

Background: Borrelia burgdorferi OspE protein recruits complement regulator FH onto the bacteria for immune evasion.
Results: We solved the structure of OspE and the OspE·FH complex by NMR and x-ray crystallography.
Conclusion: The OspE·FH structure shows how Borrelia evade complement attack by mimicking how host cells protect themselves.
Significance: This explains how the bacteria survive in the host and facilitates vaccine design against borreliosis.
Keywords: Complement, Microbial Pathogenesis, NMR, Protein Structure, X-ray Crystallography, Bb-CRASP, Borrelia, Immune Evasion, Outer Surface Protein E, Protein Complex Structure
Abstract
Borrelia burgdorferi spirochetes that cause Lyme borreliosis survive for a long time in human serum because they successfully evade the complement system, an important arm of innate immunity. The outer surface protein E (OspE) of B. burgdorferi is needed for this because it recruits complement regulator factor H (FH) onto the bacterial surface to evade complement-mediated cell lysis. To understand this process at the molecular level, we used a structural approach. First, we solved the solution structure of OspE by NMR, revealing a fold that has not been seen before in proteins involved in complement regulation. Next, we solved the x-ray structure of the complex between OspE and the FH C-terminal domains 19 and 20 (FH19-20) at 2.83 Å resolution. The structure shows that OspE binds FH19-20 in a way similar to, but not identical with, that used by endothelial cells to bind FH via glycosaminoglycans. The observed interaction of OspE with FH19-20 allows the full function of FH in down-regulation of complement activation on the bacteria. This reveals the molecular basis for how B. burgdorferi evades innate immunity and suggests how OspE could be used as a potential vaccine antigen.
Introduction
The number of patients with Lyme disease, also known as Lyme borreliosis, is increasing in the United States and Europe (1), making this disease one of the major emerging arthropod-borne infections in the world (2, 3). The disease is caused by the three main genospecies from the Borrelia burgdorferi sensu lato group: B. burgdorferi sensu stricto, Borrelia afzelii, and Borrelia garinii (4). These spirochetes are transmitted to human skin from the mid-gut of Ixodes sp. ticks if the feeding time is long enough (5). The earliest manifestation of the infection is erythema chronicum migrans, a slowly expanding skin rash around the tick bite, whereas the variable later stage manifestations are from organs where the bacteria have spread from skin mainly via blood circulation (6). These manifestations include arthritis and central nervous system disorders, depending on the Borrelia genospecies involved in the infection; B. burgdorferi sensu stricto is relatively arthrotropic, whereas B. garinii is generally neurotropic (7).
The Lyme Borrelia can survive for years inside the human body without evoking an efficient immune attack by the host. This is widely attributed to the powerful tools that these spirochetes use to evade host innate and acquired immune responses. Efficient down-regulation of complement, a major innate immune system of the host, on the surface of Borrelia is essential for survival of these pathogens in human serum and therefore central in their immune evasion (8, 9). Complement is an ancient arm of innate immunity composed of a group of plasma proteins activated by three initiation pathways. The alternative pathway initiates a non-selective attack against all surfaces in contact with host plasma and thus is responsible for the wide spectrum attack mounted by innate immunity. If a surface is unprotected, the initial attack leads to amplified activation, which results in enhanced phagocytosis, formation of lytic membrane pores, and release of chemotactic peptides (10).
Down-regulation of complement on Borrelia is mediated by recruitment of host complement regulator factor H (FH)8 onto the cell surface by two outer surface proteins, OspE (also known as the B. burgdorferi complement regulator-acquiring surface protein 3, BbCRASP-3)) and BbCRASP-1 (11–13). These proteins, unlike some other borrelial FH-binding proteins, are expressed on Borrelia in vitro at human body temperature and in vivo in a mammalian host (14). Practically all patient-derived B. burgdorferi sensu stricto strains have genes encoding OspE and/or one or more of the highly homologous FH-binding Erp proteins (the Erp paralogs) (15). OspE has been shown to be important for survival of B. burgdorferi sensu stricto and B. garinii because addition of fluid phase recombinant OspE to serum (blocking binding of FH onto the bacterial surface) results in impaired bacterial survival (13).
FH is a 150-kDa plasma glycoprotein made up of 20 globular complement control protein modules (CCP modules), each consisting of ∼60 amino acids. It is essential for keeping complement activation under control both in plasma and on host cells. The central role of FH-mediated regulation on host cells is clearly demonstrated by impaired regulation due to mutations in domains 19–20, leading to a severe systemic disease, atypical hemolytic uremic syndrome (16, 17). Although FH domains 1–4 mediate the regulatory activity (18), we and others have recently shown that the recognition of host cells results from joint binding of domain 20 to cell surface glycosaminoglycans (or heparin) and domain 19 to the main complement opsonin C3b (19, 20).
Borrelia are not the only pathogenic microbes able to bind human FH to protect themselves against the host complement attack (21–23). Two regions on FH mediate binding to practically all FH-binding pathogens, one in domains 6 and 7 and another in domains 19 and 20 (FH19-20) (24–29). Both of these regions also contain glycosylaminoglycan-binding sites (30, 31). B. burgdorferi acquires FH via FH19-20 using the surface lipoprotein OspE (27), but it is not known which part of OspE is responsible for FH binding because both N- and C-terminal OspE truncations abolish FH binding (15). To date, there are no bacterial surface protein structures solved in complex with FH19-20 and only one structure of a bacterial protein bound to FH domains 6 and 7 (32).
Currently there is no vaccine available for Lyme borreliosis because the previously used outer surface lipoprotein A (OspA)-based vaccine was withdrawn from the market a decade ago due to serious side effects (33). A vaccine would, however, be highly valuable in clinical practice. Because the recent advancement in developing a vaccine against another difficult and important bacterial target, group B meningococcus, is based on a Neisseria meningitidis protein that binds host FH, we thought that a similar strategy might work with Borrelia. We therefore decided to determine the structural basis of host FH binding by B. burgdorferi. We used both NMR and x-ray crystallography together with further biophysical analyses and mutagenesis data described elsewhere (34) to characterize the interaction between OspE and domains 19 and 20 of FH. This is the first structure of a microbial protein that binds FH20. The OspE structure in solution and in complex with FH19-20 reveals how B. burgdorferi evades complement attack and is therefore able to cause human infection, paving the way to develop OspE as a vaccine candidate.
EXPERIMENTAL PROCEDURES
Expression of Proteins and OspE·FH19-20 Complex Formation
Cloning and purification have been described previously for wild type (35) and mutant FH19-20 proteins (36, 37). For crystallography, OspE was cloned, expressed, and purified as described earlier (13). For NMR, OspE (residues 21–171) was cloned into the pHYRSF53-36 vector between the BamHI and HindIII sites with an N-terminal hexahistidine tag and a yeast Smt3 domain (38). The construct was expressed in E. coli ER2566 strain grown in M9 medium containing 15NH4Cl and d-[13C6]glucose as sole nitrogen and carbon sources, respectively. Cells were grown at 310 K, and at A600 ∼0.5, protein expression was induced by the addition of 0.5 mm isopropyl β-d-1-thiogalactopyranoside. After 4 h of protein expression, cells were harvested by centrifugation at 9,400 × g at 277 K for 10 min. The cell pellet was resuspended in 50 mm sodium phosphate buffer (pH 8.0) with 300 mm NaCl and flash-frozen in liquid nitrogen until protein purification. After being thawed, the cells were sonicated for 15 min (60% of total power) using a rod type sonicator on an ice bath for protein release. After centrifugation (45 min, 42,500 × g), the supernatant was loaded onto a 5-ml HisTrap FF column (GE Healthcare), and the bound protein was eluted with an imidazole gradient (25 ml gradient of 30–250 mm imidazole at 2 ml/min). Fractions containing His6-Smt3-OspE were digested with yeast ubiquitin-like-protein protease (Ulp1) in PBS containing 1 mm DTT at 298 K for 4 h (20 μl of Ulp1 for each 20 ml of the OspE-containing solution). The digested sample was then loaded on a 5-ml HisTrap FF column to remove His6-Smt3 and His6-Ulp1, and OspE was collected in the flow-through. After dialysis (overnight against 2 liters of 20 mm sodium phosphate, pH 6.0), the purity of OspE in SDS-PAGE was >95% (supplemental Fig. 1a).
