

Background: Stimulation by post-hydrolytic, ADP-bound conformations of SUR1 underlies current models of KATP channel activation; ATP analogs are assumed to lower activity by reducing hydrolysis.
Results: ATPγS switches conformations with lowered affinity; AMP-PxP selectively bind NBD1, preventing switching.
Conclusion: The actions of ATP analogs on KATP channels require reinterpretation.
Significance: Reduced affinities of SUR1 NBDs for ATP analogs limit conformational switching and channel activity.
Keywords: ABC Transporter, Allosteric Regulation, Ligand-binding Protein, Nucleoside Nucleotide Analogs, Potassium Channels, ABCC8, ATP Analogs, KATP Channels, Neonatal Diabetes, Sulfonylureas
Abstract
Neuroendocrine-type KATP channels, (SUR1/Kir6.2)4, couple the transmembrane flux of K+, and thus membrane potential, with cellular metabolism in various cell types including insulin-secreting β-cells. Mutant channels with reduced activity are a cause of congenital hyperinsulinism, whereas hyperactive channels are a cause of neonatal diabetes. A current regulatory model proposes that ATP hydrolysis is required to switch SUR1 into post-hydrolytic conformations able to antagonize the inhibitory action of nucleotide binding at the Kir6.2 pore, thus coupling enzymatic and channel activities. Alterations in SUR1 ATPase activity are proposed to contribute to neonatal diabetes and type 2 diabetes risk. The regulatory model is partly based on the reduced ability of ATP analogs such as adenosine 5′-(β,γ-imino)triphosphate (AMP-PNP) and adenosine 5′-O-(thiotriphosphate) (ATPγS) to stimulate channel activity, presumably by reducing hydrolysis. This study uses a substitution at the catalytic glutamate, SUR1E1507Q, with a significantly increased affinity for ATP, to probe the action of these ATP analogs on conformational switching. ATPγS, a slowly hydrolyzable analog, switches SUR1 conformations, albeit with reduced affinity. Nonhydrolyzable AMP-PNP and adenosine 5′-(β,γ-methylenetriphosphate) (AMP-PCP) alone fail to switch SUR1, but do reverse ATP-induced switching. AMP-PCP displaces 8-azido-[32P]ATP from the noncanonical NBD1 of SUR1. This is consistent with structural data on an asymmetric bacterial ABC protein that shows that AMP-PNP binds selectively to the noncanonical NBD to prevent conformational switching. The results imply that MgAMP-PNP and MgAMP-PCP (AMP-PxP) fail to activate KATP channels because they do not support NBD dimerization and conformational switching, rather than by limiting enzymatic activity.
Introduction
Neuroendocrine ATP-sensitive K+ channels, (SUR1/Kir6.2)4, couple membrane electrical activity with cell metabolism in neurons and many endocrine cells, including pancreatic islet α-, β-, and δ-cells (see Ref. 1 for review). These channels respond to changes in the levels of MgATP and ADP, which have both inhibitory and stimulatory actions. Nucleotides bind to Kir6.22 to reduce the channel open probability, whereas ATP interactions with SUR1 antagonize this inhibitory effect. These nucleotide effects are modulated by other factors including phosphoinositides (2–9) and long-chain acyl-CoAs (10–16). SUR1, the channel regulatory subunit, is an enzyme, a member of the ATP-binding cassette (ABC) family of proteins that utilize the energy of ATP binding and hydrolysis to transport substrates across cell membranes (17, 18). A current model of KATP channel regulation assumes that a post-hydrolytic, ADP-bound enzymatic intermediate or conformation of SUR1 stimulates channel openings, i.e. that hydrolysis is essential for activation (Ref. 19 and reviewed in (20–22). This model is based in part on studies demonstrating that nonhydrolyzable ATP analogs such as AMP-PNP and AMP-PCP fail to stimulate channel activity, whereas the slowly hydrolyzable ATPγS stimulates less efficiently than ATP (23–30).
Contrary to the current model, we observed that the binding of ATP, without hydrolysis, is sufficient to switch SUR1 from conformations with high affinity for glibenclamide (GBC), a KATP channel antagonist, to conformations with reduced antagonist affinity, but a markedly enhanced affinity for the channel agonist diazoxide, i.e. from nonstimulatory to stimulatory states (31). ATP4−, in the absence of Mg2+ required for enzymatic activity (17), efficiently switched the conformations of wild-type (WT) SUR1 and mutant receptors known to hyperactivate Kir6.2 pores and thus cause neonatal diabetes (ND).
The ND mutant receptors, SUR1Q1178R and SUR1R1182Q, substitutions outside the nucleotide-binding domains (NBDs), have increased affinities for MgATP and ATP4−, consistent with their spending more time in stimulatory conformations able to increase channel activity. Structural studies on multiple ABC proteins show that ATP binding to the NBDs induces the formation of an NBD dimer with two ATP molecules sandwiched in the dimer interface. Dimerization drives reconfiguration of the transmembrane helix bundles from inward-to-outward-facing conformations (reviewed in Refs. 32 and 33). Therefore we proposed that ATP binding and NBD dimerization, without hydrolysis, drive SUR1 from a nonstimulatory, inward-facing conformation with highest affinity for GBC to an outward-facing stimulatory state with >10-fold reduced affinity for GBC and >100-fold increased affinity for channel agonists (31).
AMP-PNP stabilizes NBD dimers of symmetric ABC proteins in outward-facing configurations with two bound nucleotides (34, 35). On the other hand, the structure of an asymmetric ABC protein, TM287/288 from Thermotoga maritima, shows a single AMP-PNP bound to an inward-facing conformation (36). Therefore the action of AMP-PNP and AMP-PCP on ATP-induced switching of SUR1, which like several other ABC proteins has asymmetric NBDs (Refs. 37–39 and see Ref. 40 for review), was analyzed. The sensitivity of the analysis was improved by using SUR1 substitutions with increased affinities for ATP. These include SUR1Q1178R identified with hyperactive KATP channels in an ND patient (41, 42) and SUR1E1507Q. The Glu → Gln substitution, in the highly conserved catalytic glutamate of SUR1 NBD2, dramatically reduces enzymatic activity (≥500-fold) in other ABC proteins (43, 44) and is used in structural studies to stabilize or trap ABC proteins in outward-facing, nucleotide-bound conformations with dimerized NBDs (45–47). Under equilibrium conditions, we found that SUR1E1507Q binds ATP4− nearly 150 times more tightly than WT, showing that charge at position 1507 in the conserved Walker B phosphate-binding domain is a significant determinant of nucleotide affinity. For comparison, the affinity of SUR1Q1178R, a substitution outside the NBDs, is intermediate between WT and E1507Q. The inclusion of Mg2+ increases the affinity for ATP by ≥100-fold.
