

Background: To date, the Smad cofactor involved in cell motility induced by transforming growth factor-β (TGF-β) has not been identified.
Results: Knockdown of oligodendrocyte transcription factor-1 (Olig1), as well as inhibition of the Olig1-Smad interaction, resulted in attenuation of TGF-β-induced cell motility.
Conclusion: Olig1 is involved in TGF-β-induced cell motility.
Significance: This study enhances understanding of the regulation of TGF-β-induced cell motility.
Keywords: Cell Motility, Prolyl Isomerase, Signal Transduction, Smad Transcription Factor, Transforming Growth Factor-β
Abstract
Transforming growth factor (TGF)-β plays crucial roles in embryonic development and adult tissue homeostasis by eliciting various cellular responses in target cells. TGF-β signaling is principally mediated through receptor-activated Smad proteins, which regulate expression of target genes in cooperation with other DNA-binding transcription factors (Smad cofactors). In this study, we found that the basic helix-loop-helix transcription factor Olig1 is a Smad cofactor involved in TGF-β-induced cell motility. Knockdown of Olig1 attenuated TGF-β-induced cell motility in chamber migration and wound healing assays. In contrast, Olig1 knockdown had no effect on bone morphogenetic protein-induced cell motility, TGF-β-induced cytostasis, or epithelial-mesenchymal transition. Furthermore, we observed that cooperation of Smad2/3 with Olig1 is regulated by a peptidyl-prolyl cis/trans-isomerase, Pin1. TGF-β-induced cell motility, induction of Olig1-regulated genes, and physical interaction between Smad2/3 and Olig1 were all inhibited after knockdown of Pin1, indicating a novel mode of regulation of Smad signaling. We also found that Olig1 interacts with the L3 loop of Smad3. Using a synthetic peptide corresponding to the L3 loop of Smad3, we succeeded in selectively inhibiting TGF-β-induced cell motility. These findings may lead to a new strategy for selective regulation of TGF-β-induced cellular responses.
Introduction
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that regulates various cellular responses, including growth, motility, differentiation, and apoptosis, in a wide variety of target cells. Aberrant TGF-β signal transduction often leads to progression of diseases including cancer, allergy, and fibrosis (1–4). Understanding of TGF-β signaling would thus aid in elucidating the pathogenic mechanisms of such diseases. TGF-β can play opposing roles in tumorigenesis, depending on timing and cellular context (5, 6): in the early stages of tumorigenesis, it suppresses tumors via cytostasis, maintenance of genome stability, and induction of apoptosis; in advanced stages, it promotes tumor progression via enhancement of tumor cell motility, invasion, and survival, as well as induction of epithelial-mesenchymal transition (EMT)2 and suppression of the host immune system (5–7). In addition, TGF-β regulates cancer stem cells positively or negatively, depending on tumor type: it suppresses stomach cancer stem cells (8), whereas it maintains tumorigenicity of tumor-initiating cells in glioma (9, 10) and leukemia (11). Contradictory results were reported for breast cancer (12, 13). Recently, inhibitors of TGF-β signaling, including receptor kinase inhibitors, neutralizing antibodies, and antisense oligonucleotides have been developed for possible clinical use. Thus far, all of them comprehensively inhibit TGF-β signaling. To avoid possible side effects, it would be desirable to develop inhibitors of TGF-β signaling that are selective for specific cellular responses.
TGF-β signals are mediated through two types of transmembrane receptors, type I and type II, which possess intrinsic serine/threonine kinase activity. Upon ligand binding, the receptors form a tetraheteromeric complex, in which constitutively active type II receptor phosphorylates type I receptor at its glycine/serine-rich domain, thereby activating it. The activated type I receptor in turn phosphorylates the cytoplasmic effector molecules Smad2 and Smad3 (receptor-regulated Smads, R-Smads). Phosphorylated Smad2/3 then form a trimeric or dimeric complex with Smad4; this complex translocates into the nucleus, where it regulates expression of target genes positively or negatively in cooperation with co-activators as well as co-repressors (14). The activated Smad complex usually requires DNA-binding transcription factors, so-called Smad cofactors, for its regulation of target genes (15, 16).
The wide variety of cellular responses induced by TGF-β can, to some extent, be attributed to the particular Smad-cofactor complexes that are active in target cells. Thus, one possible way to achieve cellular response-selective regulation of TGF-β signaling would be to target cooperation of Smad and Smad cofactor(s). Previously, we found that a helix-loop-helix protein named Maid (also called human homologue of murine maternal Id-like molecule, HHM) regulates TGF-β signaling in this fashion (17, 18). Maid inhibits TGF-β-induced cytostasis and cell motility, but not EMT, via sequestration of Smad cofactors (17). It is reasonable to predict that the set of Maid-binding transcription factors should include the Smad cofactors involved in cytostasis and cell motility.
In this study, we found that Olig1, which we previously identified as a Maid-interacting protein, is a Smad cofactor involved in TGF-β-induced cell motility. Cooperation of Olig1 with Smad2/3 was regulated by a peptidyl-prolyl cis/trans-isomerase, Pin1. Furthermore, we succeeded in selectively inhibiting Olig1-mediated Smad signaling by disrupting the Olig1-Smad complex. These findings facilitate further understanding of the regulation of TGF-β-induced cell motility and open the way for development of novel methods for controlling TGF-β signaling in a cellular response-selective fashion.
EXPERIMENTAL PROCEDURES
Cell Culture
NMuMG, BT549, and COS-7 cells were obtained from the American Type Culture Collection. All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 50 units/ml penicillin, and 50 μg/ml streptomycin. For culture of NMuMG cells, media were also supplemented with 10 μg/ml insulin.
Antibodies
The antibodies used were as follows: anti-Smad2/3 (BD Bioscience), anti-phospho-Smad2 (138D4; Cell Signaling Technology), anti-phospho-Smad1 (9511; Cell Signaling Technology), anti-phospho-Smad3 (9520; Cell Signaling Technology), anti-Smad4 (B-8; Santa Cruz Biotechnology), anti-Olig1 (Novus Biologicals), anti-Pin1 (Calbiochem), anti-FLAG (M2; Sigma-Aldrich); anti-Myc (9E10, Millipore); anti-α-tubulin (DM1A; Sigma-Aldrich); anti-E-cadherin and anti-N-cadherin (BD Bioscience).
Chemicals
Human recombinant TGF-β1 and bone morphogenetic protein (BMP)-4 were obtained from R&D Systems. Juglone was purchased from Calbiochem. cDNA constructs were described previously (17, 19). Flavopiridol was obtained from Sigma-Aldrich, and LiCl was from Wako Pure Chemicals.
