

Background: Xanthohumol may be beneficial for obesity-related conditions, but mechanisms are unknown.
Results: XN lowers ROS and dysfunctional lipid metabolism in animals, and XN uncouples respiration and induces oxidant defense systems in myocytes.
Conclusion: XN may ameliorate metabolic syndrome by these mechanisms.
Significance: Metabolomics helped reveal potential XN anti-obesity mechanisms.
Keywords: Flavonoids, Mass Spectrometry (MS), Metabolism, Metabolomics, Obesity
Abstract
Mild, mitochondrial uncoupling increases energy expenditure and can reduce the generation of reactive oxygen species (ROS). Activation of cellular, adaptive stress response pathways can result in an enhanced capacity to reduce oxidative damage. Together, these strategies target energy imbalance and oxidative stress, both underlying factors of obesity and related conditions such as type 2 diabetes. Here we describe a metabolomics-driven effort to uncover the anti-obesity mechanism(s) of xanthohumol (XN), a prenylated flavonoid from hops. Metabolomics analysis of fasting plasma from obese, Zucker rats treated with XN revealed decreases in products of dysfunctional fatty acid oxidation and ROS, prompting us to explore the effects of XN on muscle cell bioenergetics. At low micromolar concentrations, XN acutely increased uncoupled respiration in several different cell types, including myocytes. Tetrahydroxanthohumol also increased respiration, suggesting electrophilicity did not play a role. At higher concentrations, XN inhibited respiration in a ROS-dependent manner. In myocytes, time course metabolomics revealed acute activation of glutathione recycling and long term induction of glutathione synthesis as well as several other changes indicative of short term elevated cellular stress and a concerted adaptive response. Based on these findings, we hypothesize that XN may ameliorate metabolic syndrome, at least in part, through mitochondrial uncoupling and stress response induction. In addition, time course metabolomics appears to be an effective strategy for uncovering metabolic events that occur during a stress response.
Introduction
Obesity results from an energy imbalance where intake exceeds expenditure. Given the increasing demand placed on health care systems by a rapidly growing obese population, there is great interest in developing a therapy to combat obesity and related disorders such as type 2 diabetes. To effectively reduce bodyweight and prevent obesity, therapeutic intervention must either increase energy expenditure or decrease energy intake or both. Mitochondria utilize macronutrients and downstream products to transport protons across the mitochondrial inner membrane and create an electrochemical potential, ΔΨ, which drives ATP synthesis. Uncoupling the dissipation of ΔΨ from ATP synthesis allows for the unchecked catabolism of dietary macronutrients and formation of heat. In the early 20th century, the mitochondrial uncoupler 2,4-dinitrophenol was effectively used as an anti-obesity agent given to patients at doses ranging from 3 to 5 mg/kg daily (1). 2,4-Dinitrophenol was eventually not deemed safe for clinical use due to a narrow therapeutic window and serious risks from overdose, including death. However, given the effectiveness of general mitochondrial uncouplers and quickly rising rates of obesity worldwide, there is renewed interest in developing a safe alternative to 2,4-dinitrophenol.
Separate from expending energy, an increase in basal mitochondrial uncoupling reduces reactive oxygen species (ROS)2 generation and is associated with longevity (2–4). Mild, acute uncoupling increases the expression of the transcriptional coactivator, peroxisome proliferator-activated receptor-coactivator 1α (PGC-1α), a dominant regulator of mitochondrial homeostasis in many tissues and a critical component of a cellular adaptive stress response (ASR) (5, 6). When a cell is challenged with an acute stress, it must quickly respond to counteract that stress to survive. The molecular adjustments made to cope with acute stress and the subsequent overcompensations together constitute an ASR. In the case of acute uncoupling, ATP levels drop but are normalized through increased expression of PGC1α (5). Low doses of toxic chemicals (e.g. 2,4-dinitrophenol) often activate ASR pathways but do not induce cell damage, allowing for a heightened ability to respond to subsequent stressors (7).
Mild ROS formation, reduced glutathione (GSH) depletion, and electrophilic stress are all triggers for induction of an ASR (8–10). In fact, there is recent evidence suggesting ROS and GSH depletion, under the right conditions, may actually prevent type 2 diabetes and increase longevity (11–14). Xanthohumol (XN) is a prenylated chalcone found in hops and beer and has an electrophilic group capable of inducing a stress response (10, 15). XN is perhaps best known for its cancer chemopreventive properties and its ability to trigger apoptosis through ROS (16, 17). Several reports are now starting to describe various anti-obesity properties of XN but with no well accepted mechanisms.
Our group recently reported that XN treatment decreased body weight gain and fasting plasma glucose in high fat fed Zuckerfa/fa rats, an established model of obesity and metabolic syndrome (18) (supplemental Fig. S1). In the present study, we sought to elucidate the mechanism(s) behind these changes using untargeted metabolomics. We analyzed the fasting plasma from the same Zucker rats treated with XN and found a global reduction in products of dysfunctional lipid metabolism, specifically dicarboxylic (DC) fatty acids and acylcarnitines (ACs). We also observed a decrease in hydroperoxy (HP) fatty acids, markers of oxidative stress. During fasting, DC fatty acids and ACs in plasma serve as reflections of incomplete fatty acid β-oxidation in skeletal muscle and are well characterized markers for assessing dysfunctional lipid metabolism (19, 20). These results prompted us to investigate the effects of XN on energy metabolism in muscle cells.