ATPγS fully supports switching with or without Mg2+, but SUR1 has a reduced affinity for this analog versus ATP. In contrast, MgAMP-PNP and MgAMP-PCP (AMP-PxP) alone do not affect GBC binding at high concentration, but fully reverse the action of added MgATP in a dose-dependent manner. Additionally, AMP-PCP displaces 8-azido-[32P]ATP from the noncanonical NBD1 of SUR1. The results are consistent with studies on two other asymmetric ABC proteins that demonstrate that AMP-PNP selectively binds the noncanonical NBD1 of cystic fibrosis transmembrane conductance regulator (CFTR) with high affinity, but binds NBD2 with much lower affinity (48, 49), and interacts selectively with the noncanonical NBD of TM287/288 to stabilize an inward-facing conformation (36). We propose that AMP-PxP act on SURs to reduce channel activity by stabilizing an inward-facing conformation, and thus the actions of AMP-PxP reflect the asymmetry of SURs, rather than inhibition of ATP hydrolysis per se.
EXPERIMENTAL PROCEDURES
The procedures used to express WT and mutant SUR1 in Pichia pastoris, to isolate membranes, and to carry out 3H-labeled GBC binding experiments were described previously (31). An additional wash step was added in the membrane isolation protocol to reduce carryover of endogenous nucleotides.
[3H]GBC Binding Inhibition Experiments
3H-labeled GBC was held fixed at 1 nm, and the reactions included the indicated concentrations of nucleotides. Experiments with MgATP (free [Mg2+] > 1 mm) included a creatine phosphokinase-based ATP-regenerating system to maintain a constant concentration of ATP over the 30-min incubation (50). The stability of ATP levels was verified using luciferase assays (Sigma). Experiments with MgADP included 10 mm AMP to inhibit endogenous adenylate kinases to reduce ATP production. Experiments with MgADP and MgATP analogs, where a regenerating system was not required, included 1 mm free Mg2+. Mg2+-free experiments included 1 mm EDTA. Nonspecific binding was determined in the presence of 1 μm unlabeled GBC and was typically 15% of total binding. The results are plotted as
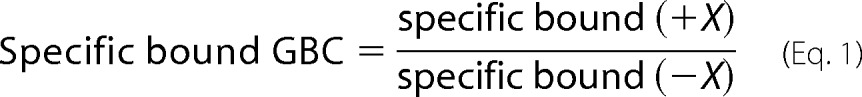 |
where X is the reagent whose effect is being assayed, e.g. ± ATP4−, etc.
Photolabeling
8-azido-[32P]ATP photolabeling and densitometry were done as described previously (31).
Allosteric Analysis
The equations for the equilibrium models were derived following Wyman and Gill (51); the algebraic manipulations were done using Mathematica (Wolfram Research, Champaign, IL). The binding equations were derived from the binding partition functions, the sum of the contributions of the different species relative to one reference species, taken here as the unliganded receptor, R. The eight-state allosteric model shown in Fig. 4C is used as an example. The model describes the linkage between the equilibrium binding of MgAMP-PNP (A), 3H-labeled GBC (G), and MgATP (T) on SUR1E1507Q (R). KA is the equilibrium dissociation constant for binding of A, KG is the constant for G, and K1 and K2 are the constants for the binding of T to NBD1 and NBD2, respectively. α, β, and γ are allosteric constants. α = γ = 1 because previous data indicate that binding of nucleotide to NBD1 alone does not alter GBC binding (31). β is the allosteric constant reflecting the linkage between GBC binding and structural changes associated with occupation of both NBDs. For β > 1, nucleotide binding reduces the affinity of SUR1 for sulfonylureas. The partition function for the eight-state model is
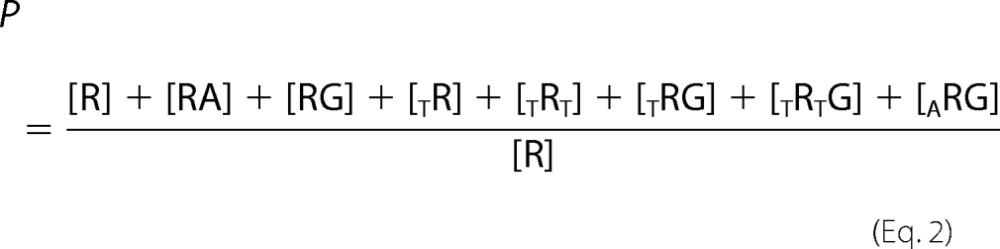 |
where TR and TRT indicate binding at NBD1 and both NBD1 and NBD2, respectively. Substituting dissociation constants gives the binding polynomial.
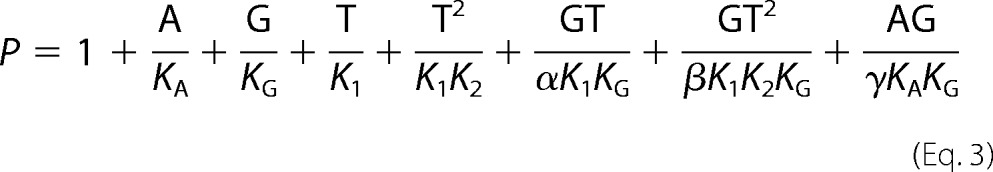 |
Ḡ, the amount of 3H-labeled GBC specifically bound per mole of SUR1, which is dependent on [G], [T], and [A], is given by
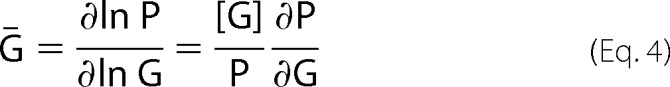 |
where
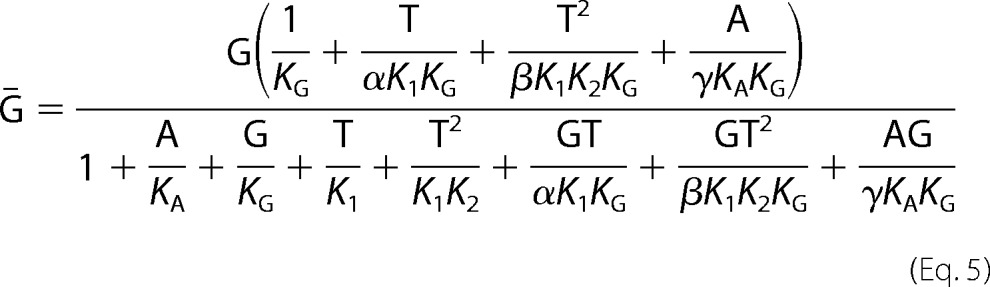 |
The plotted experimental variable, specific bound GBC, is defined as Ḡ /Ḡno ATP. Parameters were estimated using nonlinear fit models in the Mathematica environment with α = γ = 1 and predetermined values of β, KG, and G.