RNA Interference
Small interfering RNAs (siRNAs) specific for mouse Olig1 (sense, 5′-UGCCGAGUAGGGUAGGAUAACUUCG-3′ or sense, 5′-UGUAACCCACCAGCUCAUACAGCGA-3′), human Olig1 (sense, 5′-AAUCGAAACUGACUACGUAUGUAGC-3′), mouse Pin1 (sense, 5′-AUUUAAUGGAAGGUGCGUAGGGUGC-3′), mouse Smad2 (sense, 5′-CAGGACGGUUAGAUGAGCUUGAGAA), mouse Smad3 (sense, 5′-CCUGCUGGAUUGAGCUACACCUGAA) negative control oligonucleotide (Stealth siRNA 12935–200) were purchased from Invitrogen. siRNAs were introduced into cells using HiPerFect transfection reagent (Qiagen) at a final RNA concentration of 5 nm.
DNA Transfection, Cell Lysis, Immunoprecipitation, and Immunoblotting
NMuMG cells were transiently transfected using FuGENE6 or X-treamGENE 9 transfection reagent and incubated for 24 h before analysis. Cells were lysed in a buffer containing 1% Nonidet P-40, 20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm PMSF, 1% aprotinin, and 5 mm EDTA. For immunoprecipitation, cleared lysates were incubated with anti-Smad2/3, anti-Myc or anti-FLAG M2 antibody for 1 h at 4 °C. Proteins in immunoprecipitates or cleared cell lysates were subjected to SDS-PAGE and transferred to FluoroTrans W membrane (Pall) (20); immunoblotting was performed using the indicated antibodies. For detection of endogenous Olig1 in NMuMG cells, we solubilized cells with SDS-PAGE sample buffer because Olig1 could not be efficiently recovered in lysis buffer containing Nonidet P-40. Glutathione S-transferase (GST) fusion proteins for pulldown assays were prepared as described previously (19).
Luciferase Reporter Assay
Luciferase assays were performed as described previously (21) using a TGF-β-responsive reporter (CAGA)12-MLP-Luc (22). Values were normalized to activity of Renilla luciferase expressed under the control of thymidine kinase promoter.
Cell Proliferation Assay
NMuMG cells were seeded in triplicate at a density of 5 × 104 cells/well in 12-well plates, and cultured for 24 h. After treatment with 1 ng/ml TGF-β for 48 h, cells were trypsinized and harvested. Cells were counted using a hemocytometer.
Immunofluorescence Labeling
Immunocytochemical analyses were performed as described previously (23). Fluorescence was examined by confocal laser scanning microscopy (Olympus).
Cell Motility Assay
Chamber migration and wound healing assays were performed as described previously (17).
Quantitative Real-time PCR
Total RNA was extracted using TRIzol (Invitrogen). First-strand cDNA was synthesized using PrimeScript reverse transcriptase (Takara Bio) and oligo(dT)12–18 primers (Invitrogen). Quantitative real-time PCR analysis was performed using Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) and the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Specificity of detected signals was confirmed via dissociation protocol. All samples were run in triplicate in each experiment. Values were normalized against the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. The primers used were as follows: mouse PAI-1 (sense, 5′-CCACAAAGGTCTCATGGACCAT-3′; antisense, 5′-TGAAAGTGTTGTGCCCTCCAC-3′); mouse GAPDH (sense, 5′-TGCAGTGGCAAAGTGGAGATT-3′; antisense, 5′-TGCCGTTGAATTTGCCGT-3′); mouse Snail (sense, 5′-CCACTGCAACCGTGCTTTT-3′; antisense, 5′-GTGCTTGTGGAGCAAGGACAT-3′); mouse Smad7 (sense, 5′-CCTTAGCCGACTCTGCGAACTA-3′; antisense, 5′-CCAGATAATTCGTTCCCCCTGT-3′); p21WAF (sense, 5′-GCGACTGTGATGCGCTAATG-3′; antisense, 5′-CCAGTGGTGTCTCGGTGACA-3′).
DNA Microarray Analysis
NMuMG cells were transfected with siRNAs (siControl, siOlig1, or siPin1). Cells were treated with or without TGF-β for 1 h, harvested, and subjected to DNA microarray analysis using the Mouse Gene 1.0 ST Array (Affymetrix) according to the manufacturer's instructions. The microarray data were normalized using the Robust Multiarray Average (RMA) algorithm. The raw data have been deposited in the Gene Expression Omnibus (GEO) database with an accession number GSE46405.
Smad Mutant Proteins
Smad3 mutants, Smad3–4A (Thr-179, Ser-204, Ser-208, and Ser-213 are mutated to Ala), MH1(1–145), MH1+L(1–219), L+MH2(146–425), and MH2(220–425) were described previously (17, 19). Construction of human Smad1/3 chimeric proteins was described previously (24). In brief, the MH2 domains of Smad1 and Smad3 were divided into four regions (regions 1–4) and individually swapped between proteins. Region 1 was 270–300 in Smad1, 239–261 in Smad3. Region 2 was 300–354 in Smad1, 261–314 in Smad3. Region 3 was 354–412 in Smad1, 314–372 in Smad3. Region 4 was 412–465 in Smad1, 372–425 in Smad3. Other mutants were constructed using a PCR-based approach.
Peptide Transfection
Peptides were introduced into cells in culture using XfectTM protein transfection reagents (Clontech). Cells were plated onto a 6-well plate (2 × 105 cells/well). Twelve h later, cells were transfected with 5 μg of peptides in serum-free medium, followed by medium change after 1 h. Cells were then cultured for 4–48 h, harvested, and used for each assay.
RESULTS
Olig1 Is Involved in TGF-β-induced Cell Motility
We previously found that the basic helix-loop-helix protein Olig1 interacts with Smad2/3 in response to TGF-β stimulation. The Olig1-Smad2/3 complex then regulates expression of a subset of TGF-β target genes through association with their promoter regions (17). To examine the roles of Olig1 in TGF-β-induced cellular responses, we knocked down endogenous Olig1 in NMuMG cells using siRNA. Knockdown was successful, as determined by immunoblotting (Ref. 17 and Fig. 1A). Knockdown of Olig1 affected neither phosphorylation of Smad2 (Fig. 1B) nor TGF-β-induced activity of the luciferase reporter (CAGA)12-MLP-Luc (Fig. 1C). These results indicate that Olig1 is not involved in the principal events of the TGF-β signal transduction pathway.
FIGURE 1.