Light Scattering and Size Exclusion Chromatography Analysis
We used size exclusion chromatography coupled with multiangle light scattering to find the optimal proportions of FH19-20 and OspE to form the complex in solution (supplemental Fig. 1b). The proteins alone and their mixtures at two different apparent molar ratios (1:1 and 1:1.25) were run over a Superdex200 column attached to HPLC equipment (Shimadzu). A MiniDAWN TREOS light-scattering detector and Optilab rEX refractive index detector together with ASTRA version 5.3.4.20 software (Wyatt Technology Corp.) were used to calculate masses of proteins and complexes eluted from the column. A size exclusion analysis of OspE and FH19-20 alone and the OspE·FH19-20 complex was done by running the preparates through a Superdex200 column attached to an ÄKTA purifier HPLC system (GE Healthcare) (Fig. 2a). All samples were eluted in PBS at a 0.5 ml/min flow rate at 22 °C.
FIGURE 2.
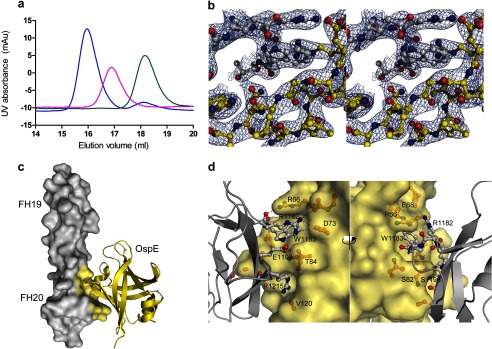
OspE·FH19-20 complex formation and the x-ray crystal structure. a, gel filtration analysis of OspE (magenta), FH19-20 (green), and OspE·FH19-20 complex (blue). b, a detail of the electron density map (2FO − FC) of the OspE·FH19-20 complex in stereo. Carbon atoms of FH19-20 are shown in gray, and those of OspE are shown in yellow. c, crystal structure of OspE (yellow schematic) in complex with FH19-20 (gray surface model, interacting atoms in yellow). d, interface between FH domain 20 (gray schematic) and OspE (yellow surface model) from two directions about 180° apart with the interacting residues shown in a ball-and-stick representation and the main interacting residues annotated.
NMR Spectroscopy and Resonance Assignment of OspE
All NMR measurements were performed on Varian Inova 600-MHz or Varian Inova 800-MHz spectrometers, both equipped with a triple resonance cold probe. For structure determination of OspE, measurements were performed at 298 K on a 1 mm 15N- and 13C-labeled protein sample in 20 mm phosphate buffer (pH 6.0). Sequence-specific resonance assignment was based on a series of standard spectra: 15N heteronuclear single quantum correlation (HSQC), aliphatic constant time 13C HSQC, HNCA, HNcoCA, HNCACB, CBCAcoNH, HNcaCO, HNCO, HBHAcoNH, 15N-edited total correlation spectroscopy (TOCSY) with 50-ms mixing time, CCCcoNH, HcCH COSY, and HcCH TOCSY with 50-ms mixing time. Aromatic rings were assigned based on (HB)CB(CD)HD, (HB)CB(CDCE)HE, and aromatic constant time 13C HSQC. For structure calculation, 15N-edited NOE NOESY-HSQC and 13C-edited NOESY-HSQC were recorded with 80-ms mixing time at a 1H frequency of 800 MHz (Table 1). All spectra were processed using NMRPipe (39), and specific resonance assignment was performed using the CCPNMR analysis suite software version 2.2.1 (40). The chemical shifts and the unassigned NOE peak lists were used as inputs for NMR structure calculation with the program CYANA version 3.0 (41, 42). Twenty structures with the lowest CYANA target functions were selected for energy refinement in a 5 Å water shell using the AMBER force field. Ramachandran plot was generated using PROCHECK-NMR (43), and it indicated that 81.9% of the residues were in the most favored and 18.1% in the additional allowed regions.
TABLE 1.
NMR data and structure statistics of the OspE structure
| NMR distance constraints | |
| Distance constraints | |
| Total | 3,446 |
| Intraresidue | 736 |
| Interresidue | 2,710 |
| Sequential (|i − j| = 1) | 809 |
| Medium range (|i − j| < 4) | 479 |
| Long range (|i − j| ≥ 5) | 1,422 |
| Structure statistics | |
| Violations (mean and S.D.) | |
| Distance constraints (Å) | 0.0012 ± 0.0001 |
| Maximum distance constraint violation (Å) | 0.22 ± 0.04 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.0097 ± 0.0001 |
| Bond angles (degrees) | 1.857 ± 0.016 |
| Impropers (degrees) | 0.623 ± 0.016 |
| Average pairwise r.m.s.d.a (Å) | |
| Heavy | 0.73 ± 0.06 |
| Backbone | 0.38 ± 0.05 |
a Pairwise r.m.s.d. was calculated among 20 refined structures of residues 42–171.
All relaxation measurements were performed at a 1H frequency of 600 MHz. T1 relaxation rates were determined using the T1 relaxation times of 10, 20, 30, 40, 50, 60, 70, 90, 110, 130, 150, and 210 ms. T2 relaxation rates were determined using a Carr-Purcell-Meiboom-Gill sequence (CPMG)-based pulse sequence with a 1.3-ms refocusing interval, and the used T2 relaxation times were 10, 30, 50, 70, 90, 110, 130, and 150 ms (44). Heteronuclear 15N{1H}-NOEs were determined by recording [1H,15N] HSQC spectra with and without 3.5 s of 1H saturation (supplemental Fig. 2). Peak intensities from the recorded spectra were used for data analysis.
Monitoring OspE and FH19-20 Interaction by NMR Spectroscopy
The 15N-labeled OspE was titrated with unlabeled FH19-20, and monitoring the [1H,15N] transverse relaxation optimized spectroscopy (TROSY) spectra indicated that OspE and FH19-20 formed a complex. Backbone resonance assignments of the OspE in complex with FH19-20 were performed on 0.3 mm 15N- and 13C-labeled OspE saturated with FH19-20, using TROSY variants of HNCA, HN(CO)CA, HNCO, and HN(CA)CO spectra recorded at 308 K. The chemical shift differences between free OspE and OspE·FH19-20 complex were calculated for each residue using the equation,
 |
where ΔδHN and ΔδN are the chemical shift differences for backbone HN and N atoms (supplemental Fig. 3).
