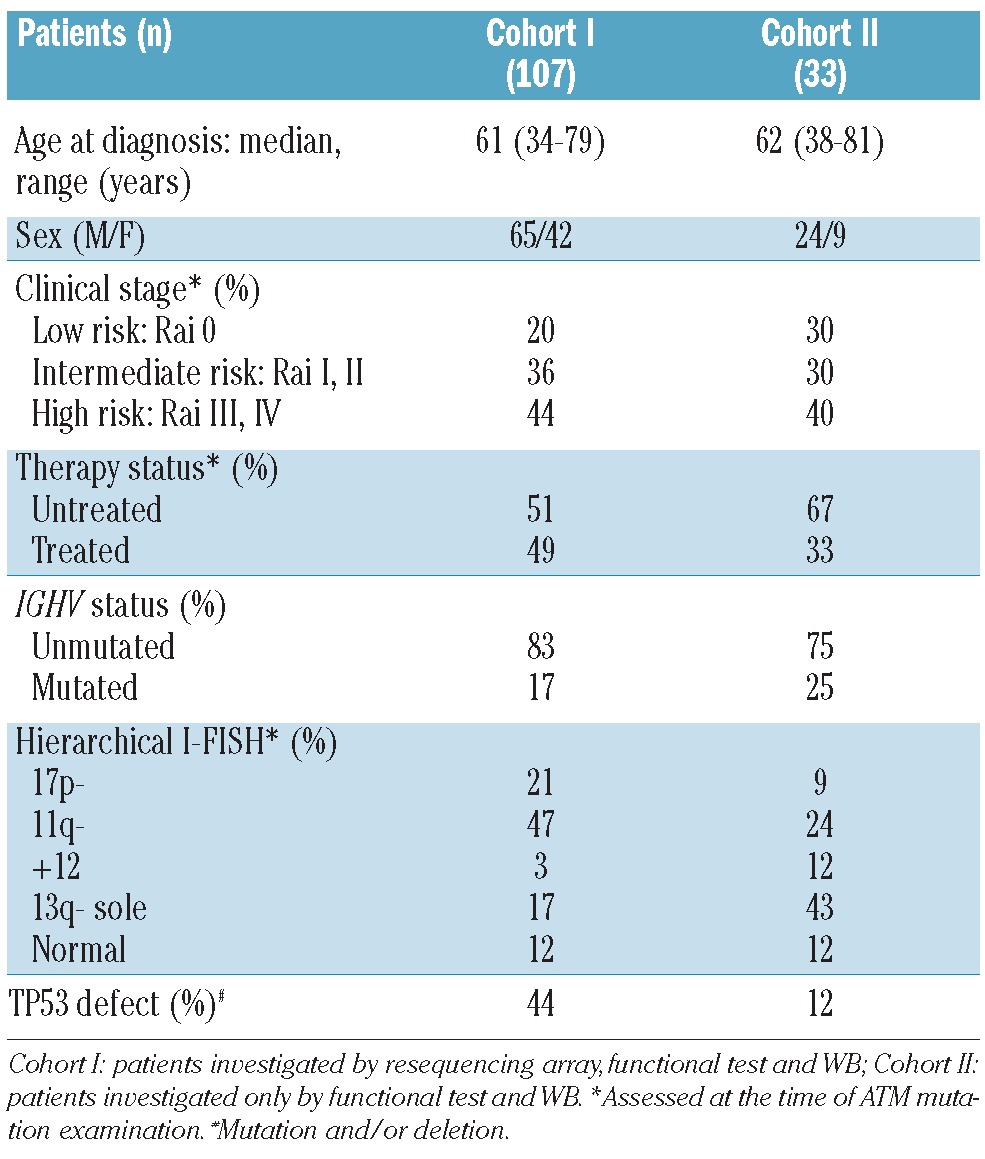
Peripheral blood mononuclear cells (PBMNC) of 140 CLL patients were processed after obtaining informed consent in accordance with the Declaration of Helsinki under protocols of the University Hospital Brno (Ethics Committee approval NS10439-3). The proportion of leukemic cells (CD5+/CD19+) exceeded 80% in all samples. Basic characteristics of the cohort are summarized in Table 1. The cohort consisted of predominantly high-risk CLL patients (harboring TP53 defect and/or 11q-and/or unmutated IGHV), and 45% of patients were treated with various regimens before ATM mutation analysis (Online Supplementary Table S1).
Table 1.
Clinical and biological characteristics of CLL patients.
ATM mutation analysis
Custom resequencing microarray (Affymetrix, CA, USA) was used to detect 1-nt substitutions in all 62 coding exons and splicing sites of the ATM gene. The resequencing procedure was carried out according to the manufacturer’s protocol (Affymetrix GeneChip Custom Resequencing Array Protocol). The resequencing principle is based on allele-specific hybridization. The hybridization of fluorescently labeled DNA fragments to particular positions determined the nucleotides in sequence with the ability to distinguish between homozygous and heterozygous state. Final sequence data was acquired using The GeneChip Sequence Analysis Software (GSEQ) processing fluorescence intensity files.