Knockdown of Olig1 does not affect the principal events in TGF-β signaling. A, knockdown of Olig1 in NMuMG mouse mammary epithelial cells. Cells were transfected with a negative control RNA or Olig1 siRNA (siControl or siOlig1, respectively). Sixteen h later, cells were stimulated with TGF-β for 24 h and harvested. Expression of Olig1 was determined by immunoblotting. Expression of tubulin is also shown as a loading control. B, effects of Olig1 knockdown on TGF-β-induced phosphorylation of Smad2. NMuMG cells were transfected with control or Olig1 siRNA. Thirty-six h later, cells were stimulated with TGF-β for 0.5–1 h and harvested. Levels of C-terminally phosphorylated Smad2, as well as total Smad2/3, were determined by immunoblotting. C, effects of Olig1 knockdown on TGF-β-induced transactivation of (CAGA)12-MLP-Luc. NMuMG cells were transfected with control or Olig1 siRNA. Thirty h later, cells were transfected with luciferase reporter constructs, stimulated with TGF-β for 24 h, and harvested. Error bars represent S.D.
We next examined the effect of silencing Olig1 expression on various cellular responses. TGF-β-induced cytostasis was not affected by Olig1 knockdown (Fig. 2A). We also performed immunoblotting for E-cadherin and N-cadherin, which are, respectively, down- and up-regulated during EMT (Fig. 2B). Neither protein level was altered by the Olig1 knockdown; consistent results were obtained via immunocytochemical staining for E-cadherin (data not shown). Thus, Olig1 does not affect TGF-β-induced EMT. These results are consistent with our previous observations of the effects of Olig1 knockdown on target gene expression regulated by TGF-β: knockdown had no effect on up-regulation of p21WAF and p15INK4b and down-regulation of c-myc, which are involved in TGF-β-induced cytostasis, or on up-regulation of Snail and down-regulation of the E-cadherin gene, which are involved in EMT (17).
FIGURE 2.

Knockdown of Olig1 selectively impairs cell motility induced by TGF-β. Cells were transfected with control or Olig1 siRNA. After 16 h, cells were used in the indicated assays. A, effects of Olig1 knockdown on TGF-β-induced cytostasis in NMuMG cells. Cells were treated with TGF-β (1 ng/ml) for 48 h and counted. siControl denotes a negative control oligonucleotide. Error bars represent S.D. B, effects of Olig1 knockdown on TGF-β-induced EMT. Cells were stimulated with TGF-β (1 ng/ml) for 24 h after knockdown of Olig1. Protein expression of a mesenchymal marker, N-cadherin (top panel) and an epithelial marker, E-cadherin (middle panel), are depicted. The bottom panel shows expression level of tubulin protein, as a loading control. C and E, chamber migration assay. Cells were stimulated with TGF-β (1 ng/ml, C) or BMP-4 (10 ng/ml, E). D and F, wound healing assay. Cells were stimulated with TGF-β (1 ng/ml, D) or BMP-4 (10 ng/ml, F). Quantitations are shown in the right. p values were determined by Student's t test. *, p < 0.01. G, effect of Smad2/3 knockdown on TGF-β-induced cell motility in chamber migration assay. Cells were transfected with control or Smad2/3 siRNA. After 48 h, cells were used in chamber migration assay.
On the other hand, TGF-β-induced cell motility was affected by knockdown of Olig1. In chamber migration assays, knockdown of Olig1 inhibited the effect of TGF-β (Fig. 2C). Similar results were also obtained in wound healing assays. In Olig1-knockdown cells, wound closure was delayed relative to control cells (Fig. 2D). We confirmed these effects of Olig1 knockdown by using another siRNA duplex targeting Olig1 (data not shown). The inhibitory effect of Olig1 knockdown on cell motility was also observed in the human breast carcinoma cell line BT549 (data not shown). Importantly, knockdown of Olig1 did not affect BMP-4-induced cell motility in chamber migration or wound healing assays (Fig. 2, E and F), indicating that the general machinery for cell motility/cell migration was not affected by knockdown of Olig1. We further examined the effect of knockdown of Smad2/3. It inhibited TGF-β-induced cell migration (Fig. 2G). These findings support our idea that the Smad-Olig1 complex is involved in these cell responses. We therefore concluded that Olig1 is a Smad cofactor involved in TGF-β-induced cell motility.
Pin1 Activity Is Required for Cooperative Action of Olig1 with Smad
Pin1 is a peptidyl-prolyl cis/trans-isomerase that plays regulatory roles in Smad signaling (19, 25). Pin1 promotes TGF-β-induced cell migration (in PC3 prostate carcinoma cells), but not cytostasis (in HaCaT immortalized keratinocytes) (25). This earlier report, together with our present findings, prompted us to hypothesize that cooperation of Olig1 with Smad proteins is regulated by Pin1 activity.
We first examined whether knockdown of Pin1 also affects cell migration in NMuMG cells. Pin1 protein was so stable in this cell line that it took 48 h before we could observe down-regulation sufficient for loss-of-function experiments (Fig. 3A). Because of the delayed time course of silencing, we had difficulty in examining the effects of Pin1 knockdown on cell motility in wound healing assays. In chamber migration assays, knockdown of Pin1 abrogated TGF-β-induced cell migration (Fig. 3B). Importantly, Pin1 knockdown did not inhibit BMP-4-induced cell migration (Fig. 3B), indicating that knockdown of Pin1 does not affect cell motility in general.
FIGURE 3.

Pin1 regulates cooperative action of Smad and Olig1. A, knockdown of Pin1 in NMuMG cells. Cells were transfected with control or Pin1 siRNA and harvested at the indicated times. Expression of Pin1 and α-tubulin (as a loading control) was analyzed by immunoblotting. In the following experiments, cells transfected for 48 h are used in each assay. B, knockdown of Pin1 inhibiting TGF-β-induced cell migration in a chamber migration assay. Cells were treated with TGF-β (1 ng/ml) or BMP-4 (10 ng/ml) for 12 h and subjected to chamber migration assays. Quantification is shown in the panel below. C, effects of Pin1 knockdown on Smad-Olig1 interaction. NMuMG cells were transfected with FLAG-tagged Olig1. Twenty-four h later, cells were stimulated with TGF-β (1 ng/ml) for 1 h and harvested. Cell lysates were subjected to immunoprecipitation (IP) with anti-Smad2/3 antibody, and co-precipitated Olig1 or Smad4 was visualized by immunoblotting (IB). D, effects of juglone, a Pin1 inhibitor, on Smad-Olig1 interaction. Cells were transfected with FLAG-tagged Olig1, treated with juglone (0.5–1.0 μm) for 24 h, stimulated with TGF-β (1 ng/ml) for 1 h, and harvested. Co-precipitation assay was performed as in C. E, effects of Pin1 knockdown on expression of TGF-β target genes. After knockdown of Pin1, cells were treated with TGF-β (1 ng/ml) for 1 h and harvested; mRNA levels of target genes were measured by quantitative real-time PCR. p values were determined by Student's t test. *, p < 0.05; **, p < 0.01. F, heat map of TGF-β-induced expression of target genes. Cells were transfected with control, Olig1, or Pin1 siRNA. After stimulation with TGF-β for 1 h, cells were harvested and subjected to DNA microarray analysis. Genes induced by TGF-β >1.6-fold in the siControl sample (151 genes) are shown. G, Venn diagram showing the overlap of Olig1-regulated genes and Pin1-regulated genes. Genes whose -fold induction by TGF-β were decreased by <0.7 after knockdown of Olig1 or Pin1 (siOlig1 or siPin1 fold/siControl fold < 0.7) are classified as Olig1-regulated genes or Pin1-regulated genes, respectively.