Crystallizing the OspE·FH19-20 Complex and Solving Its Structure
FH19-20 tends to crystallize as a homotetramer (35); therefore, we used FH19-20 with mutations D1119G and Q1139A to prevent this happening, as we have done before (20). The FH19-20D1119G,Q1139A protein has similar binding affinity for OspE as the wild type FH19-20 (mean IC50 of 0.55 versus 0.42 μm) (supplemental Fig. 4). The FH19-20D1119G,Q1139A·OspE complex was crystallized at 293 K from sitting drops in the presence of 2 m ammonium sulfate, 0.1 m citric acid, and 0.2 m sodium chloride at pH 5.5. Crystals appeared within 4 days and were cryoprotected in the mother liquor supplemented with 25% glycerol. The diffraction data (to 2.83 Å) were collected at the ESRF ID14-4 beam line at 100 K on a Q315r ADSC CCD detector at 0.97372 Å. The data were indexed and scaled using XDS (45). The structure of the OspE·FH19-20 complex was initially solved by structure-optimized molecular replacement with Rosetta (46). A molecular replacement solution was obtained using the wild type FH19-20 structure (Protein Data Bank code 2G7I) (35) and a truncated NMR structure of OspE (without the loops) with an R-factor of 0.3. After successive rounds of building with Coot (47) and refinement with REFMAC (48) or PHENIX (49), we could identify one OspE molecule bound to a single FH19-20. The loops were modeled during real space refinement using “phenix.refine” software without any applied restraints. The final R-factors (Rwork/Rfree) of the refined complex structure are 19.3/25.5 (%) (shown in Table 2). The last refinement cycles were done using TLS parameters (nine TLS groups). In the Ramachandran plot, 94.0% of the residues in the structure are in the most favored regions. The structure illustrations have been prepared using PyMOL software (version 1.3, Schrödinger, LLC, New York). The interface between FH19-20 and OspE was analyzed using the PISA (50) server.
TABLE 2.
Crystallographic data and refinement of the OspE·FH19-20 complex structure
| Data collection | |
| Space group | P3121 |
| Cell dimensions | |
| a, b, c (Å) | 86.4, 86.4, 107.8 |
| α, β, γ (degrees) | 90, 90, 120 |
| Rmerge | 17.0 (109.8)a |
| I/δI | 9.99 (2.45) |
| Completeness (%) | 99.2 (100) |
| Redundancy | 7.05 (7.08) |
| Refinement | |
| Resolution range (Å) | 19.5–2.83 (2.93–2.83) |
| No. of reflections (unique/observed) | 11,501/8,0778 |
| Rwork/ Rfree (%) | 19.3/25.5 |
| No. of atoms | |
| Protein | 1,913 |
| Ligand/SO4 | 5 |
| Water | 15 |
| B-factors | |
| Protein | 64.6 |
| Ligand/ion | 58.8 |
| Water | 49.2 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.012 |
| Bond angles (degrees) | 1.37 |
a Data of the highest resolution shell is shown in parentheses.
Sequence Alignments
Sequence alignments were done using the ClustalW software (51) for Fig. 4 and DALI software (52) for supplemental Fig. 5.
FIGURE 4.

Sequence alignment of the FH binding region of OspE with the homologous parts of the Erps. Sequence alignment between OspE from the N40 strain of B. burgdorferi (used in this study) and Erp paralog proteins encoded by other FH-binding B. burgdorferi strains (B31, BL206, 297, and Sh-2-82) and single strains of B. afzelii and B. garinii is shown. Residues of OspE forming hydrogen bonds with FH19-20 are annotated, their alignments are shown with boxes, and identical residues are highlighted in boldface type.
Statistical Analyses
Values are expressed as means ± S.D. All statistical analyses were performed using GraphPad Prism version 5.0.
RESULTS
OspE Consists of an Unexpected Globular Domain with a Flexible N Terminus
The solution structure of OspE was solved by NMR spectroscopy (Table 1 and Fig. 1a). Residues 42–171 of OspE form a globular single-domain protein with a backbone root mean square deviation (r.m.s.d.) of 0.38 ± 0.05 Å indicating a well defined structure. OspE has a rigid a + b fold of eight β-strands and two short α-helices arranged in a repeating topology of four β-strands followed by an α-helix (β1-β2-β3-β4-αI-β5-β6-β7-β8-αII). The β-strands form antiparallel β-sheets, where β1–β4 and β5–β8 are orientated almost perpendicularly. β1 and β8 are held together by hydrogen bonds, thus forming an up-down squashed asymmetric β-barrel. It can be viewed as a β-barrel with two distinctive sides, one uniform and another non-uniform. The β2 and β6 strands are highly twisted and deviate from the formation of classic β-barrel strands. β1, β2, and β6 are very long strands (10, 12, and 11 residues, respectively), and β5 along with β8 are the smallest β-strands (6 residues each).
FIGURE 1.
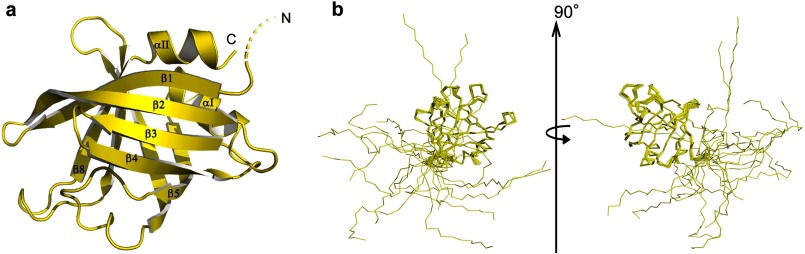
The NMR structure of OspE. a, schematic representation of the mean NMR structure of OspE (residues 21–171, dashed line indicating the flexible N terminus). b, overlay of 20 energy-minimized NMR structures of OspE with the backbone of residues 42–171 being superimposed. Two orientations are shown rotated 90° from each other.
The N-terminal region (residues 20–41) of OspE is structurally flexible, as shown by superimposition of residues 42–171 from 20 NMR structures (Fig. 1b). The 15N relaxation data of OspE (supplemental Fig. 2) also show that these residues are moving freely, because of the low T1/T2 and 15N{1H}-NOEs ratio. OspE is attached to the outer membrane via an N-terminal lipid anchor putatively bound to the N-terminal cysteine at position 20 (53). The anchoring topology of OspE is not known, but we can safely assume that the flexible N terminus allows the protein to move freely about its lipid anchor. To assess the tightness of OspE binding to the membrane, we incubated killed Borrelia in a buffer for up to 24 h and after centrifugation analyzed the detached OspE from the supernatant. The amount of OspE in the supernatant was low and did not increase as a function of time (supplemental Fig. 6), indicating tight anchoring of OspE to the bacterial surface.
A DALI (52) search showed that the globular domain of OspE resembles the SsgA-like protein (SALP) family of proteins (54), certain DNA/RNA-binding proteins (55–58), some fatty acid-binding proteins (55, 59, 60), and the “Homer” family of proteins (61, 62) (supplemental Fig. 5). The fold has never, to our knowledge, been reported in any extracellular or immune evasion-related molecule from any microbe. OspE has less than 15% sequence identity to any member of the structural family (supplemental Fig. 5). The mammalian PUR-α protein, which binds nucleic acids, has been reported to be involved in immune evasion of viruses like John Cunningham virus and human immunodeficiency virus (HIV) (58). The nucleic acid binding proteins that were found to be structural relatives of OspE were mainly from the “Whirly” protein group, so named because of the resemblance of their quaternary structure to a whirligig.