EXPERIMENTAL PROCEDURES
Chemicals
XN was isolated from female inflorescences of hops (Humulus lupulus) during previous work (21). Tetrahydroxanthohumol (TXN) was also from previous work (22). 8-Prenylnaringenin (8PNG) was synthesized by prenylation of naringenin (23). MnTMPyP (manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin) and EUK 134 were from ENZO Life Sciences (Farmingdale, NY). Oligomycin A and N-acetylcysteine (NAC) were from Sigma. Metabolite standards were from TCI America (Portland, OR) and, when unavailable, Sigma.
Zucker Rat Study
Rat plasma utilized in this study was obtained from a previous investigation that examined the effects of chronic dosing of XN on metabolic syndrome (18). In brief, 4-week-old Zucker obese fa/fa male and female rats received daily oral doses of XN at 0, 1.86, 5.64, and 16.9 mg/kg body weight for 6 weeks. All rats were maintained on a high fat (60% kcal) AIN 93G diet for 3 weeks to induce severe obesity followed by a normal AIN-93G (15% kcal fat) diet for the last 3 weeks of the study. Plasma was collected at sacrifice via a cardio-puncture after fasting overnight. All procedures were approved by and in accordance with ethical standards of Oregon State University's Institutional Animal Use and Care Committee (Protocol #3689).
LC-MS/MS-based Metabolomics
To extract plasma for metabolomics, plasma samples were thawed on ice. 80 μl of plasma was aliquoted into tubes, 240 μl of ice-cold 50:50 (v:v) MeOH:EtOH was added, and tubes were quickly vortexed 4 s. Samples were spun for 15 min at 13,000 × g at 4 °C, and the supernatant was analyzed by LC-MS/MS. To extract myocytes for time course metabolomics, media was aspirated, and cells were quickly washed once with warm Hanks' balanced salt solution. After aspirating Hanks' balanced salt solution, 400 μl of ice-cold 50:50 (v:v) MeOH:EtOH was quickly added to each well (6-well plate), then the plate was covered and placed in −80 °C freezer overnight. Cells were scraped then spun down for 15 min at 13,000 × g at 4 °C, and the supernatant was analyzed by LC-MS/MS. Metabolic profiling was performed as previously described, except column temperature was held at 50 °C (24). Importantly, the metabolite identification process was rigorous. Most metabolites were identified by mass, isotope distribution, MS/MS fragmentation pattern, and when standards were available, retention time. Further details can be found in the supplemental information.
Cell Culture and Treatment
Mouse C2C12 skeletal muscle cells and preadipocytes were first propagated in 75-cm2 flasks using a culture medium consisting of DMEM (Invitrogen), 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1 mm pyruvate. Brown preadipocytes were a kind gift from Dr. Bruce Spiegelman (Harvard Medical School) and harvested as described in Uldry et al. (25). 3T3-L1 preadipocytes and C2C12 cells were from ATCC (ATCC, Manassas, VA). For metabolomics experiments, C2C12 myocytes were plated in 6-well plates at a density of 500,000 cells per well. 24 h later the medium was replaced with fetal bovine serum (FBS)-free XF medium (Seahorse Bioscience, North Billerica, MA) with 10 mm glucose (25 mm glucose for preadipocytes) and 1 mm pyruvate, and cells were allowed to incubate 1 h at 37 °C. Cells were then treated with XN or vehicle, 0.5% EtOH. For all cell based assays, XF medium was replaced with complete medium 4 h after XN addition to maintain cell viability for later time points. The final concentration of FBS after XN treatment was 0.1%. For cell viability experiments, the cells obtained from the culture flask after trypsinization were seeded in 96-well plates at a density of 10,000 cells per well using the same growth medium. After 24 h of incubation at 37 °C in a humidified atmosphere of 5% CO2, the medium was removed, and the cells were rinsed once with Hanks' balanced salt solution (Sigma). Freshly prepared XF assay medium containing 10 mm glucose and 1 mm pyruvate was then added to each well, 180 μl/well. After 1 h of incubation at 37 °C, 20 μl of culture media (9 parts XF assay medium and 1 part complete DMEM medium) containing various concentrations of XN were added to the wells. Ethanol was added to the control wells not to exceed a final concentration of 0.5%. The final concentrations of XN were 2, 5, 8, 25, and 70 μm, all in quadruplicate wells, in the 96-well plate.