We next examined the physical interaction between Smad2/3 and Olig1, which is required for the cooperative action of Smad and Olig1. Because the endogenous expression level of Olig1 in NMuMG cells is not high enough to allow detection of the endogenous protein interaction with Smad2/3, we transfected Olig1 into the cells. Interaction of Smad2/3 with Olig1 was decreased upon Pin1 knockdown, whereas the interaction with Smad4 was not (Fig. 3C). Thus, Pin1 selectively regulates the interaction of Olig1 with Smad2/3. We also found that juglone, an inhibitor of Pin1 enzymatic activity, suppressed the interaction (Fig. 3D), suggesting that the interaction of Smad2/3 and Olig1 is regulated by peptidyl-prolyl cis/trans-isomerase activity of Pin1. Consistent with these findings, induction of two Olig1-regulated target genes of TGF-β, PAI-1 and Smad7, was attenuated upon knockdown of Pin1, whereas induction of non-Olig1-regulated genes, including p21WAF and Snail, was not (Fig. 3E). We further performed DNA microarray analysis to compare genes affected by knockdown of Olig1 and that of Pin1. After stimulation of NMuMG cells with TGF-β for 1 h, 151 genes were up-regulated >1.6-fold. Knockdown of Olig1 attenuated induction of 40 genes, 35 of which were similarly affected by Pin1 knockdown (Fig. 3, F and G). Therefore, we conclude that Pin1 activity is required for induction of genes regulated by Smad and Olig1.
Pin1 Targets Smad3 but Not Olig1
Smad2 and Smad3 are substrates of Pin1; Pin1 interacts with Smad2/3 through the phosphorylated linker region (19). In contrast, Olig1 does not interact with Pin1. We found that GST-Pin1 failed to interact with Olig1 whereas it does with Smad3 (Fig. 4A). Consistently, Olig1 lacks a consensus Pin1-target motif. We also examined physical interaction between Olig1 and a Smad3 mutant that does not interact with Pin1, Smad3–4A, in which linker phosphorylation sites are mutated (19). Olig1 interacted with Smad3–4A only weakly (Fig. 4B), suggesting that linker phosphorylation of Smad3 is required for successful interaction of Smad3 with Olig1.
FIGURE 4.

Pin1 targets linker-phosphorylated Smad3. A, in vitro interaction of GST-Pin1 with Olig1. NMuMG cells were transfected with FLAG-Smad3, Smad3–4A, or Olig1 24 h before harvest. Cell lysates were incubated with GST-Pin1 or GST and subjected to GST pulldown followed by immunoblotting (IB) with an anti-FLAG antibody. The top two panels display input protein expression and the interaction. The bottom panel shows GST-Pin1 or GST visualized by Coomassie Brilliant Blue (CBB) staining. B, effect of mutation in linker phosphorylation sites of Smad3 on its interaction with Olig1. NMuMG cells were transfected with the indicated plasmids. Twenty-four h later, cells were stimulated with TGF-β (1 ng/ml) for 1 h and harvested. Cell lysates were subjected to immunoprecipitation (IP) with an anti-FLAG antibody, followed by immunoblotting. The top two panels display precipitated proteins, and the bottom panel shows expression of Olig1. C, effects of kinase inhibitors on Smad-Olig1 interaction. Cells were transfected with FLAG-tagged Olig1, treated with 1 μm flavopiridol or 10 mm LiCl for 1 h, stimulated with 1 ng/ml TGF-β for 1 h, and harvested. Cell lysates were subjected to immunoprecipitation with anti-Smad2/3 antibody, and co-precipitated Olig1 or Smad4 was visualized by immunoblotting. D, effects of Pin1 knockdown on interaction between Olig1 and Smad3 truncated mutants. NMuMG cells were transfected with control or Pin1 siRNA. Twenty-four h later, cells were then transfected with indicated plasmids together with constitutively active forms of TGF-β type I receptor (ALK-5). Twenty-four h later, cells were harvested and cell lysates were subjected to immunoprecipitation with an anti-Myc antibody, followed by immunoblotting. The top two panels display precipitated proteins, and the bottom three panels show expression of Smad4, Olig1, or Pin1.
Several protein kinases have been reported to be involved in linker phosphorylation of Smad3. CDK8/9 phosphorylates Thr-179, Ser-208, and Ser-213, creating binding sites for Pin1 and glycogen synthase kinase-3β that additionally phosphorylates Ser-204 of the Smad3 linker region (26). We thus examined effects of inhibitors of these kinases (Fig. 4C). Flavopiridol (CDK8/9 inhibitor) effectively inhibited Smad-Olig1 interaction whereas LiCl (glycogen synthase kinase-3β inhibitor) did not, further supporting the idea that linker phosphorylation of Smad3 is required for Pin1-assisted interaction of Smad3 with Olig1.
We next examined the physical interaction between Olig1- and Smad3-truncated mutants in the presence or absence of Pin1 (Fig. 4D). Consistent with our previous findings (17), Olig1 interacted with the MH2 domain of Smad3. Importantly, a Smad3 mutant containing the MH2 domain alone does not require Pin1 for its interaction with Olig1, whereas a Smad3 mutant containing the MH2 domain plus the linker region interacted with Olig1 only in the presence of Pin1. Their interaction with Smad4 was not affected by Pin1 knockdown. These findings suggest that the linker region has a negative impact on the interaction between Olig1 and Smad3, and Pin1-induced conformational change of the linker region appears to have a de-repressing role. Taken together, we concluded that Pin1 interacts with linker-phosphorylated Smad2/3, changes its conformation, and allows association and cooperative action of Smad3 with Olig1.