The OspE·FH19-20 Complex Reveals the Interacting Surfaces
Optimization of heterodimer formation by gel filtration analysis of OspE and FH19-20 (Fig. 2a) enabled us to crystallize OspE in complex with FH19-20 (supplemental Fig. 1b). Using the optimized 1:1 molar ratio of the proteins, we obtained rod-shaped co-crystals of OspE and FH19-20 in the presence of 2 m ammonium sulfate, 0.1 m citric acid, and 0.2 m sodium chloride (pH 5.5) at 20 °C. The crystal structure of the OspE·FH19-20 complex was solved at 2.83 Å resolution (Table 2 and Fig. 2b) using our previously solved FH19-20 structure (35) and the NMR structure of OspE (described above) as models in molecular replacement (Fig. 2b). In the complex, OspE binds to domain 20 of FH with a contact surface area of 691 Å. The complex is held together by an E68OspE-R1182FH-D73OspE ion triple, which is also buttressed by a sulfate group forming an Arg1182FH-SO42−-Arg66OspE ion triple, whereas Arg1182FH also forms an ion pair with Glu1198FH (Fig. 2, c and d). These interactions form the top part of a pocket for Trp1183FH, which makes hydrophobic contacts with the aliphatic side chain of Arg1182FH and with the OspE backbone around Gly75 and Ala83 (Table 3).
TABLE 3.
Hydrogen bonds at the OspE·FH19-20 interface in the crystal structure along with the chemical shift perturbations of the concerned residues
| FH19-20 residue (atom) | Distance | OspE residue (atom) | Chemical shift perturbation in 1H,15N HSQC spectra |
|---|---|---|---|
| Å | ppm | ||
| Arg1182 (NH) | 3.50 | Asp73 (OD2) | 0.71 |
| Arg1182 (NH2) | 2.93 | Asp73 (OD2) | 0.71 |
| Arg1182 (NH2) | 3.25 | Glu68 (OE1) | 0.13 |
| Arg1182 (O) | 2.91 | Arg66 (NH2) | —a |
| Trp1183 (O) | 2.96 | Asn77 (ND2) | 0.72 |
| Ser1191 (OG) | 2.69 | Gly80 (O) | — |
| Glu1195 (OE2) | 3.50 | Tyr114 (OH) | 0.58 |
| Ser1196 (O) | 2.89 | Ser82 (N) | 1.01 |
| Glu1198 (N) | 2.93 | Ser82 (O) | 1.01 |
| Glu1198 (OE1) | 2.89 | Thr84 (N) | — |
| Glu1198 (OE1) | 2.51 | Thr84 (OG1) | — |
| Arg1215 (NH2) | 2.72 | Val120 (O) | 0.20 |
a —, atoms with larger than 0.4-ppm perturbation were considered to be significantly perturbed upon interaction of OspE with FH19-20.
The OspE structures in solution (NMR) and in complex with FH19-20 (x-ray) were very similar (backbone r.m.s.d. of 1.43 Å/Cα) (supplemental Fig. 7a). The structure of FH19-20 in complex with OspE was also practically identical to the published structures of FH19-20 in homotetramers (35) (backbone r.m.s.d. 1.43 Å/Cα) or in the FH19-20·C3d complex (20) (backbone r.m.s.d. 1.15 Å/Cα) (supplemental Fig. 7b). The sulfate ion found in the OspE·FH19-20 interface did not affect the orientation of the side chain of FH R1182 (supplemental Fig. 8, a and b). When the region of OspE close to the sulfate ion was compared between the NMR ensemble and the crystal complex structure, it was observed that orientation of Glu68 was similar, whereas that of Arg66 was slightly different, most probably due to a hydrogen bond between the NH2 of Arg66 and the oxygen of Arg1182 (shown in red in supplemental Fig. 8a).
The Interaction Site on OspE Confirmed by NMR Analysis
To verify the observed interface between OspE and FH19-20, we analyzed chemical shift perturbations in the NMR spectrum of OspE upon the addition of saturating concentrations of wild type FH19-20 (Fig. 3a and supplemental Fig. 3). The residues of OspE that shifted most (>0.4 ppm) are clustered on β-strands 1–4, which form the core of the OspE·FH19-20 interface in the crystal structure (Fig. 3b). Of the nine OspE residues involved in hydrogen bonding, six could be assigned in the assay, and five of those were among the residues that shifted most (>0.4 ppm) (Table 3). The other three residues involved in binding (Arg66, Gly80, and Thr84) could not be assigned in the spectra of the FH19-20-bound OspE, probably due to large changes in the microenvironment caused by formation of the complex. We confirmed the interface of OspE from the FH19-20 side using mutants of FH19-20 that we have reported elsewhere (34).
FIGURE 3.

The interaction site between OspE and FH19-20. a, chemical shift perturbation of OspE residues in an HSQC titration NMR experiment upon the addition of FH19-20 (residues not assigned indicated with asterisks). b, residues with larger that 0.4 ppm (dotted line) perturbation considered to interact with FH19-20 are colored red in the schematic representation of OspE, whereas residues not observed in the OspE-FH19-20 complex are colored black. The β-strands involved are indicated (β1–β4). c, surface representation of FH domain 20 with the OspE-binding residues found in the crystal structure highlighted in green (left), the heparin-binding residues highlighted in blue (middle), and the endothelial cell-binding residues highlighted in orange (right). The key heparin and endothelial cell binding residues have been annotated.
The OspE Binding Site Overlaps with the Heparin and Endothelial Cell Binding Sites on FH
The OspE binding site was next compared with the previously mapped binding sites for heparin or endothelial cells on FH domain 20 (Fig. 3c). Mutations in residues Arg1182, Lys1186, Lys1188, Arg1203, Arg1206, Arg1210, Arg1215, and Lys1230 impair binding of FH19-20 to heparin (37, 63, 64). Of these eight residues, five are on the same side of FH19-20 as the binding site for OspE, and two (Arg1182 and Arg1215) directly interact with OspE. This indicates that the heparin/endothelial cell binding site and the OspE binding site on FH domain 20 overlap.
The OspE Residues Mediating Binding to FH19-20 Are Conserved
The sequence identity between OspE and the other FH-binding Erp paralog proteins (i.e. the OspE or BbCRASP-3 protein family) is more than 85% (Fig. 4). The sequence alignment shows that, considering only the subset of Erp paralog proteins from B. burgdorferi sensu stricto that bind FH, eight of the nine OspE residues that form hydrogen bonds with FH19-20 in the crystal structure are conserved. Arg66, Glu68, Asn77, Gly80, Thr84, and Tyr114 are identical in all of the FH-binding Erp paralog proteins (Fig. 4), and Asp73 and Ser82 were conserved, being replaced by the conservative D73E and S82T mutations in some of the proteins. Seven of the nine residues were also conserved in the OspEs from B. afzelii and B. garinii (all except for Ser82 and Val120) (Fig. 4).
The OspE Binding Site Is Available in FH19-20 Bound to C3b
To analyze if the OspE binding site is available on FH19-20 bound to its physiological ligand C3b, we superimposed our OspE·FH19-20 complex structure on the previously solved structure of FH19-20 in complex with C3d (20) by superimposing FH19-20 structures. The r.m.s.d. of Cα for the superimposed FH19-20 structures was only 0.9 Å, and there were no steric clashes between OspE and C3d. Next we generated an OspE·FH19-20·C3b complex by superimposing the C3d from our OspE·FH19-20·C3d superimposition model on the structure of C3b (containing the C3d domain, also called the thioester domain) (65). In this model, OspE had no steric clashes with any part of C3b, and most importantly, OspE and the thioester site of C3b faced the same direction without blocking each other (Fig. 5b). This indicates that FH bound to OspE on the borrelial surface is sterically able to bind a C3b molecule deposited onto the same surface. This is consistent with the role of OspE in mediating complement evasion of B. burgdorferi.