Direct Sanger sequencing (3130xl Genetic Analyzer, Applied Biosystems) was used to: i) confirm ATM alterations detected by microarray analysis; ii) identify other mutations indicated by functional test and/or Western blot (WB) through screening of all 62 coding exons and splicing sites. A search was made for all identified sequence variations in appropriate databases of single nucleotide polymorphisms (SNPs) and mutations.
Variations detected by the resequencing array but not confirmed by Sanger sequencing were evaluated by next-generation deep sequencing technology (GS Junior, Roche) to distinguish subclonal ATM alterations occurring under direct sequencing detection limit from the array false positivity.
Western blotting
Protein level analysis was performed using the following antibodies (Cell Signaling Technology): anti-ATM (mAb D2E2), anti-ATM-Ser1981 (mAb D6H9), anti-p53-Ser15 (mAb 16G8), and anti-β-actin (mAb 13E5). Total p53 level was detected by DO-1 mAb (a gift from Dr. Vojtesek, MMCI, Brno). For ATM analysis PBMNC were lysed in RIPA buffer and protein lysates (50 ug for ATM-Ser1981, 100 μg for total ATM) were run on NuPAGE Novex 3–8 % Tris-Acetate Gel (Invitrogen).
Induction of p53-downstream target gene expression after DNA damage
Primary CLL cells were subjected to IR (5 Gy in total, 0.3 Gy/min) or treated with fludarabine (Bayer-Schering, 3.6 μg/mL) or doxorubicin (Teva, 0.25 μg/mL). Cells were seeded in 6-well plates (2.5 × 107 cells per well, volume 5 mL) and harvested after 2, 10 and 24 h in case of IR or after 24 h drug exposure. Real-time polymerase chain reaction (PCR) was performed using TaqMan technology and 7300 Real-Time PCR System (Applied Biosystems). Primer and probe set was specific for the CDKN1A (p21), BBC3 (PUMA), BAX, and GADD45 genes (Applied Biosystems).
Cell viability after doxorubicin and fludarabine administration
CLL cells were seeded in quadruplicates using 96-well plates (5 x 105 cells per well) and treated for 48 h with chemotherapeutics at four different concentrations. The viability was assessed by the metabolic WST-1 assay (Roche) using spectrophotometer 1420 Multilabel Counter Victor (PerkinElmer).
Results
ATM mutations identification by resequencing microarray
Chronic lymphocytic leukemia samples from 107 patients were investigated for ATM mutations using the resequencing array as a pre-screening method. This cohort (Cohort I) was intentionally selected and biased towards high-risk CLL patients (Table 1). In total, 46 different ATM alterations were identified in a variable number of patients, and 29 of these variants were readily confirmed by direct Sanger sequencing. Next-generation deep sequencing applied to unconfirmed variants revealed only two real substitutions that were present in 16% and 10% of molecules, which is under the direct sequencing detection limit. The rest represented array false positivity (Online Supplementary Table S2).