Olig1 Interacts with the Surface-exposed L3 Loop of Smad3
Because TGF-β-induced cell motility is implicated in tumor invasion and metastasis, its selective inhibition would be potentially useful as a therapeutic strategy for treatment of cancer. From the data described above, it appeared likely that inhibition of the Smad-Olig1 association would lead to attenuation of TGF-β-induced cell motility. To develop a method for inhibiting Olig1-Smad2/3 interaction, we first identified the Olig1-binding region in Smad3.
We previously found that Olig1 interacts with Smad3, but not Smad1, through its MH2 domain (17). The overall identity of the MH2 domains of Smad1 and Smad3 is ∼80%, with some regions highly conserved and other regions more divergent. We thus constructed a series of Smad1-Smad3 chimeric proteins in which the MH2 domains were divided into four regions (regions 1–4) and swapped (24), and we examined their physical interaction with Olig1 (Fig. 5A). Smad1 did not interact with Olig1 (lane 2), but a chimeric protein containing the MH2 domain from Smad3 did (lane 10). Further investigation revealed that only chimeric proteins containing region 4 from Smad3 were able to interact with Olig1 (lanes 3–5).
FIGURE 5.
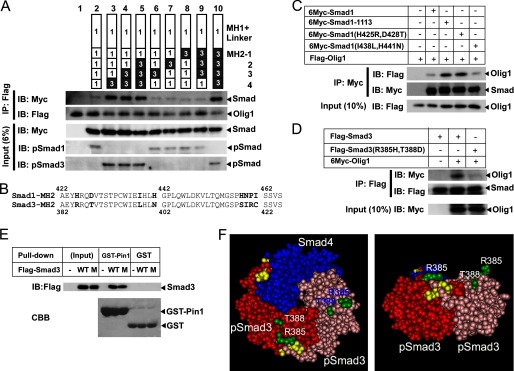
Mapping of the Olig1-binding region in Smad3. A, physical interaction of Olig1 with Smad1/3 chimeric proteins. COS-7 cells were transfected with FLAG-Olig1 and 6Myc-Smad1/3 chimeric proteins, together with constitutively active forms of BMP receptor type IB (ALK-6) and TGF-β type I receptor (ALK-5). Chimeric proteins used are schematically presented in the top panel. Domains or regions derived from Smad1 are shown as open boxes; those from Smad3 are shown as filled boxes. Smad was not transfected into the sample in lane 1. Olig1 was immunoprecipitated (IP) with anti-FLAG antibody; co-precipitated Smad1/3 chimeric proteins were visualized by immunoblotting (IB, middle panels). Input and C-terminal phosphorylation of Smad1/3 chimeric proteins are also shown (bottom panel). B, alignment of amino acid sequences of the region 4 of Smad1 and Smad3. Diverged residues are shown in bold. C, physical interaction of Olig1 with Smad1 mutants, Smad1–1113 (Smad1 with region 4 of the MH2 domain from Smad3), Smad1(H425R,D428T) or Smad1(I438L,H441N). COS-7 cells were transfected with FLAG-Olig1 and 6Myc-Smad1 mutants, together with constitutively active TGF-β type I receptor (ALK-5) and BMP receptor type IB (ALK-6). Smad1 mutants were immunoprecipitated with anti-Myc antibody; co-precipitated Olig1 was visualized by immunoblotting. Input of Olig1 is also shown (bottom panel). D, physical interaction of Olig1 with a Smad3 mutant (R385H,T388D). COS-7 cells were transfected with FLAG-Smad3 or Smad3(R385H,T388D) and 6Myc-Olig1, together with constitutively active TGF-β type I receptor (ALK-5) and BMP receptor type IB (ALK-6). Smad3 was immunoprecipitated with anti-FLAG antibody; co-precipitated Olig1 was visualized by immunoblotting. Input of Olig1 is also shown (bottom panel). E, in vitro interaction of GST-Pin1 with a Smad3 mutant (R385H,T388D). COS-7 cells were transfected with FLAG-Smad3 or Smad3 mutant (R385H,T388D) 24 h before harvest. Cell lysates were then incubated with GST-Pin1 or GST, and subjected to GST pulldown followed by immunoblotting with an anti-FLAG antibody. The top panel displays input protein expression and the interaction. The lower panel shows GST-Pin1 or GST visualized by Coomassie Brilliant Blue (CBB) staining. F, three-dimensional structural model of the MH2 domain of the phospho-Smad3–phospho-Smad3–Smad4 trimeric complex (Protein Databank code 1U7F). The left panel shows a view from the C-terminal side of the trimeric complex. The right panel shows a side view (the C-terminal side is placed upward). The C-terminally phosphorylated serine residues are shown in yellow; the Olig1-binding determinant is shown in green.
We next compared amino acid sequences of region 4 from Smad1 (residues 422–465) and Smad3 (residues 382–425). Region 4 is highly conserved between Smad1 and Smad3 (Fig. 5B). We first focused on four divergent amino acid residues in Smad3 region 4: Arg-385, Thr-388, Leu-398, and Asn-401. We then constructed two mutants in which these residues were introduced pairwise into the corresponding positions in Smad1 and examined their interaction with Olig1 (Fig. 5C). A mutant Smad1(H425R,D428T) interacted with Olig1, whereas a mutant Smad1(I438L,H441N) did not. In addition, a mutant Smad3 (R385H,T388D) failed to interact with Olig1 (Fig. 5D) although it maintains interaction with Pin1 (Fig. 5E). Thus, we concluded that Arg-385, Thr-388, or both in Smad3 are the Olig1-binding determinants. These two amino acid residues are also conserved in Smad2, another R-Smad that interacts with Olig1. These two residues are located in the L3 loop, which forms a protruding structure on the surface of the activated Smad complex (Fig. 5F) and plays an important role in the specific recognition of receptor-regulated Smads by type I receptors (27).
The L3 Loop Peptide Derived from Smad3 (P-L3S3) Selectively Inhibits TGF-β-induced Cell Motility
We next chemically synthesized a peptide containing the Olig1-binding determinant in Smad3. To promote successful folding of this peptide into the active conformation, we included the 29-residue region surrounding the determinant (Thr-371–His-399, Fig. 6A). The peptide was covalently linked to a fluorescent dye (fluorescein isothiocyanate) in the N terminus, to monitor its incorporation into cells, and a nuclear localization signal in the C terminus. We named the peptide P-L3S3 (peptide corresponding to the L3 loop of Smad3). We also synthesized a peptide containing the corresponding region from Smad1 (P-L3S1), and used it as a negative control (Fig. 6A).
FIGURE 6.