FIGURE 4.

AMP-PxP reverse ATP-induced conformational switching. A and B, MgAMP-PNP alone does not affect the conformational state of SUR1Q1178R (A) or SUR1E1507Q (B). MgAMP-PNP reverses the conformational shift induced by MgATP (30 μm) in both SURs. MgAMP-PCP and AMP-PNP4− also reverse, but the apparent affinities are lower. The dashed lines are logistic curves through the data points; the parameters are S0.5 = 530 ± 124 μm for SUR1Q1178R (A). The SUR1E1507Q S0.5 values in B are 573 ± 103, 3360 ± 410, and 31,100 ± 2240 μm for MgAMP-PNP, MgAMP-PCP, and AMP-PNP4−, respectively. C, the solid curve in B for SUR1E1507Q is a fit to an eight-state model with the following parameters: α = γ = 1, β = 40, KG = 0.63 nm, K1 = 0.7 ± 0.2 μm, K2 = 1.5 ± 0.1 μm, KA = 1.2 ± 0.2 μm.
The equation for the four-state equilibrium model is.
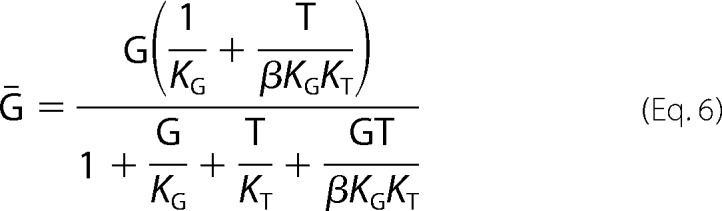 |
Fittings were estimated using predetermined values for KG.
Statistics
Where indicated, the IC50 values were estimated by fitting a logistic equation. The means ± S.E. are plotted; the number of replicate experiments varies, but n ≥ 3 in all cases.
RESULTS
ATP-induced Conformational Switching of WT and Mutant SUR1
The effect of ATP on SUR1 conformation was determined using GBC as a conformational probe. Fig. 1 shows that ATP, with or without Mg2+, has a strong negative effect on GBC binding. ATP effectively switches WT and substituted SURs from inward-facing conformations with highest affinities for GBC to outward-facing states with reduced affinities as shown schematically. The apparent differences in affinity, judged for example by their IC50 values (panel A and Table 1), is ∼430-fold for WT versus SUR1E1507Q; the value for SUR1Q1178R, characterized previously, is intermediate (31). Comparison of panels A and B shows that Mg2+ significantly increases the apparent affinity of all three receptors for ATP.
FIGURE 1.

ATP-induced conformational switching of SURs. A, the effect of MgATP on GBC binding. Dashed curves are fits to a logistic equation used to estimate the IC50 values (in μm): WT: 1163 ± 439, slope = 0.8 ± 0.1; Q1178R: 17 ± 3, slope = 0.8 ± 0.1; E1507Q: 2.7 ± 0.4, slope = 0.8 ± 0.1. IC50 mean ± S.E., slope mean ± S.E., n = 3–20. The solid curve for SUR1E1507Q is the best fit to the four-state model shown in the inset. The parameters are KT = 1 μm and β = 40 with a defined value for KG = 0.63 nm. B, the effect of ATP4− on GBC binding. The curves are the best fits to the four-state model. The parameters are KT = 94, 2550, and 13,400 μm and β = 40, 9.2, and 15.1 with KG defined as 0.63, 1.0, and 0.25 nm, for E1507Q, Q1178R, and WT, respectively. Here and in the following figures, the graphics suggest the conformational changes in SUR1 upon binding of nucleotides.
TABLE 1.
Binding parameters (KG, IC50, and KT values) for WT, Q1178R, and E1507Q
KG values are nm; all IC50 and KT values are μm. —, not detectable.
| KG | IC50 |
KT |
|||||
|---|---|---|---|---|---|---|---|
| MgATP | MgADP | MgATP (β) | ATP4− (β | MgATPγS (β) | ATPγS4− (β) | ||
| WT | 0.25 ± 0.02 | 1163 ± 439 | 161 ± 28 | — | 13,400 ± 3360 (15.1 ± 9.4) | — | — |
| Q1178R | 1.0 ± 0.1 | 17 ± 3 | 25 ± 2 | — | 2550 ± 290 (9.2 ± 1.4) | — | — |
| E1507Q | 0.63 ± 0.16 | 2.7 ± 0.4 | — | 1 ± 0.2 (40 ± 3) | 94 ± 8 (40 ± 1) | 29 ± 7 (11.6 ± 3) | 234 ± 31 (3.2 ± 0.1) |
The E1507Q and Q1178R substitutions do not have large effects on the affinities of the apo-receptors for GBC, given by the dissociation constants (KG), which are determined in independent experiments (Table 1). To characterize the negative allosteric linkage between binding sites in SUR1E1507Q, we assume that hydrolysis is negligible both in the presence and in the absence of Mg2+ (43, 44). A four-state equilibrium model (Fig. 1, inset) was used to estimate the affinities for ATP (KT) and the allosteric constants (β). The results are tabulated in Table 1 along with the IC50 values. The four-state model has not been used to analyze the MgATP effects on WT and SUR1Q1178R, which are potentially in an enzymatic steady state. In Fig. 1, the solid lines are the best-fit curves derived from the four-state model. The fits were constrained using the values for KG, the dissociation constants for GBC binding to the apo-receptors (Table 1). The product of the allosteric constant, β, and KG determines the plateau values at saturating concentrations of nucleotide and is the dissociation constant for GBC binding to the fully ATP-liganded receptor. The estimates of β for WT and SUR1Q1178R are consistent with fully ATP-liganded, outward-facing receptors having approximately a 10-fold lower affinity for GBC (31). The value for SUR1E1507Q, 40, is significantly greater, implying that the affinity of fully ATP-liganded SUR1E1507Q for GBC is considerably reduced, i.e. 0.63 versus 25 nm, unliganded versus liganded, respectively.