In total, only 29 properly selected and clearly confirmed alterations were further considered as real ATM substitutions. Among them 13 represented different known SNPs, and their frequencies in our cohort are listed in the Online Supplementary Table S3. The remaining 16 alterations detected in 13 patients (P1-P13 in Table 2) included two nonsense mutations, one known splicing mutation28 leading to null expression of normal transcript (data not shown), and missense mutations.
Table 2.
ATM mutations identified in CLL patients.
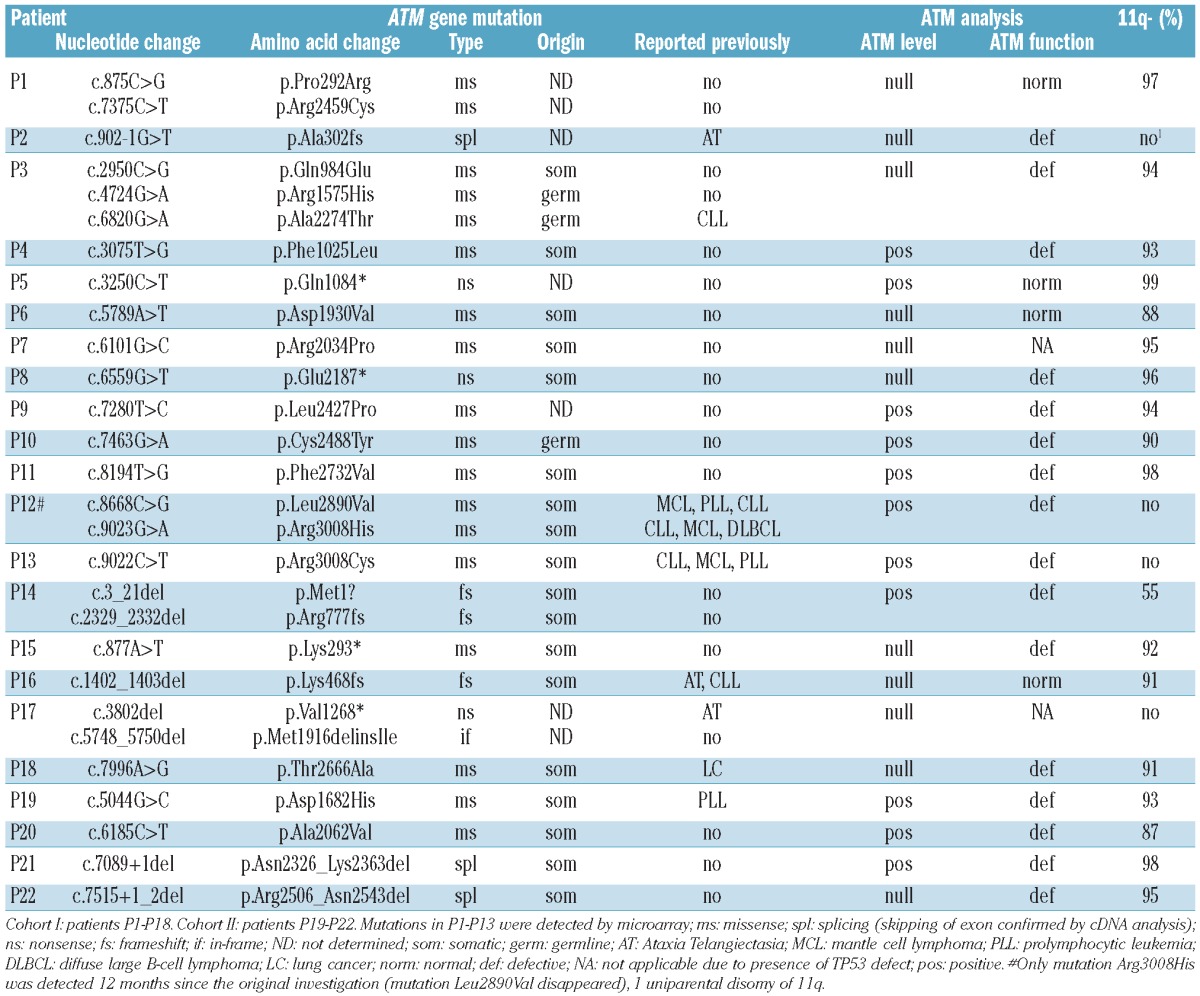
With regards to allele composition, 10 of 13 mutated patients (77%) manifested 11q- in addition to ATM mutation. Another patient (P2) having homozygous mutation and no 11q- demonstrated uniparental disomy of 11q arm (detected by CytoScan HD array, Affymetrix) (data not shown). Two remaining patients exhibited heterozygous mutations, and we searched for a second potential mutation by direct sequencing. Such a mutation was disclosed in patient P12, while the last patient (P13) harboring a well known pathogenic mutation Arg3008Cys in heterozygous state did not exhibit another mutation. This mutation may, therefore, have a predicted dominant-negative effect (DNE) on wt allele.18 Interestingly, patient P12 with two mutations recorded during the original investigation manifested only mutation Arg3008His in a sample obtained 12 months later. This further supports the notion that alterations in this mutable codon are connected with DNE.
Functional test and Western blot reveal ATM protein inactivation in mutated patients
In order to directly assess overall ATM status in affected patients, samples from all 13 patients (P1–P13) with array-identified mutation were subjected to the ATM protein functional analysis and to ATM protein level assessment.
We designed the functional test based on parallel treatment of CLL cells with doxorubicin and fludarabine followed by CDKN1A (p21) gene expression monitoring. The impaired p21 induction as an ATM dysfunction marker was selected on the basis of its demonstrated ATM-dependent upregulation following DSBs after IR.25 We presumed that: i) doxorubicin as a radiomimetic drug has a similar effect to IR; ii) mutations leading to ATM protein dysfunction should impair p21 induction during response to DSBs imposed by doxorubicin,29 while p21 activation should be preserved after fludarabine, creating a broader spectrum of DNA (and also RNA) damage;30 iii) TP53 defects should result in reduced p21 induction after exposure to both drugs. The proposed principle of our functional test was proven by testing 3 TP53-wt/ATM-wt samples with and without the presence of ATM-specific inhibitor KU5593331 (Figure 1).
Figure 1.
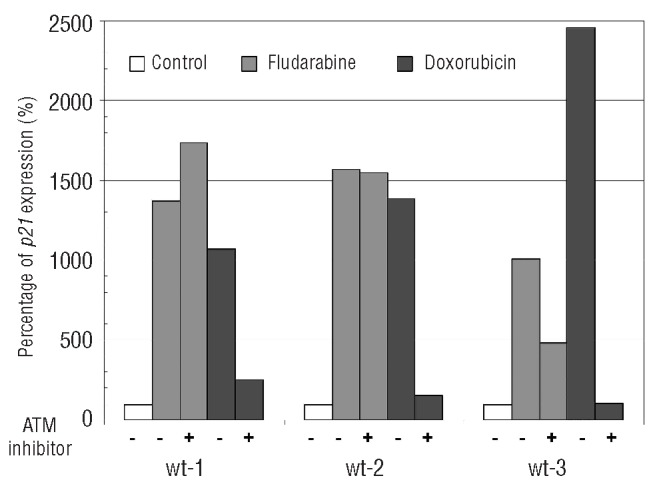
The p21 expression induction after ATM inhibition. CLL cells were pre-treated with ATM inhibitor (KU55933; 10 μM) for 1 h and exposed to fludarabine (3.6 μg/mL) or doxorubicin (0.25 μg/mL) for 24 h. The level of p21 expression was determined by real-time PCR in comparison with untreated control set to 100%. Sample’s Ct values were subjected to 2−ΔΔCt analysis.