A peptide blocker P-L3S3 inhibits the Smad-Olig1 interaction. A, primary sequences of peptide blockers P-L3S3 and P-L3S1. Diverged amino acid residues are shown in bold. The nuclear localization signal attached to the C terminus is underlined. B, phosphorylation of Smad2 in response to TGF-β stimulation in the presence of peptide blockers. NMuMG cells were transfected with P-L3S1 and P-L3S3. Four h later, cells were stimulated with TGF-β (1 ng/ml) for 1 h followed by immunoblotting analysis. C, effects of peptide blockers on physical interaction of Olig1 with Smad2/3. NMuMG cells were transfected with FLAG-tagged Olig1, and 12 h later they were transfected with peptides. Eleven h later, cells were stimulated with TGF-β for 1 h and harvested. Cell lysates were subjected to immunoprecipitation with anti-Smad2/3 antibody; co-precipitated Olig1 and Smad4 were visualized by immunoblotting (IB).
We then introduced these peptides into NMuMG cells using a protein transfection reagent. Both of these peptides were detected at similar levels in the cytoplasm as well as the nucleus, as assessed by fluorescence microscopy (data not shown). Neither P-L3S3 nor P-L3S1 inhibited TGF-β-induced Smad2 phosphorylation (Fig. 6B), indicating that these peptides do not interfere with the interaction of the type I receptor with Smad2/3. We then examined the Smad2/3-Olig1 interaction in the presence of these peptides (Fig. 6C). P-L3S3, but not P-L3S1, inhibited the association of Olig1 with Smad2/3, whereas neither peptide affected the Smad2/3-Smad4 interaction.
Finally, we examined the effects of P-L3S3 on various cellular responses induced by TGF-β. P-L3S3 inhibited TGF-β induced cell migration in chamber migration and wound healing assays, but the negative control peptide P-L3S1 did not (Fig. 7, A and B). P-L3S3 did not affect BMP-4-induced cell motility (Fig. 7, C and D), TGF-β-induced EMT, or cytostasis (Fig. 7, E and F). Consistent with these findings, P-L3S3 did not affect induction of p21WAF and Snail, which are involved, respectively, in TGF-β-induced cytostasis and EMT (Fig. 7G). In contrast, the induction of Smad-Olig1 regulated genes PAI-1 and Smad7 was attenuated by P-L3S3 (Fig. 7G). Thus, P-L3S3 appears to selectively inhibit cell motility, but not other TGF-β-induced cellular responses, via suppression of Smad-Olig1-regulated genes.
FIGURE 7.

P-L3S3 selectively inhibits TGF-β-induced cell motility. Cells were transfected with peptide P-L3S3 or P-L3S1 using a protein transfection reagent. Four h later, cells were subjected to the indicated assays. A and C, chamber migration assay. Cells were stimulated with TGF-β (1 ng/ml, A) or BMP-4 (10 ng/ml, C). B and D, wound healing assay. Cells were stimulated with TGF-β (1 ng/ml, B) or BMP-4 (10 ng/ml, D). Quantitations are shown at the right. NT denotes no-treatment control (without peptide transfection). E, TGF-β-induced EMT. Expression of N-cadherin (a mesenchymal marker, top panel), and E-cadherin (an epithelial marker, middle), and α-tubulin (a loading control, bottom) is shown. F, TGF-β-induced cytostasis. Cells were treated with TGF-β for 48 h and counted. G, expression of target genes of TGF-β. Cells were treated with 1 ng/ml TGF-β for 1 h, harvested, and mRNA levels of target genes were measured by quantitative real-time PCR. p values were determined by Student's t test. *, p < 0.05; **, p < 0.01. Error bars, S.D.
DISCUSSION
Smad-mediated transcriptional regulation is specified and/or modified by cooperation with other DNA-binding transcription factors, so-called Smad cofactors, which form complexes with Smad proteins. The DNA-binding affinity of Smad proteins to the canonical Smad binding element is not high enough to allow Smads to bind on their own; interaction with Smad cofactors allows the complex to interact with specific target sites on genomic DNA. Moreover, a wide variety of cellular contexts are generated by the distinct profiles of Smad cofactors in target cells (16); expression as well as activity of Smad cofactors affects transcriptional regulation by Smad proteins. Thus far, several Smad cofactors, including AP-1 (28), FoxO (29), p53 (30), and Ets1 (31), have been identified. The cofactors affecting TGF-β-induced cytostasis have been studied extensively. In mouse embryonic fibroblasts and hematopoietic progenitor cells, TGF-β failed to induce cytostasis in cells derived from p53−/− mice (30). In HaCaT cells, knockdown of Ets1 resulted in resistance of cells to TGF-β-induced cytostasis (31). In addition, expression of p21WAF and p15INK4b is affected by knockdown of FoxO in HaCaT cells (29). However, the Smad cofactors involved in other cellular responses have not been elucidated. In this study, we found that Olig1, a basic helix-loop-helix transcription factor, is involved in TGF-β-induced cell migration.
Thus far, we have not identified the Olig1-target genes that enhance cell motility. Vasilaki et al. previously reported that up-regulation of RhoB is involved in TGF-β-induced cell motility (32). However, we observed induction of RhoB even after knockdown of Olig1.3 Thus, Olig1 regulates expression of other genes required for TGF-β-induced cell motility. Intriguingly, knockdown of Olig1 resulted in reduced up-regulation of PAI-1 and Smad7 after TGF-β stimulation (Fig. 3E). These genes have recently been implicated in TGF-β-induced cell motility (33, 34). Attenuated up-regulation of these genes may collectively affect cell motility in response to TGF-β stimulation. Alternatively, the relevant genes may not be direct targets of the Smad-Olig1 transcriptional complex because TGF-β enhances cell motility not in the early phase but rather in the late phase.
Another outstanding question pertains to the mechanisms of regulation of the cooperative actions of Smad and specific Smad cofactors. Previously, we found that cooperative action of Olig1 and Smad2/3 is regulated by a helix-loop-helix protein, Maid, which sequesters Olig1 and inhibits its association with Smad2/3 (17). In this study, we found that Pin1 also regulates the cooperative action of Smad2/3 and Olig1. Pin1 is a peptidyl-prolyl cis/trans-isomerase that isomerizes phosphorylated Ser/Thr-Pro bonds, thereby inducing conformational changes in substrate proteins (35). Pin1 interacts with Smad2/3 through the phosphorylated linker region and induces a conformational change. The conformational change is required for association of Smad3 with Olig1, probably through releasing the L3 loop of the MH2 domain from repression by the linker region, leading to enhancement cell motility.