ATP4− switches the conformation of WT and mutant receptors. Fig. 1B shows a significant difference, ∼27-fold, in the affinities of SUR1E1507Q versus SUR1Q1178R for ATP4−. The effect of nucleotide on WT is not saturated, but the estimated affinity of SUR1E1507Q is ∼150-fold greater than WT (Table 1). The structural difference between SUR1E1507Q and WT (Glu-1507) is the elimination of the negative charge at position 1507. The results imply that electrostatic interactions between phosphate residues and the carboxyl group at position 1507 is a significant determinant of nucleotide affinity. Neutralizing the charge significantly increases the binding energy by ∼3 kcal/mol at 37 °C, SUR1E1507Q versus WT. The general finding that added Mg2+, bound to the β and γ phosphates, increases the apparent affinity supports this idea.
Comparing the affinities of SUR1E1507Q for MgATP versus ATP4− shows that the receptor binds MgATP ∼100-fold more tightly than ATP4−. Thus the addition of Mg2+ contributes ∼2.8 kcal/mol to the binding energy at 37 °C. The WT and SUR1Q1178R receptors show a semiquantitatively similar Mg2+ effect when comparing the IC50 values for MgATP versus KT for ATP4− (Table 1). It is worth noting that nucleotide binding and GBC binding are negatively linked allosteric functions; thus the GBC (1 nm) used to assess conformational changes reduces the apparent affinity for nucleotide, i.e. ”right shifts“ the response curves. This is apparent, for example, for SUR1E1507Q where the IC50 is ∼3-fold greater than the estimated KT value.
MgADP-induced Switching
Fig. 2 shows that SUR1E1507Q has a reduced affinity for MgADP when compared with WT SUR1. The membrane preparations have significant endogenous adenylate kinase activity able to convert MgADP to ATP. This activity is suppressed by the addition of AMP (10 mm). ATP measurements using luciferase show that the concentration of ATP is ∼4 μm when the concentration of MgADP is 300 μm (31). Thus at the highest concentration, 1 mm MgADP, a fraction of the switching is due to ATP.
FIGURE 2.

MgADP-induced conformational switching of SURs. The solid lines are fits to a logistic equation: IC50 values: WT: 161 ± 28, slope = 1.7 ± 0.4; Q1178R: 25 ± 2, slope = 1.5 ± 0.1. Values are means ± S.E., n > 10. Dashed lines are drawn through the data points for SUR1E1507D and SUR1E1507Q. The apparent affinity for this receptor is difficult to estimate. Its affinity for MgADP is lower than WT, whereas its affinity for MgATP is significantly higher, and thus a significant fraction of the switching above 300 μm ADP may reflect the conversion of ADP to ATP by endogenous adenylate kinase in the membrane preparations.
It is worth noting that although the E1507Q substitution has not been associated with disease, the E1507D substitution, also expected to have impaired ATPase activity, is a cause of ND. The SUR1E1507D/Kir6.2 channels exhibit a comparable reduced sensitivity to stimulation by MgADP (52). Interestingly, although the SUR1E1507Q substitution has a reduced affinity for MgADP, SUR1Q1178R, which also hyperactivates Kir6.2 pores to produce ND, has a significantly higher apparent affinity for MgADP (31) (Fig. 2).
ATPγS, a Slowly Hydrolyzable ATP Analog, Switches SUR1 Conformations with Reduced Affinity
ATPγS, an analog of ATP with one oxygen of the γ-phosphate replaced by a sulfur atom, is slowly utilized by a variety of kinases and ATPases (53). In the current regulatory model, reduced enzymatic activity would slow transition to a post-hydrolytic stimulatory conformation. This idea was tested by assessing the action of ATPγS on GBC binding under steady state and equilibrium conditions (± Mg2+). Fig. 3A compares the action of MgATP versus MgATPγS on conformational switching. SUR1E1507Q has a higher affinity for MgATP versus MgATPγS, estimated using a four-state equilibrium model. The dissociation constants (KT) for ATP are 1.0 ± 0.2 versus 29 ± 7 μm, MgATP versus MgATPγS, respectively. To support the use of the four-state model and to assess the binding of ATPγS4− directly, i.e. without Mg2+ present, the affinities of SUR1E1507Q for ATP4− and ATPγS4− were compared (Fig. 3B). The estimated KT values were 94 ± 8 versus 234 ± 31 μm, for ATP4− versus ATPγS4−, respectively. It is worth noting that the allosteric constants, β values, for the two nucleotides are significantly different. The final plateau at saturating concentrations of nucleotide determines this parameter. The β values used to specify the curves in Fig. 3B were 3.2 ± 0.1 versus 40 ± 1, for ATPγS4− versus ATP4−, respectively. The results show that ATPγS supports conformational switching of SUR1 and that the affinity for this analog is less than for ATP, with or without Mg2+.
FIGURE 3.

SURs have reduced affinity for ATPγS versus ATP. A, comparison of MgATPγS versus MgATP effects. The curves are best fits to a four-state model (Fig. 1B, inset). The best-fit dissociation constants are KT = 1 and 29 μm for MgATP and MgATPγS, respectively. The β values are 40 and 11.6 for MgATP and MgATPγS, respectively. The dissociation constant, KG = 0.63 nm for GBC, was determined independently. B, comparison of the affinities of SUR1E1507Q for ATP4− versus ATPγS4−. The solid curves are the best fits to the four-state model; the parameters are KT = 94 versus 234 μm; β = 40 versus 3.2, ATP4− versus ATPγS4−, respectively. The dissociation constant for GBC, KG = 0.63 nm, was fixed during fitting.