All samples with an array-identified mutation excepting one (n=12) showed ATM dysfunction using the described functional test and/or manifested null ATM protein level on Western blot (WB) (Table 2 and Online Supplementary Figure S1); the last sample (P5) harboring nonsense mutation and 11q- exhibited no defect in these tests due to incomplete mutation selection (data not shown). Therefore, all patients with ATM mutation identified by resequencing microarray (which is neutral regarding identified mutation function) can be considered as having disturbed ATM activity. Furthermore, in 2 samples with detectable ATM level (P9 and P13), we performed additional Western blots analyzing ATM function. ATM-Ser1981 autophosphorylation was obviously diminished in one sample, while p53-Ser15 phosphorylation and p53 accumulation was completely lost in both samples, confirming the elimination of ATM activity towards p53 pathway activation (Online Supplementary Figure S2).
By contrast, all tested samples with only ATM polymorphism and no mutation (11 of 13 detected polymorphisms were tested) (Online Supplementary Table S3) displayed normal profile in the functional test.
Functional test and Western blot disclosed other patients with ATM mutation
In order to identify other potential causal mutations undetectable by resequencing microarray (nonmissense mutations), we employed the functional test and WB analysis on samples from 64 patients with no detected mutation and with material available. Either one or both of these tests indicated a mutation presence in 7 patients (11%), and in 5 cases the mutation(s) were indeed found by direct sequencing. In 2 patients (P14, P17) there were two short deletions, and another (P16) harbored one short deletion accompanied by 11q-. Two other patients (P15, P18) manifested nonsense and missense mutation that was not recognized by array, again together with 11q-. The situation of the last 2 patients remains unclear as no mutation was found. In total, using resequencing microarray, functional test, and WB, we identified 18 patients (17%) in Cohort I having ATM mutation confirmed by Sanger sequencing and exhibiting ATM dysfunction.
The additional analysis performed on 33 randomly selected samples (Cohort II; Table 1) was carried out to evaluate the efficiency of our functional test. The test indicated ATM mutation in 4 patients, and in all cases a mutation was identified by direct sequencing (P19–P22) (Table 2). The final cut-off value for ATM dysfunction was determined using receiver operation characteristic analysis (Online Supplementary Figure S3) applied on selected samples with known ATM mutation status (omitting the TP53-defective group). The p21 expression induction of less than 300% in comparison with untreated control (100%) after doxorubicin treatment defined dysfunctional cases. The functional test showed 80% sensitivity and 97% specificity in this setting. Using a dilution series of wt cells and ATM mutated cells, we determined that this test is able to detect mutation, if present in at least 80% of cells (Online Supplementary Figure S4); our test is, therefore, suitable for detection of properly selected ATM mutations.
Based on our retrospective analysis, the sample should be screened for TP53 defect if p21 induction does not exceed 400% after exposure to each drug.
ATM mutations are unevenly distributed in genetic groups and frequently occur upon CLL diagnosis
Collectively in Cohorts I and II, we detected ATM mutation(s) in 16% (22 of 140) of CLL patients. Mutated patient proportions in groups divided according to high-risk genetic features were as follows: 34% (17 of 50) in patients with 11q-; 4% (2 of 51) in patients having TP53 mutation and/or deletion 17p; and 8% (3 of 39) in patients without these defects. The association between ATM mutation presence and IGHV mutation status was not assessed because of predominant 11q- occurrence in IGHV-unmutated samples.
We did not observe any tendency towards ATM mutation accumulation in previously treated patients; the frequency of ATM mutations was 18% (14 of 77) in untreated and 13% (8 of 63) in treated patients. The retrospective analysis of 9 mutated patients disclosed a mutation presence at the time of diagnosis or up to one year after in 8 cases. Moreover, the ATM mutation germline origin was disclosed in 2 (P3, P10) of 17 analyzed patients with available material (buccal swab). These observations all indicate that the genesis of a substantial proportion of ATM mutations can be considered primarily as an early event in CLL pathogenesis.
Cells with ATM mutation exhibit impaired overall response to doxorubicin
The strong association between presence of ATM mutation and dysfunction status prompted us to compare p53-downstream gene induction and overall cell viability after doxorubicin and fludarabine exposure in the following genetic categories: cells without any TP53 or ATM abnormality (“wt”), with sole 11q- (“del-ATM”), with ATM mutation regardless of 11q- presence or absence (“mut-ATM”), and TP53 defect (“def-TP53”).
Data concerning p21 induction after cell exposure to doxorubicin and fludarabine is summarized in Figure 2A and B, respectively. It is evident that mut-ATM (n=20) and def-TP53 (n=30), but not del-ATM (n=23) samples exhibited significantly impaired induction after doxorubicin exposure in comparison with wt cells (n=31). By contrast, after treating the same samples with fludarabine, we observed that mut-ATM samples manifested similar p21 induction compared to wt and del-ATM groups. The def-TP53 samples exhibited diminished induction similarly to doxorubicin exposure.