Importantly, Pin1 appears to be required for only a subset of TGF-β target genes, because its knockdown affected induction of neither p21WAF nor Snail (Fig. 3E). DNA microarray analysis using NMuMG cells 1 h after TGF-β stimulation also revealed considerable overlap between Olig1-regulated genes and Pin1-regulated genes (Fig. 3F). Matsuzaki et al. previously reported that expression of MMP-9 requires linker phosphorylation of Smad2 (36). Phosphorylation of Smad2/3 in the linker region may affect regulation of TGF-β target genes via Pin1-dependent association with a subset of Smad cofactors.
We previously reported that Pin1 negatively regulates Smad signaling by promoting proteasome-mediated degradation of Smad proteins, in MDA-MB-231, HT1080, and human embryonic kidney 293 cells (19). Subsequently Matsuura et al. reported that Pin1 is required for cell migration, but not cytostasis, induced by TGF-β, suggesting cell response-selective effects of Pin1 (25). In this study, we elucidated the mechanism by which Pin1 preferentially affects cell migration. Together, these findings indicate that activation by Pin1 allows Smad2/3 to cooperate with Smad cofactors, including Olig1, and simultaneously sensitizes it for proteasomal degradation. Depending on contexts of target cells, however, one of these effects may dominate. Pin1 would more efficiently down-regulate Smad2/3 signaling in cells where the activity of ubiquitin ligases targeting Smad2/3 is very high. Levels of Pin1 in different cell lines may also affect the balance of these effects.
Although inhibitors of TGF-β signaling, including receptor kinase inhibitors, neutralizing antibodies, and antisense oligonucleotides have been developed for possible clinical uses, these compounds inhibit TGF-β signaling comprehensively and may therefore adversely affect beneficial as well as detrimental outcomes of TGF-β signaling. To avoid such side effects, it would be useful to develop TGF-β inhibitors selective for specific cellular responses (13). In this study we successfully inhibited TGF-β-induced cell migration/motility by using a peptide derived from the L3 loop of Smad3 (P-L3S3). Cui et al. previously described a Smad-binding aptamer based on Smad-binding sequences derived from FoxH1, Lef1, and CREB-binding protein (37). They observed attenuated induction of PAI-1, but not Smad7, by an aptamer containing the Smad-interacting motif from Xenopus FoxH1, but they did not examine effects of the aptamers on Smad-Smad cofactor interaction or specific cellular responses. These earlier data, in conjunction with our findings from this study, strongly suggest that blocking interactions between Smad proteins and their cofactors represents a promising strategy for selective regulation of TGF-β signaling.
The L3 loop is also involved in recognition of type I receptors by R-Smad proteins (27). Thus, it appeared possible that the P-L3S3 peptide might competitively inhibit Smad phosphorylation by TGF-β type I receptor (ALK-5). We found, however, that the peptide did not affect TGF-β-induced Smad phosphorylation. This result suggests that the interaction between Smad2/3 and ALK-5 is strong enough to overcome competition from the peptide, possibly because the interaction between Smad2/3 and ALK-5 is enhanced by the Smad2/3-presenting membrane protein, SARA (Smad anchor for receptor activation) (38).
Chen et al. previously identified the determinant in Smad2 responsible for physical interaction with FoxH1 as a motif spanning residues Gln-364–Arg-365–Tyr-366 (corresponding to residues 322–324 in Smad3) (39). We also identified the c-Ski-interacting region in Smad3 as Ser-266–Glu-267 (24). Smad2 and Smad3 thus interact with cofactors, co-activators, and co-repressors through distinct regions on their surfaces (Fig. 8). c-Ski interacts with the Smad3 MH2 toroid at the upper (linker side) surface, whereas FoxH1 does so from the side (24), and Olig1 associates at the bottom surface. Because the FoxH1-binding site is shared by other transcription factors, Mixer and Milk (40), it is possible that the L3 loop is involved in interaction with Smad cofactors other than Olig1. Therefore P-L3S3 may not be a completely specific blocker of Smad-Olig1 cooperativity. Further investigation will be required to develop specific inhibitors of Smad2/3-Olig1 interaction.
FIGURE 8.

Interaction surfaces with cofactors on Smad3. Three-dimensional model of the MH2 domain of the phospho-Smad3–phospho-Smad3–Smad4 trimeric complex (Protein Data Bank ID code 1U7F) in ribbon format. Smad4 is not shown here. Smad3 proteins are shown in pink and dark pink. The N-terminal residue of the MH2 domain is shown in blue. Binding sites for FoxH1, c-Ski, and Olig1 are shown in yellow, red, and green, respectively. Dotted lines denote the amino acids residues of the linker region.
In this study, we validated the idea that selective regulation of TGF-β-induced cellular responses can be achieved by targeting Smad cofactors. Identification of Smad cofactors involved in other cellular responses that promote tumor malignancy, including EMT or maintenance of tumorigenicity of tumor-initiating cells, remains an important task for the future.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant 40209896, JSPS Core-to-Core Program “Cooperative International Framework in TGF-β Family Signaling” and the Fugaku Memorial Foundation.
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE46405).
M. Motizuki and K. Miyazawa, unpublished observation.
- EMT
- epithelial-mesenchymal transition
- BMP
- bone morphogenetic protein
- Olig1
- oligodendrocyte transcription factor 1
- PAI-1
- plasminogen activator inhibitor-1
- R-Smad
- receptor-regulated Smad.