AMP-PNP and AMP-PCP Reverse the ATP-induced Conformational Switching of SUR1
Nonhydrolyzable ATP analogs, specifically MgAMP-PNP and MgAMP-PCP, are unable to activate KATP channels (19, 25, 54–56) and will reduce channel activity when MgATP is present (Ref. 19, but see Ref. 57). MgAMP-PNP and MgAMP-PCP are assumed not to hydrolyze and thus prevent the transition of SUR to post-hydrolytic, stimulatory conformations (for review, see Refs. 20 and 21). MgAMP-PNP does dimerize the NBDs of symmetric ABC proteins (34, 35); thus we anticipated that it could switch SUR1. However, Fig. 4 shows that MgAMP-PNP at concentrations >100 times the IC50 values for MgATP (Fig. 1A) has no significant effect on GBC binding, i.e. AMP-PNP alone does not support conformational switching of either SUR1Q1178R (Fig. 4A) or SUR1E1507Q (Fig. 4B). AMP-PCP alone also has no effect. This could imply that AMP-PNP does not interact with SUR1 as suggested by others (57). To test this possibility, receptors were incubated with 30 μm MgATP (to “preswitch” them) and increasing concentrations of AMP-PNP or AMP-PCP. AMP-PNP concentration-dependently reverses the effect of MgATP on GBC binding, similar to an early observation by Schwanstecher et al. (58). Fig. 4 (A and B) shows the results for SUR1Q1178R and SUR1E1507Q, respectively. As an additional control, these experiments were performed with and without the ATP-regenerating system present with similar results. Fig. 4B suggests that the affinity of SUR1E1507Q for AMP-PCP is ∼10-fold less than for AMP-PNP. The reversal effect of AMP-PNP is not dependent on Mg2+. AMP-PNP4− effectively reverses the action of 500 μm ATP4− (Fig. 4B).
An eight-state equilibrium model was used to estimate the linkage between the binding of AMP-PNP, ATP, and GBC on SUR1E1507Q (Fig. 4C, graphic). The model includes the binding of two ATP (T) molecules at the NBDs of SUR1 and the binding of a single GBC molecule (G). Based on the reversal experiments, and the observation of Hohl et al. (36) that AMP-PNP interacts with a single NBD in an asymmetric ABC protein to stabilize the inward-facing conformation, we assume that AMP-PNP (A) binds to NBD1 to stabilize inward-facing conformations of SUR1 with the highest affinity for GBC. The estimated binding parameters are reasonably consistent with the results in Fig. 1. The dissociation constant (K2) for MgATP at NBD2 is ∼1.5 μm, in agreement with ∼1 μm determined in Fig. 1B. The dissociation constant (K1) for MgATP at NBD1 is estimated at 0.7 μm, broadly consistent with estimates of IC50, 10–40 μm, for 8-azido-[32P]ATP4− binding at NBD1 (31) and the observation that adding Mg2+ increases the affinity of nucleotide binding at NBD2 by ∼100-fold. The fitting suggests that the dissociation constant (KA) for MgAMP-PNP at NBD1 is similar to that for MgATP. The allosteric constant, β = 40, agrees with the estimates in Fig. 1. In Fig. 4 (A and B), the dashed lines are curves drawn through the points; no attempt has been made to fit an eight-state model to the partial saturation data for AMP-PCP and AMP-PNP4− or for SUR1Q1178R, which is potentially in steady state. The binding parameters are summarized in Tables 1 and 2.
TABLE 2.
Binding parameters (S0.5 and K1, K2, and KA values) for Q1178R and E1507Q
Values are μm; K1 and K2, the affinities for MgATP at NBD1 and NBD2, respectively and KA, the affinity for MgAMP-PNP at NBD1, were estimated using an eight-state model. —, not detectable.
| SUR1 | S0.5 |
K1 | K2 | KA | ||
|---|---|---|---|---|---|---|
| MgAMP-PNP | MgAMP-PCP | AMP-PNP4− | ||||
| Q1178R | 530 ± 124 | — | — | — | — | — |
| E1507Q | 573 ± 103 | 3360 ± 410 | 31100 ± 2240 | 0.7 ± 0.2 | 1.5 ± 0.1 | 1.2 ± 0.2 |
AMP-PCP Interacts with NBD1
The model in Fig. 4 assumes that AMP-PxP bind to the asymmetric NBD1 of SUR1. To support this assumption, nucleotide competition experiments were carried out in the absence of Mg2+, conditions where NBD1 of SUR1 has a significantly greater affinity for ATP (Refs. 59 and 60, and see also Ref. 31). An IC50 of 232 ± 25 μm was determined for the heterologous displacement of 8-azido-[α-32P]ATP4− by AMP-PCP4− (Fig. 5). A Ki value (= 95 μm) was estimated using the Cheng-Prusoff relation (61) and KD for azido-ATP4− of ∼0.7 μm estimated by homologous displacement of 8-azido-[α-32P]ATP4− by unlabeled 8-azido-ATP4− (data not shown) The results are consistent with the idea that AMP-PxP bind to the asymmetric NBD1 of SUR1 and stabilize the inward-facing conformation.
FIGURE 5.

Interaction of AMP-PCP with NBD1 of SUR1. Membranes were incubated with 1 μm 8-azido-[α-32P]ATP and increasing concentrations of unlabeled AMP-PCP and then analyzed as described previously (31). EDTA (2 mm) was added to chelate Mg2+. Images of the scanned autoradiographs are shown. IC50 value was estimated by fitting a logistic equation. The IC50 value is 232 ± 25 μm, slope = 1.04 ± 0.1.
DISCUSSION
ATP has inhibitory and stimulatory actions on KATP channels. Nucleotide binding to the Kir pore reduces the probability of channel openings, whereas interactions with SUR1 antagonize this inhibition to stimulate channel activity. SUR1 is an ABC protein, and like other ABC proteins, is reported to have Mg2+-dependent ATPase activity (17, 18). An ADP-bound, post-hydrolytic conformation of SUR1 is proposed to be the enzymatic intermediate that stimulates openings of ATP-inhibited Kir6.2 pores. By contrast, we observe that ATP binding, in the absence of Mg2+ needed for hydrolysis, is sufficient to switch the conformations of WT SUR1 and SUR1 ND mutants. The ATP-bound states are presumed to be stimulatory conformations based on pharmacologic criteria, i.e. their reduced affinity for GBC, a channel antagonist, and increased affinity for diazoxide, a channel agonist (31). The structures of multiple apo- and ATP-bound ABC proteins (reviewed in Refs. 33 and 35) imply that ATP binding switches SUR1 from inward-facing, nonstimulatory apo states to ATP-liganded, outward-facing stimulatory configurations. This is shown as a graphic in Fig. 1. The increased affinity of the ND mutant receptors for ATP implies that they will spend more time in stimulatory conformations and thus produce the hyperactive KATP channels characteristic of neonatal diabetes.