Figure 2.

The p21 expression induction in individual genetic groups. Medians of p21 expression and the significance (Kruskal-Wallis ANOVA test) related to wt group were following: (A) after doxorubicin treatment – wt: 815%; del-ATM: 731%; non-significant (NS); mut-ATM: 182%, P<0.001; def-TP53: 250%, P<0.001. (B) after fludarabine treatment – wt: 718%; del-ATM: 639%, NS; mut-ATM: 617%, NS; def-TP53: 190%, P<0.001.
In addition to impaired p21 induction, ATM mutated samples displayed also identically disturbed induction of pro-apoptotic genes BBC3 (PUMA) and BAX, and DNA damage response gene GADD45 after doxorubicin exposure (Online Supplementary Figure S5). Similarly to doxorubicin, a series of the samples with different ATM mutations also exhibited defective p53-downstream gene induction after IR exposure, which is commonly used for DSBs induction and for ATM function testing. However, the difference in gene expression was obvious only for three out of the four studied genes (i.e. p21, PUMA, GADD45) at certain times after radiation (Online Supplementary Figure S6).
In agreement with the data regarding p53-downstream gene induction, we observed significantly higher resistance of mut-ATM (n=8) and def-TP53 (n=5) samples to doxorubicin when compared with wt samples (n=5); del-ATM samples (n=9) behaved similarly to the wt group (Figure 3A). The contrasting negligible effect of ATM mutations on p53-downstream gene induction elicited by fludarabine was also in compliance with cell viability in the tested groups (Figure 3B). Only def-TP53 (n=13), but neither mut-ATM (n=14) nor del-ATM samples (n=8), showed higher resistance to fludarabine compared with the wt group (n=8).
Figure 3.

CLL cell viability after doxorubicin and fludarabine exposure. Significance (two-factorial ANOVA with subsequent Dunnett post-hoc test) related to wt group was: (A) after doxorubicin treatment -del-ATM: non-significant (NS), mut-ATM: P<0.001; def-TP53: P<0.001; (B) after fludarabine treatment -del-ATM: NS; mut-ATM: NS, def-TP53: P<0.001. Vertical lines represent 95% confidence intervals.
Patients having ATM mutation and those with sole 11q- have identical time to first treatment
The above data confirm that cells with sole 11q- (“del-ATM”) have preserved ATM activity,17 while defects involving ATM mutation (“mut-ATM”) in our study exclusively led to ATM dysfunction. We, therefore, focused on a correlation between these defects and the time to first treatment (TTFT). We limited our analysis to patients with unmutated IGHV locus (n=41) because of the strong association with ATM defects and to those with ATM abnormality observed at the diagnosis or up to one year after. TTFT medians in the group mut-ATM (n=8), del-ATM (n=15), and wt (n=18) were as follows: 7, 9.5, and 27 months (Figure 4). Both defective groups exhibited significantly reduced TTFT in comparison with wt patients (both P=0.04) and did not differ mutually (P=0.6).
Figure 4.
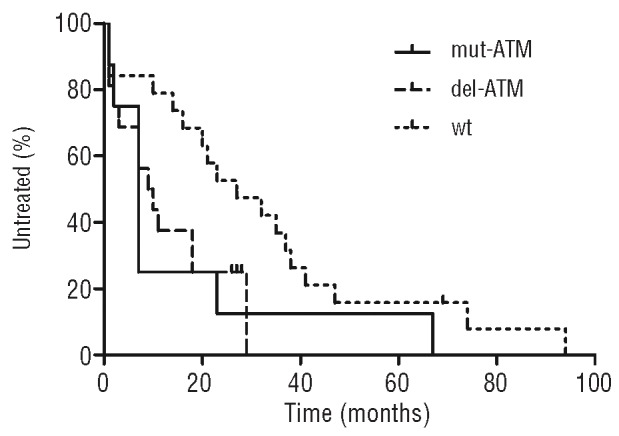
Time to first treatment analysis limited to patients with unmutated IGHV. Mut-ATM and del-ATM groups related to wt patients: both P=0.04. Kaplan-Meier curves were created and statistically evaluated using the GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
We also performed a progression-free survival (PFS) analysis after the first therapy consisting of chemoimmunotherapy (76%, 25 of 33 patients) and chemotherapy (24%, 8 of 33 patients) with a similar proportion in individual genetic groups. PFS medians were 9, 16 and 16.5 months in the mut-ATM, del-ATM and wt group, respectively, with no statistical significance among tested groups (Online Supplementary Figure S7). Overall survival analysis was not performed due to short median follow up.
Discussion
ATM defects are commonly assessed as 11q- presence, but sole deletion does not mean ATM inactivation if the other allele remains intact.17,24 Resulting ATM activity depends on the defect composition (monoallelic vs. biallelic) and, additionally, on functional consequences of individual ATM mutations, which have a decisive impact according to type and position.19,20 Thus, it is difficult to properly distinguish patients with clear ATM dysfunction. With the advent of next-generation sequencing technologies that provide a more sensitive test for ATM mutation, a function assessment will be crucial to set aside real pathogenic mutations.