FIGURE 5.

Superimposition based models of OspE in complex with FH19-20 and C3d or C3b. a, superimposition of the OspE·FH19-20 structure with the FH19-20·C3d structure (20), indicating no steric clashes in the OspE·FH19-20·C3d complex. b, model of the spatial organization of the OspE·FH19-20 complex bound to C3b (65) on the target surface as based on superimposition of the OspE·FH19-20·C3d complex model with the structure of C3b (containing the C3d domain). The location of the thioester site is indicated in orange.
DISCUSSION
This study resolves the underlying molecular mechanism of complement evasion by B. burgdorferi. It reveals the structural fold of borrelial OspE, identifies the binding surfaces and residues used for the interaction between OspE and host FH, shows the similarity in binding of FH19-20 to the host endothelial cell surface via the heparin binding site and to the microbial OspE protein, and finally explains how FH recruited by OspE onto the borrelial surface is sterically able to eliminate the central complement component C3b on the same surface.
A previous computational analysis suggested that the structure of OspE was composed of coiled coil motifs (66). Our structure, however, clearly shows that this is wrong; OspE has an eight-stranded up-down β-barrel globular structure (residues 42–171) linked to the membrane by a very flexible 21-amino acid tail (Fig. 1b). A DALI (52) search indicated that the structural fold has been found in a trimeric protein from the SALP family, several proteins from the “nucleic acid-binding protein” group, some proteins from the “fatty acid-binding protein” family, and a few proteins from the Homer family (supplemental Fig. 5). The sequence homology between OspE and its structural relative proteins is less than 15%, explaining why the structural similarity has not been identified earlier. As far as we know, this is the first time this fold has been identified in any extracellular or immune evasion-related protein of microbial origin. Of the OspE distant homologues, the mammalian transcriptional activator PUR-α appears to be involved in viral immune evasion (58) by acting as the host factor for viral replication. Distant structural homologues of OspE are also found among the Homer family of proteins. Certain members of this family are involved in neuronal plasticity by binding to and regulating metabotropic glutamate receptors (67), whereas others function by binding to members of the nuclear factor of activated T cells (NFAT) family of proteins and down-regulating T-cell activation (62). There is, however, no similarity in the ligand-binding residues between PUR-α or Homer proteins and OspE, which is hardly surprising given the difference in the charge properties of the two ligands. The fatty acid-binding proteins that have a β-sheet structure similar to that of OspE mainly interact with the fatty acids via the residues inside the hydrophobic core of the protein (55, 59, 60).
The fold of the globular domain seems to be stable and fairly rigid because the r.m.s.d. of the globular domain of the 20 NMR structures was only 0.38 ± 0.05 Å for the backbone atoms, and the structures of the liganded (x-ray) and unliganded (NMR) OspE were very similar (r.m.s.d. of 1.43 Å for Cα atoms) (supplemental Fig. 7a). The length of the extended, structurally flexible tail is more than 50 Å. Although the anchor topology of OspE is not known, trypsinolysis studies on the anchor topologies of borrelial OspA and Vsp1 proteins indicate that in both OspA (N-terminal tethering domain of just 12 residues) and Vsp1 (tethering domain of 21 residues), the tail is on the outside. In OspA, residues in positions 6–11 can be cleaved by trypsin, whereas in Vsp1, residues in positions 7–14 can be cleaved (68, 69); i.e. most of the N-terminal tail in both proteins is surface-exposed and thus cannot be part of a protein membrane anchor but is instead presumably part of a flexible linker. Consequently, the most plausible anchoring topology for OspE is that it binds through a triacyl cysteine anchor, and the N-terminal region provides flexibility. Our results on incubation of killed Borrelia (supplemental Fig. 6) indicate that the majority of OspE is tightly bound to the borrelial surface and suggests that it is bound very firmly to the OM, indeed. Such a lipid anchor would provide OspE with good lateral mobility in the membrane, whereas the longer tethering domain would allows OspE the freedom to rotate and tilt. OspE can obviously bind to host FH on the cell surface because the binding site is far from the tethering domains.
In our OspE·FH19-20 structure, four residues (Arg1182, Trp1183, Glu1198, and Arg1215) in the FH C terminus play a key role in binding to OspE. The central role of these residues is supported by the chemical shift perturbations in the NMR spectrum of OspE upon the addition of saturating concentrations of wild type FH19-20 (Fig. 3a and supplemental Fig. 3) and by a competition binding assay with 14 mutant FH19-20 proteins (34). We have previously proposed, on the basis of alanine scanning mutations to OspE peptides, that multiple lysines in OspE might be required to bind FH19-20 (27). None of the peptides used in that work, however, could inhibit OspE binding to FH19-20 (27), indicating that binding of the linear OspE peptides to FH19-20 was weak. In our OspE·FH19-20 structure, there are no OspE lysines in the interface, so our previous results with peptides did not reflect the true OspE·FH19-20 interaction but an artificial binding of positively charged short linear synthetic peptides to a negatively charged patch on FH19-20.
A sulfate ion was found in the OspE·FH19-20 interface (supplemental Fig. 8a). In principle, a sulfate ion could potentially influence orientation of side chains in its vicinity either by steric hindrance or by forming hydrogen bonds. Superposition of the OspE·FH19-20 complex structure with the OspE NMR structure and the WT FH19-20 structure separately (supplemental Fig. 8, b and c) showed hardly any difference in the side chains close to the sulfate ion. Therefore, the sulfate ion itself obviously has not disturbed the arrangement of the residues within the interface.
Is it possible that the OspE·FH19-20 structure we see is a crystallographic artifact? The NMR-based perturbation assay clearly excludes that possibility because the chemical shift of the OspE residues that interact with FH19-20 in the crystal structure changed when wild type FH19-20 was bound to OspE in solution (Fig. 3a). Radioligand binding assays using 14 point-mutated FH19-20 proteins also support our results. Single point mutations of five (R1182A, W1183L, L1189R, E1198A, and R1215Q) of seven key residues in the OspE·FH19-20 interface caused clear impairment in binding of FH19-20 to OspE (34) (the other two were not in the mutation panel). Deletion of the C-terminal 15 residues of the highly homologous OspE paralog p21 impairs, but does not abolish, binding to FH, whereas deletion of the N-terminal 35 residues has no effect (27). On the basis of the OspE·FH19-20 structure, it is possible that the effect of the C-terminal OspE deletion is either due to disturbance of the globular OspE fold, because the last 15 residues in OspE form α-helix 2 and part of β-sheet 8, or due to a local effect of the disruption of the β-sheet 8 on the loop between β-sheets 3 and 4 and thereby on the FH-binding residues on those sheets. On the basis of the structure (only part of the β-sheet 8 needed for the barrel was missing) and because the deletion did not abolish binding to FH19-20, it is likely that the deletion did not disrupt the fold completely. Our results thus explain well the previous data with deletion mutants of an OspE homolog.