The inhibitory actions of ATP analogs have been used to support the current regulatory model by assuming that reduction of the rate of transition to a post-hydrolytic stimulatory conformation would reduce KATP channel openings. To test this assumption, we analyzed the mechanism(s) by which ATPγS and AMP-PxP affect conformation switching. Substituted receptors with increased affinities for ATP were used to facilitate the analysis. Substitution of the SUR1 catalytic glutamate, Glu-1507, with glutamine, expected to drastically reduce enzymatic activity and thus conversion to ADP-bound intermediates, significantly increased the affinity for ATP, which enhanced, rather than impaired, conformational switching (Fig. 1).
The results show that ATPγS can efficiently switch the conformation of substituted SUR1, albeit the affinities for this analog are weaker than for ATP. The affinity of SUR1E1507Q for MgATPγS is ∼30-fold weaker than for MgATP. Mg2+ is not required for switching, and thus reduced rates of hydrolysis are not a factor. The ATP γ-phosphate has multiple interactions with the NBDs, and thus the reduced affinities for ATPγS versus ATP imply that substitution of a sulfur atom for oxygen weakens one or more of these interactions. We suggest that the reduced stimulatory action of MgATPγS is a reflection of the reduced affinity of SUR1 for this analog rather than a slow rate of hydrolysis.
The interactions with AMP-PNP and AMP-PCP are more complex. Several studies showed that these nonhydrolyzable analogs alone fail to stimulate KATP channels, will inhibit ATP stimulated channels, and fail to support the action of channel agonists (23–30). The usual interpretation has been that these nonhydrolyzable analogs bind to SUR1 and prevent transition to ADP-bound stimulatory states. We thought initially that like ATPγS, AMP-PNP and AMP-PCP would bind to the SUR1 NBDs, support dimerization, and thus support conformational switching. However, an early study showed that AMP-PNP alone did not affect GBC binding, but could partially rescue or reverse ATP-induced switching (58). Extending these observations showed that MgAMP-PNP alone, at concentrations far in excess of the IC50 for MgATP, does not support conformational change, but will reverse ATP-induced switching. These results are consistent with a structural study of an asymmetric bacterial ABC protein, TM287-TM288 from T. maritima, which shows that one molecule of AMP-PNP binds to the noncanonical NBD, where it impairs subsequent NBD dimerization and thus switching to outward-facing conformations (36) and with data on cystic fibrosis transmembrane conductance regulator showing preferential binding at NBD1 (48, 49). An eight-state model (Fig. 4C) was used to make a semiquantitative estimate of the affinity of AMP-PNP for SUR1E1507Q. Based on the structure of TM287-TM288 with a single AMP-PNP bound to the noncanonical NBD, the eight-state model assumes binding only at NBD1 at concentrations below 10 mm. Equilibrium conditions are assumed to hold for SUR1E1507Q. A dissociation constant (KA) ∼1.2 μm for MgAMP-PNP binding to NBD1 was obtained.
The analysis of switching in SUR1E1507Q provides insight into factors affecting ATP binding. Mg2+ significantly increases the apparent affinity of SUR1 for nucleotides (see Ref. 31) (compare Fig. 1A with Fig. 1B), consistent with Mg2+ interacting with Glu-1507 and the γ-phosphate directly or via a bridging water molecule as observed in other ABC proteins (35, 62). To quantify the effect of Mg2+ on ATP binding in the absence of enzymatic activity, a four-state equilibrium model was used to compare the affinities of SUR1E1507Q for MgATP versus ATP4−. The estimated dissociation constants (KT) are 1 versus 94 μm, ± Mg2+, respectively, i.e. Mg2+ increases the affinity ∼100-fold, adding ∼2.8 kcal/mol of binding energy.
A second observation is worth noting. The allosteric constant, β, in the four-state model is significantly greater for SUR1E1507Q than WT or any of the other mutant receptors. Typical values range from ∼7 to 15, but the value for SUR1E1507Q is ∼40 with or without Mg2+ present. The product, βKG, reflects the affinity of the fully ATP-liganded receptor for GBC. Previous estimates for the affinities for GBC of WT, SUR1Q1178R, and SUR1R1182Q in the ATP-bound outward-facing state showed that they were 10–12 times weaker than for the inward-facing conformation, i.e. ∼10 nm versus 1 nm for the apo-receptors (31). Thus the affinity of ATP-bound SUR1E1507Q is ∼25 nm (40 × 0.63 nm), reflected in the lower plateau values at saturating concentrations of ATP. The structural difference(s) responsible for this change are not clear, but we speculate that the SUR1E1507Q NBD dimer may be more compact, allowing a greater “twist” of the transmembrane helical domains, TMD1 and TMD2, that determine the GBC-binding pocket.
The E1507Q substitution has not been identified with disease to date, but its high affinity for ATP predicts that it would produce hyperactive channels. SUR1E1507Q has a somewhat reduced affinity for MgADP versus WT, but a more precise estimate is difficult because its affinity for ATP is high and it is hard to suppress the endogenous adenylate kinase in our membrane preparations. The results are consistent with electrophysiologic data on the E1507D substitution that, when assembled with Kir6.2, produces hyperactive KATP channels that are less sensitive to stimulation by MgADP (52).
In summary, the present results demonstrate the need to reinterpret early results that suggested that ATP analogs modulate KATP channel activity by affecting ATP hydrolysis. The present data suggest that ATPγS is less effective than ATP because SUR1 binds it less tightly, whereas AMP-PNP and AMP-PCP interact with asymmetric SUR1 via selective binding to NBD1 that prevents NBD dimerization and thus conformational switching.
This work was supported by American Diabetes Association Grant ADA 1-10-BS21 (to J. B.).