We detected ATM mutation(s) in 16% (22 of 140) of the patients. Since we analyzed predominantly high-risk CLL patients, this frequency is not representative of CLL cohorts in general. The occurrence of ATM mutated patients in individual genetic groups matched previously reported observations. The highest frequency was noted in the 11q- group (34%), which is nearly identical to the 36% presented in the study by Austen et al.17 Mutations were rare in patients with TP53 defects (4%), confirming mutual exclusivity of p53 and ATM inactivation.24,25 Interestingly, in 3 patients experiencing progression after therapy (P10, P17, P19), during routine TP53 examinations we observed an emergence of new mutations accompanying ATM dysfunction. This suggests that TP53 mutation may provide further advantage to an ATM mutated clone under therapy pressure.
Using several complementary methodologies proved to be beneficial for effective ATM status assessment. The functionally neutral resequencing array enabled fast analysis of point mutations, but false positivity reached 30% and false negativity 11%. Also, its inability to detect other mutations is a clear drawback32 since the proportion of short deletions detected by functional approach was high (25%, 7 of 28 mutations). In our study, all mutated patients exhibited obvious ATM dysfunction, which was predominantly caused by biallelic defects (95%, 21 of 22) but also in one case by sole heterozygous mutation in codon 3008 (P13). In addition, alteration in the same codon was found as the only selected mutation in the second investigation of patient P12, who originally harbored two mutations. Interestingly, both of these samples with sole mutation in codon 3008 showed null p53 downstream gene induction after both IR and doxorubicin, confirming their presumed DNE. Our proposed functional test is, therefore, suitable for common identification of all mutation types that lead to ATM dysfunction. By contrast, none of the SNPs detected in our study disturbed ATM activity, supporting the view they do not contribute to adverse prognosis in CLL patients.33 Our observations conclusively suggest that complete disruption of ATM function in DDR is prominently selected among high-risk CLL patients.
ATM mutation occurrence during the disease course is an important issue to clarify. ATM activation in DDR is a critical defensive mechanism already in early cancerogenesis when genomic instability begins.34 The recurrent presence of 11q- in patients with monoclonal B-cell lymphocytosis35 can indicate a similar ATM role in early CLL development. ATM mutations may already be present in germline8,10 or in hematopoietic precursor cells,24 and may contribute to rapid disease progression through loss of the other allele.36 At the same time, ATM mutations have been observed as clonal variants contributing further to the 11q-subclone expansion.17 The considerable proportion of ATM mutations in our study can be regarded as an early event in the CLL course, which is supported by their frequent presence before first therapy, at the time of diagnosis or even in germline form. Nonetheless, Landau et al.37 recently reported that ATM mutations also frequently occur in subclonal form, contributing later to disease progression.
The defects in ATM-p53-p21 pathway integrating response to DNA damage have shown independent prognostic value in CLL,38 and effective functional testing of this pathway is, therefore, desirable. Initially, two basic defect types after IR exposure were defined as “type A”, which associates with TP53 mutation, and “type B” connected with ATM mutation.25 Later, “type C” that was correlated with polymorphism in p21 gene39 and “type D”, having unclear association with any gene defect38 were described. The functional test based on IR may not, however, be convenient for all laboratories, and use of radiomimetic drugs seems to be a reasonable alternative approach.27 In our study, we show that CLL cells with ATM mutation(s) have an analogously poor response to IR and doxorubicin; both agents should primarily create DNA DSBs.25,40 Our functional test using doxorubicin and fludarabine in parallel enables effective identification of ATM mutations (sensitivity 80%, specificity 97%) and distinguishing between ATM and TP53 defects based on different DNA damaging mechanisms of these drugs (data not shown). In our study, this test failed in several cases for unknown reasons. We also assume that samples without ATM mutation that were approaching the cut-off value could potentially harbor other defect types in the ATM-p53-p21 pathway, e.g. “type C” defect.
ATM dysfunction might play a role in patient response to the conventionally used DNA damaging drugs. In this respect, our data suggest that doxorubicin, which is included in CHOP and R-CHOP regimens employed in lymphoproliferative disorders,41 is most probably ineffective in ATM mutated patients. Notably, low ATM expression level was associated with resistance to doxorubicin in breast cancer patients.42 With regards to fludarabine, the situation remains to some extent more elusive. It was reported that ATM deficiency leads to impaired ATM-mediated phosphorylation17 and higher in vitro resistance of CLL cells to this drug.43 Despite recorded observations, we have demonstrated that cells with obvious ATM dysfunction had preserved response to fludarabine; i.e. normal p53-downstream gene induction and similar in vitro sensitivity as wt and 11q- cells. We propose that overall response to fludarabine could be mediated through other proteins involved in DDR in ATM-deficient patients, although our findings are not conclusive. Thus, primary response to fludarabine seems to be less influenced by ATM inactivation than generally anticipated.