We and others have earlier shown that binding of FH is essential for B. burgdorferi to survive in human serum and that OspE (i.e. BbCRASP-3) and BbCRASP-1 (70) are needed for this survival (8, 9, 13, 15, 27). It is obvious that the microbe benefits from the recruitment of host FH onto the microbial surface because FH acts as a cofactor for factor I in the degradation process of the central complement component C3b needed for both opsonization and propagation of the complement cascade to form membrane attack complexes (71). Therefore, it is easy to understand that acquisition of host FH onto Borrelia correlates strictly with survival of the spirochetes in non-immune serum or blood.
The C-terminal domains 19 and 20 of FH are known to mediate physiologically important binding of FH to both the C3d part of C3b (19, 20) and heparin/endothelial cells (37, 63, 64). Comparing the binding site of OspE to the previously reported binding sites for these physiological ligands showed that the OspE site overlaps with the heparin/endothelial cell binding site (residues Arg1182 and Arg1215 and the surrounding region) (37, 63, 64), whereas the C3d/C3b binding site on domain 19 is clearly distinct. This is also clear in the superimposition of the OspE·FH19-20 structure with the previously published FH19-20·C3d structure (20) (Fig. 5a). The overlap of the OspE and heparin binding sites indicates that B. burgdorferi mimics host cells in acquiring FH with OspE by binding to the same site as host cells bind with heparin. This resembles acquisition of FH by N. meningitidis via a heparin binding site on domain 7 of FH (32).
Two requirements have been suggested for physiologically relevant interaction between a microbe and host FH; the microbial binding site on FH needs to be easily accessible, and the cofactor site on FH domains 1–4 must not be disturbed (72). Our work shows that the interaction between FH and OspE fulfills both of these requirements at the structural level, because binding of OspE to FH19-20 does not block binding of FH (neither domains 1–4 nor 19–20) to C3b (20, 73), and the OspE site is fully accessible on FH, as seen in our OspE·FH19-20·C3b model (Fig. 5). Finally, this superimposition shows that the borrelial binding site on FH is directed toward the surface to which C3b is bound to via the thioester bond. This suggests that FH bound to OspE on the borrelial surface can recruit factor I not only to fluid phase C3b but also to opsonizing C3b bound to the borrelial surface, thus preventing both opsonophagocytosis and propagation of the complement cascade to form lytic membrane attack complexes.
Humoral immune response against an outer surface lipoprotein of B. burgdorferi, OspA, has been shown to protect humans from Lyme borreliosis (74). Although the OspA-based vaccine against the disease failed due to side effects (75), it indicated that a humoral immune response can protect from Lyme borreliosis. The FH-binding protein from N. meningitidis serotype B has recently been successfully used as a candidate for vaccine development against meningococcal disease and is now in phase III clinical trials (76). Therefore, studies aimed at identifying a new borrelial surface antigen, preferably one binding FH, are warranted. A suitable vaccine candidate must 1) be surface-exposed, 2) be conserved among different strains and genospecies of the pathogen, 3) be produced during human infection, 4) be necessary for development of a clinical infection, and 5) raise an immune response in humans in vivo. There is an increasing body of evidence that the FH-binding protein OspE meets all of these criteria (13). Our current study increases the acceptability of OspE as a vaccine candidate because it shows that the residues needed for the OspE·FH19-20 interaction are highly conserved among the different Borrelia genospecies and strains (Fig. 4).
Our OspE·FH19-20 structure is the first structure of a microbial protein binding to domains 19 and 20 of FH. It is known that more than 10 pathogenic microbes other than Borrelia acquire host FH using the same domains. We have studied the binding of several other microbes to FH19-20, and the data indicate a conserved pattern of microbial acquisition of FH via these domains (34). Domains 19 and 20 of FH are evidently essential for recognition of host cells and surfaces by FH because mutations in these domains lead to a life-threatening disease, atypical hemolytic-uremic syndrome. Therefore, it is logical that microbes use these domains as a key way of avoiding complement attack, and in this report, we have shown an example of how an important and emerging human pathogen, Lyme disease-causing B. burgdorferi, utilizes these domains.
Supplementary Material
Acknowledgments
We thank Miia Eholuoto and Ilkka J. T. Seppälä (University of Helsinki and Huslab, Helsinki University Central Hospital Laboratory) for providing the OspE clone. We acknowledge Marjatta Ahonen, Pirkko Kokkonen, and Kirsti Widing for excellent technical assistance. We also thank ESRF for beamtime on ID14-4, Seija Mäki, Serranda Gashi, and the Biocenter Finland crystallization facility. A. B thanks Prof. Mikael Skurnik for very fruitful scientific discussions.

This article contains supplemental Figs. 1–8.
The atomic coordinates and structure factors (codes 2M4F and 4J38) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The OspE resonance assignment has been deposited to BMRB with accession number 19001.
- FH
- factor H
- OspE and -A
- outer surface protein E and A, respectively
- HSQC
- heteronuclear single quantum correlation
- TROSY
- transverse relaxation optimized spectroscopy
- SALP
- SsgA-like protein
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Barbour A. G., Maupin G. O., Teltow G. J., Carter C. J., Piesman J. (1996) Identification of an uncultivable Borrelia species in the hard tick Amblyomma americanum. Possible agent of a Lyme disease-like illness. J. Infect. Dis. 173, 403–409 [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention (CDC) (1996) Lyme disease. United States, 1995. MMWR Morb. Mortal. Wkly. Rep. 45, 481–484 [PubMed] [Google Scholar]
- 3. Barbour A. G., Fish D. (1993) The biological and social phenomenon of Lyme disease. Science 260, 1610–1616 [DOI] [PubMed] [Google Scholar]
- 4. Steere A. C., Grodzicki R. L., Kornblatt A. N., Craft J. E., Barbour A. G., Burgdorfer W., Schmid G. P., Johnson E., Malawista S. E. (1983) The spirochetal etiology of Lyme disease. N. Engl. J. Med. 308, 733–740 [DOI] [PubMed] [Google Scholar]
- 5. Ohnishi J., Piesman J., de Silva A. M. (2001) Antigenic and genetic heterogeneity of Borrelia burgdorferi populations transmitted by ticks. Proc. Natl. Acad. Sci. U.S.A. 98, 670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steere A. C., Malawista S. E., Hardin J. A., Ruddy S., Askenase W., Andiman W. A. (1977) Erythema chronicum migrans and Lyme arthritis. The enlarging clinical spectrum. Ann. Intern. Med. 86, 685–698 [DOI] [PubMed] [Google Scholar]
- 7. van Dam A. P., Kuiper H., Vos K., Widjojokusumo A., de Jongh B. M., Spanjaard L., Ramselaar A. C., Kramer M. D., Dankert J. (1993) Different genospecies of Borrelia burgdorferi are associated with distinct clinical manifestations of Lyme borreliosis. Clin. Infect. Dis. 17, 708–717 [DOI] [PubMed] [Google Scholar]
- 8. Alitalo A., Meri T., Comstedt P., Jeffery L., Tornberg J., Strandin T., Lankinen H., Bergström S., Cinco M., Vuppala S. R., Akins D. R., Meri S. (2005) Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. Eur. J. Immunol. 35, 3043–3053 [DOI] [PubMed] [Google Scholar]