- Kir6.2
- potassium inward rectifier type 6.2
- SUR1
- sulfonylurea receptor type 1
- ABC
- ATP-binding cassette
- GBC
- glibenclamide
- ND
- neonatal diabetes
- NBD
- nucleotide-binding domain
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- AMP-PCP
- adenosine 5′-(β,γ-methylenetriphosphate)
- MgAMP-PNP
- Mg-5′-adenylyl imidodiphosphate
- MgAMP-PCP
- Mg-adenylyl (β,γ-methylene) diphosphonic acid
- AMP-PxP
- MgAMP-PNP and MgAMP-PCP.
REFERENCES
- 1. Aguilar-Bryan L., Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 [DOI] [PubMed] [Google Scholar]
- 2. Pratt E. B., Shyng S. L. (2011) ATP activates ATP-sensitive potassium channels composed of mutant sulfonylurea receptor 1 and Kir6.2 with diminished PIP2 sensitivity. Channels (Austin) 5, 314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pratt E. B., Tewson P., Bruederle C. E., Skach W. R., Shyng S. L. (2011) N-terminal transmembrane domain of SUR1 controls gating of Kir6.2 by modulating channel sensitivity to PIP2. J. Gen. Physiol. 137, 299–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shyng S. L., Barbieri A., Gumusboga A., Cukras C., Pike L., Davis J. N., Stahl P. D., Nichols C. G. (2000) Modulation of nucleotide sensitivity of ATP-sensitive potassium channels by phosphatidylinositol-4-phosphate 5-kinase. Proc. Natl. Acad. Sci. U.S.A. 97, 937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shyng S. L., Nichols C. G. (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138–1141 [DOI] [PubMed] [Google Scholar]
- 6. Baukrowitz T., Fakler B. (2000) KATP channels gated by intracellular nucleotides and phospholipids. Eur. J. Biochem. 267, 5842–5848 [DOI] [PubMed] [Google Scholar]
- 7. Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S. J., Ruppersberg J. P., Fakler B. (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282, 1141–1144 [DOI] [PubMed] [Google Scholar]
- 8. Enkvetchakul D., Loussouarn G., Makhina E., Shyng S. L., Nichols C. G. (2000) The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys. J. 78, 2334–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fan Z., Makielski J. C. (1999) Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 114, 251–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bränström R., Corkey B. E., Berggren P. O., Larsson O. (1997) Evidence for a unique long chain acyl-CoA ester binding site on the ATP-regulated potassium channel in mouse pancreatic beta cells. J. Biol. Chem. 272, 17390–17394 [DOI] [PubMed] [Google Scholar]
- 11. Bränström R., Leibiger I. B., Leibiger B., Corkey B. E., Berggren P. O., Larsson O. (1998) Long chain coenzyme A esters activate the pore-forming subunit (Kir6. 2) of the ATP-regulated potassium channel. J. Biol. Chem. 273, 31395–31400 [DOI] [PubMed] [Google Scholar]
- 12. Gribble F. M., Proks P., Corkey B. E., Ashcroft F. M. (1998) Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J. Biol. Chem. 273, 26383–26387 [DOI] [PubMed] [Google Scholar]
- 13. Bränström R., Aspinwall C. A., Välimäki S., Ostensson C. G., Tibell A., Eckhard M., Brandhorst H., Corkey B. E., Berggren P. O., Larsson O. (2004) Long-chain CoA esters activate human pancreatic beta-cell KATP channels: potential role in Type 2 diabetes. Diabetologia 47, 277–283 [DOI] [PubMed] [Google Scholar]
- 14. Schulze D., Rapedius M., Krauter T., Baukrowitz T. (2003) Long-chain acyl-CoA esters and phosphatidylinositol phosphates modulate ATP inhibition of KATP channels by the same mechanism. J. Physiol. 552, 357–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riedel M. J., Boora P., Steckley D., de Vries G., Light P. E. (2003) Kir6.2 polymorphisms sensitize beta-cell ATP-sensitive potassium channels to activation by acyl CoAs: a possible cellular mechanism for increased susceptibility to type 2 diabetes? Diabetes 52, 2630–2635 [DOI] [PubMed] [Google Scholar]
- 16. Best L., Jarman E., Brown P. D. (2011) A dual action of saturated fatty acids on electrical activity in rat pancreatic beta-cells. Role of volume-regulated anion channel and KATP channel currents. J. Physiol. 589, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Wet H., Mikhailov M. V., Fotinou C., Dreger M., Craig T. J., Vénien-Bryan C., Ashcroft F. M. (2007) Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 274, 3532–3544 [DOI] [PubMed] [Google Scholar]
- 18. Mikhailov M. V., Campbell J. D., de Wet H., Shimomura K., Zadek B., Collins R. F., Sansom M. S., Ford R. C., Ashcroft F. M. (2005) 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J. 24, 4166–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zingman L. V., Alekseev A. E., Bienengraeber M., Hodgson D., Karger A. B., Dzeja P. P., Terzic A. (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31, 233–245 [DOI] [PubMed] [Google Scholar]
- 20. Aittoniemi J., Fotinou C., Craig T. J., de Wet H., Proks P., Ashcroft F. M. (2009) Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R Soc. Lond. B Biol. Sci. 364, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alekseev A. E., Hodgson D. M., Karger A. B., Park S., Zingman L. V., Terzic A. (2005) ATP-sensitive K+ channel channel/enzyme multimer: metabolic gating in the heart. J. Mol. Cell. Cardiol. 38, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsuo M., Kimura Y., Ueda K. (2005) KATP channel interaction with adenine nucleotides. J. Mol. Cell. Cardiol. 38, 907–916 [DOI] [PubMed] [Google Scholar]
- 23. Findlay I. (1987) ATP-sensitive K+ channels in rat ventricular myocytes are blocked and inactivated by internal divalent cations. Pflugers Arch. 410, 313–320 [DOI] [PubMed] [Google Scholar]
- 24. Findlay I. (1988) Effects of ADP upon the ATP-sensitive K+ channel in rat ventricular myocytes. J. Membr. Biol. 101, 83–92 [DOI] [PubMed] [Google Scholar]
- 25. Dunne M. J. (1989) Protein phosphorylation is required for diazoxide to open ATP-sensitive potassium channels in insulin (RINm5F) secreting cells. FEBS Lett. 250, 262–266 [DOI] [PubMed] [Google Scholar]
- 26. Dunne M. J., Aspinall R. J., Petersen O. H. (1990) The effects of cromakalim on ATP-sensitive potassium channels in insulin-secreting cells. Br. J. Pharmacol. 99, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ashcroft F. M., Kakei M. (1989) ATP-sensitive K+ channels in rat pancreatic beta-cells: modulation by ATP and Mg2+ ions. J. Physiol. 416, 349–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takano M., Qin D. Y., Noma A. (1990) ATP-dependent decay and recovery of K+ channels in guinea pig cardiac myocytes. Am. J. Physiol. 258, H45–50 [DOI] [PubMed] [Google Scholar]