Although patients with sole 11q- have preserved ATM function,13,17 it has been reported that this defect is associated with reduced TTFT44 and distinctive gene expression profile.12 Our analysis recorded almost identically reduced TTFT in groups with sole 11q- and ATM mutation in comparison with wt patients. A potential explanation for the observed clinical impact of sole 11q- may possibly result from a gene dosage effect of ATM12,45,46 or other genes located in deleted region at 11q,13 or from mutations affecting genes like BIRC3 located at 11q. This gene could be an interesting candidate because its mutations are recurrent in CLL and have been recognized as mutually exclusive with TP53 defects,47 similarly to ATM mutations.25
In conclusion, ATM defects involving mutation uniformly resulted in obvious ATM dysfunction throughout our study. By contrast, sole 11q- does not affect ATM function, and simultaneous ATM mutation analysis is, therefore, warranted. Currently, prediction of mutation presence is feasible through functional testing using IR or based on our study using doxorubicin, and this will be critical for distinguishing CLL patients with unambiguously inactive ATM. Indeed, this has potential predictive value and may also offer novel therapeutic strategies utilizing the synthetic lethality concept based on DDR inhibitor application in ATM-defective patients.48 Furthermore, knowledge of ATM mutations will be important to delineate their association with mutations in newly described CLL-related genes, e.g. SF3B1.49
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by grants IGA MZ CR NT13493-4 and NT13519-4, MSMT CR projects SuPReMMe (CZ.1.07/2.3.00/20.0045), MH CZ-DRO (FNBr, 65269705), CEITEC - Central European Institute of Technology (CZ.1.05/1.1.00/02.0068), and MUNI/A/0723/2012, and by the Czech Leukemia Study Group for Life (CELL). The authors would like to thank Rich Zimmerman for English edits and corrections.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic lymphocytic leukemia. Crit Rev Oncol Hematol. 2007;64(3):234–46 [DOI] [PubMed] [Google Scholar]
- 2.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–54 [PubMed] [Google Scholar]
- 3.Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6 [DOI] [PubMed] [Google Scholar]
- 4.Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53–p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114(13):2589–97 [DOI] [PubMed] [Google Scholar]
- 5.Trbusek M, Smardova J, Malcikova J, Sebejova L, Dobes P, Svitakova M, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011;29(19): 2703–8 [DOI] [PubMed] [Google Scholar]
- 6.Döhner H, Stilgenbauer S, James MR, Benner A, Weilguni T, Bentz M, et al. 11q deletions identify a new subset of B-cell chronic lymphocytic leukemia characterized by extensive nodal involvement and inferior prognosis. Blood. 1997;89(7):2516–22 [PubMed] [Google Scholar]
- 7.Starostik P, Manshouri T, O’Brien S, Freireich E, Kantarjian H, Haidar M, et al. Deficiency of the ATM protein expression defines an aggressive subgroup of B-cell chronic lymphocytic leukemia. Cancer Res. 1998;58(20):4552–7 [PubMed] [Google Scholar]
- 8.Bullrich F, Rasio D, Kitada S, Starostik P, Kipps T, Keating M, et al. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res. 1999;59(1):24–7 [PubMed] [Google Scholar]
- 9.Schaffner C, Stilgenbauer S, Rappold GA, Döhner H, Lichter P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood. 1999;94(2):748–53 [PubMed] [Google Scholar]
- 10.Stankovic T, Weber P, Stewart G, Bedenham T, Murray J, Byrd PJ, et al. Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet. 1999;353(9146):26–9 [DOI] [PubMed] [Google Scholar]
- 11.Dickinson JD, Gilmore J, Iqbal J, Sanger W, Lynch JC, Chan J, et al. 11q22.3 deletion in B-chronic lymphocytic leukemia is specifically associated with bulky lymphadenopathy and ZAP-70 expression but not reduced expression of adhesion/cell surface receptor molecules. Leuk Lymphoma. 2006;47(2): 231–44 [DOI] [PubMed] [Google Scholar]
- 12.Guarini A, Marinelli M, Tavolaro S, Bellacchio E, Magliozzi M, Chiaretti S, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2012;97(1):47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouillette P, Fossum S, Parkin B, Ding L, Bockenstedt P, Al-Zoubi A, et al. Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double-strand breaks. Clin Cancer Res. 2010;16(3):835–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouillette P, Li J, Shaknovich R, Li Y, Melnick A, Shedden K, et al. Incidence and clinical implications of ATM aberrations in chronic lymphocytic leukemia. Gene Chromosome Cancer. 2012;51(12):1125–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skowronska A, Parker A, Ahmed G, Oldreive C, Davis Z, Richards S, et al. Biallelic ATM Inactivation Significantly Reduces Survival in Patients Treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 Trial. J Clin Oncol. 2012;30(36):4524–32 [DOI] [PubMed] [Google Scholar]
- 16.Austen B, Powell JE, Alvi A, Edwards I, Hooper L, Starczynski J, et al. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood. 2005;106(9):3175–82 [DOI] [PubMed] [Google Scholar]
- 17.Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25(34): 5448–57 [DOI] [PubMed] [Google Scholar]
- 18.Meyn MS. Ataxia-telangiectasia, cancer and the pathobiology of the ATM gene. Clin Genet. 1999;55(5):289–304 [DOI] [PubMed] [Google Scholar]