- 9. Alitalo A., Meri T., Rämö L., Jokiranta T. S., Heikkilä T., Seppälä I. J., Oksi J., Viljanen M., Meri S. (2001) Complement evasion by Borrelia burgdorferi. Serum-resistant strains promote C3b inactivation. Infect. Immun. 69, 3685–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walport M. J. (2001) Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 11. Hartmann K., Corvey C., Skerka C., Kirschfink M., Karas M., Brade V., Miller J. C., Stevenson B., Wallich R., Zipfel P. F., Kraiczy P. (2006) Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 61, 1220–1236 [DOI] [PubMed] [Google Scholar]
- 12. Kraiczy P., Hellwage J., Skerka C., Becker H., Kirschfink M., Simon M. M., Brade V., Zipfel P. F., Wallich R. (2004) Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 279, 2421–2429 [DOI] [PubMed] [Google Scholar]
- 13. Hellwage J., Meri T., Heikkilä T., Alitalo A., Panelius J., Lahdenne P., Seppälä I. J., Meri S. (2001) The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J. Biol. Chem. 276, 8427–8435 [DOI] [PubMed] [Google Scholar]
- 14. Hefty P. S., Jolliff S. E., Caimano M. J., Wikel S. K., Radolf J. D., Akins D. R. (2001) Regulation of OspE-related, OspF-related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host-specific signals. Infect. Immun. 69, 3618–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alitalo A., Meri T., Lankinen H., Seppälä I., Lahdenne P., Hefty P. S., Akins D., Meri S. (2002) Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J. Immunol. 169, 3847–3853 [DOI] [PubMed] [Google Scholar]
- 16. de Córdoba S. R., de Jorge E. G. (2008) Translational mini-review series on complement factor H. Genetics and disease associations of human complement factor H. Clin. Exp. Immunol. 151, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atkinson J. P., Goodship T. H. (2007) Complement factor H and the hemolytic uremic syndrome. J. Exp. Med. 204, 1245–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gordon D. L., Kaufman R. M., Blackmore T. K., Kwong J., Lublin D. M. (1995) Identification of complement regulatory domains in human factor H. J. Immunol. 155, 348–356 [PubMed] [Google Scholar]
- 19. Morgan H. P., Schmidt C. Q., Guariento M., Blaum B. S., Gillespie D., Herbert A. P., Kavanagh D., Mertens H. D., Svergun D. I., Johansson C. M., Uhrín D., Barlow P. N., Hannan J. P. (2011) Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol. 18, 463–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kajander T., Lehtinen M. J., Hyvärinen S., Bhattacharjee A., Leung E., Isenman D. E., Meri S., Goldman A., Jokiranta T. S. (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. U.S.A. 108, 2897–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haapasalo K., Vuopio J., Syrjänen J., Suvilehto J., Massinen S., Karppelin M., Järvelä I., Meri S., Kere J., Jokiranta T. S. (2012) Acquisition of complement factor H is important for pathogenesis of Streptococcus pyogenes infections. Evidence from bacterial in vitro survival and human genetic association. J. Immunol. 188, 426–435 [DOI] [PubMed] [Google Scholar]
- 22. Zipfel P. F., Hallström T., Hammerschmidt S., Skerka C. (2008) The complement fitness factor H. Role in human diseases and for immune escape of pathogens, like pneumococci. Vaccine 26, I67–I74 [DOI] [PubMed] [Google Scholar]
- 23. Lambris J. D., Ricklin D., Geisbrecht B. V. (2008) Complement evasion by human pathogens. Nat. Rev. Microbiol. 6, 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meri T., Hartmann A., Lenk D., Eck R., Würzner R., Hellwage J., Meri S., Zipfel P. F. (2002) The yeast Candida albicans binds complement regulators factor H and FHL-1. Infect. Immun. 70, 5185–5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho D. K., Jarva H., Meri S. (2010) Human complement factor H binds to outer membrane protein Rck of Salmonella. J. Immunol. 185, 1763–1769 [DOI] [PubMed] [Google Scholar]
- 26. Kraiczy P., Skerka C., Kirschfink M., Brade V., Zipfel P. F. (2001) Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and Factor H. Eur. J. Immunol. 31, 1674–1684 [DOI] [PubMed] [Google Scholar]
- 27. Alitalo A., Meri T., Chen T., Lankinen H., Cheng Z. Z., Jokiranta T. S., Seppälä I. J., Lahdenne P., Hefty P. S., Akins D. R., Meri S. (2004) Lysine-dependent multipoint binding of the Borrelia burgdorferi virulence factor outer surface protein E to the C terminus of factor H. J. Immunol. 172, 6195–6201 [DOI] [PubMed] [Google Scholar]
- 28. Amdahl H., Jarva H., Haanperä M., Mertsola J., He Q., Jokiranta T. S., Meri S. (2011) Interactions between Bordetella pertussis and the complement inhibitor factor H. Mol. Immunol. 48, 697–705 [DOI] [PubMed] [Google Scholar]
- 29. McDowell J. V., Harlin M. E., Rogers E. A., Marconi R. T. (2005) Putative coiled-coil structural elements of the BBA68 protein of Lyme disease spirochetes are required for formation of its factor H binding site. J. Bacteriol. 187, 1317–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackmore T. K., Hellwage J., Sadlon T. A., Higgs N., Zipfel P. F., Ward H. M., Gordon D. L. (1998) Identification of the second heparin-binding domain in human complement factor H. J. Immunol. 160, 3342–3348 [PubMed] [Google Scholar]
- 31. Blackmore T. K., Sadlon T. A., Ward H. M., Lublin D. M., Gordon D. L. (1996) Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J. Immunol. 157, 5422–5427 [PubMed] [Google Scholar]
- 32. Schneider M. C., Prosser B. E., Caesar J. J., Kugelberg E., Li S., Zhang Q., Quoraishi S., Lovett J. E., Deane J. E., Sim R. B., Roversi P., Johnson S., Tang C. M., Lea S. M. (2009) Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458, 890–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Batsford S., Rust C., Neubert U. (1998) Analysis of antibody response to the outer surface protein family in lyme borreliosis patients. J. Infect. Dis. 178, 1676–1683 [DOI] [PubMed] [Google Scholar]
- 34. Meri T., Amdahl H., Lehtinen M. J., Hyvärinen S., McDowell J. V., Bhattacharjee A., Meri S., Marconi R., Goldman A., Jokiranta T. S. (2013) Microbes bind complement inhibitor factor H via a common site. PLoS Pathog. 9, e1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jokiranta T. S., Jaakola V. P., Lehtinen M. J., Pärepalo M., Meri S., Goldman A. (2006) Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J. 25, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhattacharjee A., Lehtinen M. J., Kajander T., Goldman A., Jokiranta T. S. (2010) Both domain 19 and domain 20 of factor H are involved in binding to complement C3b and C3d. Mol. Immunol. 47, 1686–1691 [DOI] [PubMed] [Google Scholar]
- 37. Lehtinen M. J., Rops A. L., Isenman D. E., van der Vlag J., Jokiranta T. S. (2009) Mutations of factor H impair regulation of surface-bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J. Biol. Chem. 284, 15650–15658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muona M., Aranko A. S., Iwai H. (2008) Segmental isotopic labelling of a multidomain protein by protein ligation by protein trans-splicing. Chembiochem 9, 2958–2961 [DOI] [PubMed] [Google Scholar]