- 29. Treherne J. M., Ashford M. L. (1992) Extracellular cations modulate the ATP sensitivity of ATP-K+ channels in rat ventromedial hypothalamic neurons. Proc. Biol. Sci. 247, 121–124 [DOI] [PubMed] [Google Scholar]
- 30. Schwanstecher M., Sieverding C., Dörschner H., Gross I., Aguilar-Bryan L., Schwanstecher C., Bryan J. (1998) Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 17, 5529–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ortiz D., Voyvodic P., Gossack L., Quast U., Bryan J. (2012) Two Neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J. Biol. Chem. 287, 17985–17995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Higgins C. F., Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 [DOI] [PubMed] [Google Scholar]
- 33. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters: the power to change. Nat. Rev. Mol. Cell. Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dawson R. J., Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 35. Oldham M. L., Chen J. (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hohl M., Briand C., Grütter M. G., Seeger M. A. (2012) Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19, 395–402 [DOI] [PubMed] [Google Scholar]
- 37. Lubelski J., van Merkerk R., Konings W. N., Driessen A. J. (2006) Nucleotide-binding sites of the heterodimeric LmrCD ABC-multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry 45, 648–656 [DOI] [PubMed] [Google Scholar]
- 38. Procko E., Ferrin-O'Connell I., Ng S. L., Gaudet R. (2006) Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell. 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 39. Qin L., Zheng J., Grant C. E., Jia Z., Cole S. P., Deeley R. G. (2008) Residues responsible for the asymmetric function of the nucleotide binding domains of multidrug resistance protein 1. Biochemistry 47, 13952–13965 [DOI] [PubMed] [Google Scholar]
- 40. Procko E., O'Mara M. L., Bennett W. F., Tieleman D. P., Gaudet R. (2009) The mechanism of ABC transporters: general lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 23, 1287–1302 [DOI] [PubMed] [Google Scholar]
- 41. Christesen H. B. T., Sjöblad S., Brusgaard K., Papadopoulou D., Jacobsen B. B. (2005) Permanent neonatal diabetes in a child with an ABCC8 gene mutation. Hormone Res. 64, Suppl. 1, 135 [Google Scholar]
- 42. Babenko A. P. (2008) A novel ABCC8 (SUR1)-dependent mechanism of metabolism-excitation uncoupling. J. Biol. Chem. 283, 8778–8782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 44. Orelle C., Dalmas O., Gros P., Di Pietro A., Jault J. M. (2003) The conserved glutamate residue adjacent to the Walker-B motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. J. Biol. Chem. 278, 47002–47008 [DOI] [PubMed] [Google Scholar]
- 45. Oldham M. L., Khare D., Quiocho F. A., Davidson A. L., Chen J. (2007) Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450, 515–521 [DOI] [PubMed] [Google Scholar]
- 46. Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., Hunt J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moody J. E., Millen L., Binns D., Hunt J. F., Thomas P. J. (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277, 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aleksandrov L., Aleksandrov A. A., Chang X. B., Riordan J. R. (2002) The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277, 15419–15425 [DOI] [PubMed] [Google Scholar]
- 49. Aleksandrov L., Mengos A., Chang X., Aleksandrov A., Riordan J. R. (2001) Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 276, 12918–12923 [DOI] [PubMed] [Google Scholar]
- 50. Löffler-Walz C., Quast U. (1998) Binding of KATP channel modulators in rat cardiac membranes. Br. J. Pharmacol. 123, 1395–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wyman J., Gill S.J.(1990) Binding and Linkage: Functional Chemistry of Biological Macromolecules, University Science Books, Mill Valley, CA [Google Scholar]
- 52. Männikkö R., Flanagan S. E., Sim X., Segal D., Hussain K., Ellard S., Hattersley A. T., Ashcroft F. M. (2011) Mutations of the same conserved glutamate residue in NBD2 of the sulfonylurea receptor 1 subunit of the KATP channel can result in either hyperinsulinism or neonatal diabetes. Diabetes 60, 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yount R. G. (1975) ATP analogs. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 1–56 [DOI] [PubMed] [Google Scholar]
- 54. Hehl S., Neumcke B. (1994) KATP channels of mouse skeletal muscle: mechanism of channel blockage by AMP-PNP. Eur. Biophys. J. 23, 231–237 [DOI] [PubMed] [Google Scholar]
- 55. Lederer W. J., Nichols C. G. (1989) Nucleotide modulation of the activity of rat heart ATP-sensitive K+ channels in isolated membrane patches. J. Physiol. 419, 193–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwanstecher C., Dickel C., Panten U. (1994) Interaction of tolbutamide and cytosolic nucleotides in controlling the ATP-sensitive K+ channel in mouse beta-cells. Br. J. Pharmacol. 111, 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Proks P., de Wet H., Ashcroft F. M. (2010) Activation of the KATP channel by Mg-nucleotide interaction with SUR1. J. Gen. Physiol. 136, 389–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schwanstecher M., Löser S., Brandt C., Scheffer K., Rosenberger F., Panten U. (1992) Adenine nucleotide-induced inhibition of binding of sulphonylureas to their receptor in pancreatic islets. Br. J. Pharmacol. 105, 531–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matsuo M., Kioka N., Amachi T., Ueda K. (1999) ATP binding properties of the nucleotide-binding folds of SUR1. J. Biol. Chem. 274, 37479–37482 [DOI] [PubMed] [Google Scholar]
- 60. Ueda K., Inagaki N., Seino S. (1997) MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J. Biol. Chem. 272, 22983–22986 [DOI] [PubMed] [Google Scholar]
- 61. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 62. Hung L. W., Wang I. X., Nikaido K., Liu P. Q., Ames G. F., Kim S. H. (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 396, 703–707 [DOI] [PubMed] [Google Scholar]