- 19.Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet. 1998;62(2):334–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58(10):1009–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22(20):5612–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281 (5383):1677–9 [DOI] [PubMed] [Google Scholar]
- 23.Stankovic T, Hubank M, Cronin D, Stewart GS, Fletcher D, Bignell CR, et al. Microarray analysis reveals that TP53- and ATM-mutant B-CLLs share a defect in activating proapoptotic responses after DNA damage but are distinguished by major differences in activating prosurvival responses. Blood. 2004;103(1):291–300 [DOI] [PubMed] [Google Scholar]
- 24.Stankovic T, Stewart GS, Fegan C, Biggs P, Last J, Byrd PJ, et al. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood. 2002;99(1):300–9 [DOI] [PubMed] [Google Scholar]
- 25.Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood. 2001;98(3): 814–22 [DOI] [PubMed] [Google Scholar]
- 26.Carter A, Lin K, Sherrington PD, Pettitt AR. Detection of p53 dysfunction by flow cytometry in chronic lymphocytic leukaemia. Br J Haematol. 2004;127(4):425–8 [DOI] [PubMed] [Google Scholar]
- 27.Best OG, Gardiner AC, Majid A, Walewska R, Austen B, Skowronska A, et al. A novel functional assay using etoposide plus nutlin-3a detects and distinguishes between ATM and TP53 mutations in CLL. Leukemia. 2008;22(7):1456–9 [DOI] [PubMed] [Google Scholar]
- 28.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengüt S, Tolun A, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64(6):1617–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279(51):53272–81 [DOI] [PubMed] [Google Scholar]
- 30.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41(2):93–103 [DOI] [PubMed] [Google Scholar]
- 31.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24): 9152–9 [DOI] [PubMed] [Google Scholar]
- 32.Kothiyal P, Cox S, Ebert J, Aronow BJ, Greinwald JH, Rehm HL. An overview of custom array sequencing. Curr Protoc Hum Genet. 2009;Chapter 7:Unit 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozanski G, Ruppert AS, Heerema NA, Lozanski A, Lucas DM, Gordon A, et al. Variations of the ataxia telangiectasia mutated gene in patients with chronic lymphocytic leukemia lack substantial impact on progression-free survival and overall survival: a Cancer and Leukemia Group B study. Leuk Lymphoma. 2012;53(9):1743–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434(7035):864–70 [DOI] [PubMed] [Google Scholar]
- 35.Kern W, Bacher U, Haferlach C, Dicker F, Alpermann T, Schnittger S, et al. Monoclonal B-cell lymphocytosis is closely related to chronic lymphocytic leukaemia and may be better classified as early-stage CLL. Br J Haematol. 2012;157(1):86–96 [DOI] [PubMed] [Google Scholar]
- 36.Skowronska A, Austen B, Powell JE, Weston V, Oscier DG, Dyer MJS, et al. ATM germline heterozygosity does not play a role in chronic lymphocytic leukemia initiation but influences rapid disease progression through loss of the remaining ATM allele. Haematologica. 2012;97(1):142–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin K, Adamson J, Johnson GG, Carter A, Oates M, Wade R, et al. Functional Analysis of the ATM-p53–p21 Pathway in the LRF CLL4 Trial: Blockade at the Level of p21 Is Associated with Short Response Duration. Clin Cancer Res. 2012;18(15):4191–200 [DOI] [PubMed] [Google Scholar]
- 39.Johnson GG, Sherrington PD, Carter A, Lin K, Liloglou T, Field JK, et al. A novel type of p53 pathway dysfunction in chronic lymphocytic leukemia resulting from two interacting single nucleotide polymorphisms within the p21 gene. Cancer Res. 2009;69 (12):5210–7 [DOI] [PubMed] [Google Scholar]
- 40.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506 [DOI] [PubMed] [Google Scholar]
- 41.Kahl B. Chemotherapy combinations with monoclonal antibodies in non-Hodgkin’s lymphoma. Semin Hematol. 2008;45(2):90–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knappskog S, Chrisanthar R, Løkkevik E, Anker G, Ostenstad B, Lundgren S, et al. Low expression levels of ATM may substitute for CHEK2/TP53 mutations predicting resistance towards anthracycline and mitomycin chemotherapy in breast cancer. Breast Cancer Res. 2012;14(2):R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kojima K, Konopleva M, McQueen T, O’Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108(3):993–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wierda WG, O’Brien S, Wang X, Faderl S, Ferrajoli A, Do K-A, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol. 2011;29(31):4088–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kienle DL, Korz C, Hosch B, Benner A, Mertens D, Habermann A, et al. Evidence for distinct pathomechanisms in genetic subgroups of chronic lymphocytic leukemia revealed by quantitative expression analysis of cell cycle, activation, and apoptosis-associated genes. J Clin Oncol. 2005;23(16):3780–92 [DOI] [PubMed] [Google Scholar]
- 46.Mohr J, Helfrich H, Fuge M, Eldering E, Bühler A, Winkler D, et al. DNA damage-induced transcriptional program in CLL: biological and diagnostic implications for functional p53 testing. Blood. 2011;117(5):1622–32 [DOI] [PubMed] [Google Scholar]
- 47.Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119(12):2854–62 [DOI] [PubMed] [Google Scholar]
- 48.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJS, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116 (22):4578–87 [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26): 2497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]