- 39. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 40. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 41. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 42. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 43. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR. Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 44. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 45. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DiMaio F., Tyka M. D., Baker M. L., Chiu W., Baker D. (2009) Refinement of protein structures into low resolution density maps using Rosetta. J. Mol. Biol. 392, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 48. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 49. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 50. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 51. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 52. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lam T. T., Nguyen T. P., Montgomery R. R., Kantor F. S., Fikrig E., Flavell R. A. (1994) Outer surface proteins E and F of Borrelia burgdorferi, the agent of Lyme disease. Infect. Immun. 62, 290–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu Q., Traag B. A., Willemse J., McMullan D., Miller M. D., Elsliger M. A., Abdubek P., Astakhova T., Axelrod H. L., Bakolitsa C., Carlton D., Chen C., Chiu H. J., Chruszcz M., Clayton T., Das D., Deller M. C., Duan L., Ellrott K., Ernst D., Farr C. L., Feuerhelm J., Grant J. C., Grzechnik A., Grzechnik S. K., Han G. W., Jaroszewski L., Jin K. K., Klock H. E., Knuth M. W., Kozbial P., Krishna S. S., Kumar A., Marciano D., Minor W., Mommaas A. M., Morse A. T., Nigoghossian E., Nopakun A., Okach L., Oommachen S., Paulsen J., Puckett C., Reyes R., Rife C. L., Sefcovic N., Tien H. J., Trame C. B., van den Bedem H., Wang S., Weekes D., Hodgson K. O., Wooley J., Deacon A. M., Godzik A., Lesley S. A., Wilson I. A., van Wezel G. P. (2009) Structural and functional characterizations of SsgB, a conserved activator of developmental cell division in morphologically complex actinomycetes. J. Biol. Chem. 284, 25268–25279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharma A. (2011) Fatty acid induced remodeling within the human liver fatty acid-binding protein. J. Biol. Chem. 286, 31924–31928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schumacher M. A., Karamooz E., Ziková A., Trantírek L., Lukes J. (2006) Crystal structures of T. brucei MRP1/MRP2 guide-RNA binding complex reveal RNA matchmaking mechanism. Cell 126, 701–711 [DOI] [PubMed] [Google Scholar]
- 57. Desveaux D., Allard J., Brisson N., Sygusch J. (2002) A new family of plant transcription factors displays a novel ssDNA-binding surface. Nat. Struct. Biol. 9, 512–517 [DOI] [PubMed] [Google Scholar]
- 58. Graebsch A., Roche S., Niessing D. (2009) X-ray structure of Pur-α reveals a Whirly-like fold and an unusual nucleic-acid binding surface. Proc. Natl. Acad. Sci. U.S.A. 106, 18521–18526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Capaldi S., Guariento M., Saccomani G., Fessas D., Perduca M., Monaco H. L. (2007) A single amino acid mutation in zebrafish (Danio rerio) liver bile acid-binding protein can change the stoichiometry of ligand binding. J. Biol. Chem. 282, 31008–31018 [DOI] [PubMed] [Google Scholar]
- 60. Nichesola D., Perduca M., Capaldi S., Carrizo M. E., Righetti P. G., Monaco H. L. (2004) Crystal structure of chicken liver basic fatty acid-binding protein complexed with cholic acid. Biochemistry 43, 14072–14079 [DOI] [PubMed] [Google Scholar]
- 61. Beneken J., Tu J. C., Xiao B., Nuriya M., Yuan J. P., Worley P. F., Leahy D. J. (2000) Structure of the Homer EVH1 domain-peptide complex reveals a new twist in polyproline recognition. Neuron 26, 143–154 [DOI] [PubMed] [Google Scholar]
- 62. Huang G. N., Huso D. L., Bouyain S., Tu J., McCorkell K. A., May M. J., Zhu Y., Lutz M., Collins S., Dehoff M., Kang S., Whartenby K., Powell J., Leahy D., Worley P. F. (2008) NFAT binding and regulation of T cell activation by the cytoplasmic scaffolding Homer proteins. Science 319, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Herbert A. P., Deakin J. A., Schmidt C. Q., Blaum B. S., Egan C., Ferreira V. P., Pangburn M. K., Lyon M., Uhrín D., Barlow P. N. (2007) Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J. Biol. Chem. 282, 18960–18968 [DOI] [PubMed] [Google Scholar]
- 64. Ferreira V. P., Herbert A. P., Cortés C., McKee K. A., Blaum B. S., Esswein S. T., Uhrín D., Barlow P. N., Pangburn M. K., Kavanagh D. (2009) The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J. Immunol. 182, 7009–7018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Janssen B. J., Christodoulidou A., McCarthy A., Lambris J. D., Gros P. (2006) Structure of C3b reveals conformational changes that underlie complement activity. Nature 444, 213–216 [DOI] [PubMed] [Google Scholar]
- 66. McDowell J. V., Wolfgang J., Senty L., Sundy C. M., Noto M. J., Marconi R. T. (2004) Demonstration of the involvement of outer surface protein E coiled coil structural domains and higher order structural elements in the binding of infection-induced antibody and the complement-regulatory protein, factor H. J. Immunol. 173, 7471–7480 [DOI] [PubMed] [Google Scholar]
- 67. Brakeman P. R., Lanahan A. A., O'Brien R., Roche K., Barnes C. A., Huganir R. L., Worley P. F. (1997) Homer. A protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288 [DOI] [PubMed] [Google Scholar]
- 68. Chen S., Zückert W. R. (2011) Probing the Borrelia burgdorferi surface lipoprotein secretion pathway using a conditionally folding protein domain. J. Bacteriol. 193, 6724–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen S., Kumru O. S., Zückert W. R. (2011) Determination of Borrelia surface lipoprotein anchor topology by surface proteolysis. J. Bacteriol. 193, 6379–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cordes F. S., Roversi P., Kraiczy P., Simon M. M., Brade V., Jahraus O., Wallis R., Skerka C., Zipfel P. F., Wallich R., Lea S. M. (2005) A novel fold for the factor H-binding protein BbCRASP-1 of Borrelia burgdorferi. Nat. Struct. Mol. Biol. 12, 276–277 [DOI] [PubMed] [Google Scholar]
- 71. Pangburn M. K., Müller-Eberhard H. J. (1978) Complement C3 convertase. Cell surface restriction of β1H control and generation of restriction on neuraminidase-treated cells. Proc. Natl. Acad. Sci. U.S.A. 75, 2416–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ferreira V. P., Pangburn M. K., Cortés C. (2010) Complement control protein factor H. The good, the bad, and the inadequate. Mol. Immunol. 47, 2187–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wu J., Wu Y. Q., Ricklin D., Janssen B. J., Lambris J. D., Gros P. (2009) Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 10, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fikrig E., Barthold S. W., Kantor F. S., Flavell R. A. (1991) Protection of mice from Lyme borreliosis by oral vaccination with Escherichia coli expressing OspA. J. Infect. Dis. 164, 1224–1227 [DOI] [PubMed] [Google Scholar]
- 75. Rosé C. D., Fawcett P. T., Gibney K. M. (2001) Arthritis following recombinant outer surface protein A vaccination for Lyme disease. J. Rheumatol. 28, 2555–2557 [PubMed] [Google Scholar]
- 76. Giuliani M. M., Adu-Bobie J., Comanducci M., Aricò B., Savino S., Santini L., Brunelli B., Bambini S., Biolchi A., Capecchi B., Cartocci E., Ciucchi L., Di Marcello F., Ferlicca F., Galli B., Luzzi E., Masignani V., Serruto D., Veggi D., Contorni M., Morandi M., Bartalesi A., Cinotti V., Mannucci D., Titta F., Ovidi E., Welsch J. A., Granoff D., Rappuoli R., Pizza M. (2006) A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. U.S.A. 103, 10834–10839 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.