REFERENCES
- 1. Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ikushima H., Miyazono K. (2010) TGFβ signalling: a complex web in cancer progression. Nat. Rev. Cancer 10, 415–424 [DOI] [PubMed] [Google Scholar]
- 3. Yoshimura A., Wakabayashi Y., Mori T. (2010) Cellular and molecular basis for the regulation of inflammation by TGF-β. J. Biochem. 147, 781–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biernacka A., Dobaczewski M., Frangogiannis N. G. (2011) TGF-β signaling in fibrosis. Growth Factors 29, 196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts A. B., Wakefield L. M. (2003) The two faces of transforming growth factor β in carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 8621–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bierie B., Moses H. L. (2006) Tumour microenvironment. TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520 [DOI] [PubMed] [Google Scholar]
- 7. Saitoh M., Miyazawa K. (2012) Transcriptional and post-transcriptional regulation in TGF-β-mediated epithelial-mesenchymal transition. J. Biochem. 151, 563–571 [DOI] [PubMed] [Google Scholar]
- 8. Ehata S., Johansson E., Katayama R., Koike S., Watanabe A., Hoshino Y., Katsuno Y., Komuro A., Koinuma D., Kano M. R., Yashiro M., Hirakawa K., Aburatani H., Fujita N., Miyazono K. (2011) Transforming growth factor-β decreases the cancer-initiating cell population within diffuse-type gastric carcinoma cells. Oncogene 30, 1693–1705 [DOI] [PubMed] [Google Scholar]
- 9. Peñuelas S., Anido J., Prieto-Sánchez R. M., Folch G., Barba I., Cuartas I., García-Dorado D., Poca M. A., Sahuquillo J., Baselga J., Seoane J. (2009) TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15, 315–327 [DOI] [PubMed] [Google Scholar]
- 10. Ikushima H., Todo T., Ino Y., Takahashi M., Miyazawa K., Miyazono K. (2009) Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5, 504–514 [DOI] [PubMed] [Google Scholar]
- 11. Naka K., Hoshii T., Muraguchi T., Tadokoro Y., Ooshio T., Kondo Y., Nakao S., Motoyama N., Hirao A. (2010) TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463, 676–680 [DOI] [PubMed] [Google Scholar]
- 12. Tang B., Yoo N., Vu M., Mamura M., Nam J. S., Ooshima A., Du Z., Desprez P. Y., Anver M. R., Michalowska A. M., Shih J., Parks W. T., Wakefield L. M. (2007) Transforming growth factor-β can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 67, 8643–8652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyazawa K., Shinozaki M., Hara T., Furuya T., Miyazono K. (2002) Two major Smad pathways in TGF-β superfamily signalling. Genes Cells 7, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 15. Schmierer B., Hill C. S. (2007) TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 [DOI] [PubMed] [Google Scholar]
- 16. Ikushima H., Miyazono K. (2010) Cellular context-dependent “colors” of transforming growth factor-β signaling. Cancer Sci. 101, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikushima H., Komuro A., Isogaya K., Shinozaki M., Hellman U., Miyazawa K., Miyazono K. (2008) An Id-like molecule, HHM, is a synexpression group-restricted regulator of TGF-β signalling. EMBO J. 27, 2955–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishii R., Isogaya K., Seto A., Koinuma D., Watanabe Y., Arisaka F., Yaguchi S., Ikushima H., Dohmae N., Miyazono K., Miyazawa K., Ishitani R., Nureki O. (2012) Structure of a dominant-negative helix-loop-helix transcriptional regulator suggests mechanisms of autoinhibition. EMBO J. 31, 2541–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakano A., Koinuma D., Miyazawa K., Uchida T., Saitoh M., Kawabata M., Hanai J., Akiyama H., Abe M., Miyazono K., Matsumoto T., Imamura T. (2009) Pin1 down-regulates transforming growth factor-β (TGF-β) signaling by inducing degradation of Smad proteins. J. Biol. Chem. 284, 6109–6115 [DOI] [PubMed] [Google Scholar]
- 20. Nagano Y., Koinuma D., Miyazawa K., Miyazono K. (2010) Context-dependent regulation of the expression of c-Ski protein by Arkadia in human cancer cells. J. Biochem. 147, 545–554 [DOI] [PubMed] [Google Scholar]
- 21. Goto K., Kamiya Y., Imamura T., Miyazono K., Miyazawa K. (2007) Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 282, 20603–20611 [DOI] [PubMed] [Google Scholar]
- 22. Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J. M. (1998) Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shirakihara T., Saitoh M., Miyazono K. (2007) Differential regulation of epithelial and mesenchymal markers by ΔEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol. Biol. Cell 18, 3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizuide M., Hara T., Furuya T., Takeda M., Kusanagi K., Inada Y., Mori M., Imamura T., Miyazawa K., Miyazono K. (2003) Two short segments of Smad3 are important for specific interaction of Smad3 with c-Ski and SnoN. J. Biol. Chem. 278, 531–536 [DOI] [PubMed] [Google Scholar]
- 25. Matsuura I., Chiang K. N., Lai C. Y., He D., Wang G., Ramkumar R., Uchida T., Ryo A., Lu K., Liu F. (2010) Pin1 promotes transforming growth factor-β-induced migration and invasion. J. Biol. Chem. 285, 1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aragón E., Goerner N., Zaromytidou A. I., Xi Q., Escobedo A., Massagué J., Macias M. J. (2011) A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 25, 1275–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lo R. S., Chen Y. G., Shi Y., Pavletich N. P., Massagué J. (1998) The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 17, 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y., Feng X. H., Derynck R. (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature 394, 909–913 [DOI] [PubMed] [Google Scholar]
- 29. Gomis R. R., Alarcón C., He W., Wang Q., Seoane J., Lash A., Massagué J. (2006) A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 12747–12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cordenonsi M., Dupont S., Maretto S., Insinga A., Imbriano C., Piccolo S. (2003) Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with Smads. Cell 113, 301–314 [DOI] [PubMed] [Google Scholar]
- 31. Koinuma D., Tsutsumi S., Kamimura N., Taniguchi H., Miyazawa K., Sunamura M., Imamura T., Miyazono K., Aburatani H. (2009) Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol. Cell. Biol. 29, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vasilaki E., Papadimitriou E., Tajadura V., Ridley A. J., Stournaras C., Kardassis D. (2010) Transcriptional regulation of the small GTPase RhoB gene by TGFβ-induced signaling pathways. FASEB J. 24, 891–905 [DOI] [PubMed] [Google Scholar]
- 33. Dellas C., Loskutoff D. J. (2005) Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb. Haemost. 93, 631–640 [DOI] [PubMed] [Google Scholar]
- 34. Ekman M., Mu Y., Lee S. Y., Edlund S., Kozakai T., Thakur N., Tran H., Qian J., Groeden J., Heldin C. H., Landström M. (2012) APC and Smad7 link TGFβ type I receptors to the microtubule system to promote cell migration. Mol. Biol. Cell 23, 2109–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liou Y. C., Zhou X. Z., Lu K. P. (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 36, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsuzaki K., Kitano C., Murata M., Sekimoto G., Yoshida K., Uemura Y., Seki T., Taketani S., Fujisawa J., Okazaki K. (2009) Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-β signal in later stages of human colorectal cancer. Cancer Res. 69, 5321–5330 [DOI] [PubMed] [Google Scholar]
- 37. Cui Q., Lim S. K., Zhao B., Hoffmann F. M. (2005) Selective inhibition of TGF-β responsive genes by Smad-interacting peptide aptamers from FoxH1, Lef1, and CBP. Oncogene 24, 3864–3874 [DOI] [PubMed] [Google Scholar]
- 38. Tsukazaki T., Chiang T. A., Davison A. F., Attisano L., Wrana J. L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 95, 779–791 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y. G., Hata A., Lo R. S., Wotton D., Shi Y., Pavletich N., Massagué J. (1998) Determinants of specificity in TGF-β signal transduction. Genes Dev. 12, 2144–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Germain S., Howell M., Esslemont G. M., Hill C. S. (2000) Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes Dev. 14, 435–451 [PMC free article] [PubMed] [Google Scholar]