Respiration Measurements
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements were performed with a Seahorse XF 24 Analyzer (Seahorse Bioscience). Cells were plated 24 h before measurements at a density of 20,000 cells per well for C2C12 myocytes and preadipocytes and 30,000 cells per well for HepG2 hepatocytes. The ECAR measurements are highly sensitive to FBS in growth medium, and it is suggested that little or none be present. XN is highly nonpolar, and previous experiments suggest FBS should be present in media to dissolve XN. To overcome this problem, XN was first dissolved in vehicle (ethanol), and the resulting solution was diluted 20-fold in treatment medium (TM) (9 parts XF media and 1 part 10% FBS media) that was added directly to cells. The TM contained 1% FBS, and after a further 10-fold dilution into the media in wells, only 0.1% FBS was present during OCR and ECAR measurements. This amount appeared to dissolve up to 70 μm XN at 37 °C and was well below the 1% limit recommended by Seahorse Bioscience. This treatment procedure was also used for oligomycin A, and accordingly, nearly 0.2% FBS was present in experiments using both. The three antioxidants were dissolved directly into serum-free XF medium before addition to cells. XN, TXN, 8PNG, oligomycin A, MnTMPyP, EUK 134, and NAC were prepared as fresh solutions the day of each experiment.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 4.0 and the software environment, R. In regard to the statistical analysis for the Zucker rat metabolomics study, the proportional odds model used to assess the variation of the metabolite intensities across the four levels was adopted from Agresti (26).
RESULTS
XN Reduces Products of Dysfunctional Lipid Metabolism and ROS
Several studies suggest that XN treatment could be beneficial for obesity, metabolic syndrome, and type 2 diabetes. Our group recently performed a 6-week treatment study with XN to obese Zucker rats and found attenuated body weight gain and decreased fasting plasma glucose in treated rats (supplemental Fig. S1) (18). To investigate the mechanism(s) of action responsible, we performed an untargeted metabolomics experiment on fasting plasma from the Zucker rats treated with XN at four different doses: 1) 0 mg/kg/day (vehicle control), 2) 1.86 mg/kg/day (low dose), 3) 5.64 mg/kg/day (medium dose), and 4) 16.9 mg/kg/day (high dose). XN was administered orally once per day. The animal doses correspond to doses of 0, 20, 60, and 180 mg/day for a human weighing 64 kg (27). Metabolites of interest are shown in Fig. 1A. XN decreased fasting plasma levels of DC fatty acids, HP fatty acids, and one phosphatidylcholine (PC) (Fig. 1A). Amino acids were generally elevated in the high XN treatment group, but this change was specific to female rats (supplemental Fig. S2A).
FIGURE 1.

Metabolomics reveals a reduction in products of dysfunctional lipid metabolism and ROS in Zucker fatty rats treated with XN. A, significantly differing metabolites were initially defined as having a p < 0. 01 from a Student's two-tailed t test, control (n = 13) versus XN 16.9 mg/kg (n = 11). Related metabolites were identified, and statistical analysis taking into account all XN dose groups was performed. To incorporate all four groups (control, low dose, medium dose, high dose), we regarded the dose level as in an ordinal scale (control < low < medium < high) and used a proportional odds model to assess the variation of the metabolite intensities across these four levels. A likelihood ratio test was used to evaluate the significance of the metabolite variation across the four groups. The p values reported here arise from this analysis. Metabolites with a p < 0.01 from this test were considered metabolites of interest and are represented graphically with % change, XN 16.9 mg/kg versus control. PC, phosphatidylcholine. B, acylcarnitines were detected as a result of targeted data mining and respective % changes in 16.9 mg/kg XN-treated versus control Zucker rats. Fisher's method was used to combine individual p values and to test whether a group of metabolite intensities vary across different treatment groups. All acylcarnitines includes all carnitines represented here except carnitine and dehydroxy (DH) carnitine. Medium chain only includes only acylcarnitines 6:0, 8:0, 8:1, and 10:0. C, XN treatment decreased fasting plasma DC fatty acids and medium chain acylcarnitines, metabolites that result from dysfunctional lipid oxidation.
DC fatty acids in the plasma and urine are markers of dysfunctional or substrate-overloaded mitochondria during fasting. With XN treatment, we report decreases in six different DC fatty acids varying in chain length and degrees of unsaturation (Fig. 1A). Five of the six were decreased >40% compared with controls (Fig. 1A). HP fatty acids are well characterized products of ROS and precursors to reactive lipid aldehydes. XN decreased the HP fatty acids 18:2 and 18:3 by 40 and 30%, respectively, suggesting lower levels of ROS production or an enhanced capacity to scavenge ROS in treated rats (Fig. 1A).
Although DC fatty acids are associated with dysfunctional mitochondrial β-oxidation, plasma ACs are often measured instead, as their detection is sensitive with mass spectrometry due to a constitutive positive charge and because they offer easy identification through a common, dominant, collision-induced fragment near m/z 85.028. We were able to target and identify 30 previously characterized ACs in our metabolomics data and, in agreement with the DC fatty acids, found that most were decreased with XN treatment (Fig. 1B). Although no single AC had a p < 0.01 from a Student's t test, the comprehensiveness of the decrease could not be ignored. Fisher's grouping analysis revealed a significant decrease in global (p = 0.0009) as well as medium chain ACs (p = 0.0091) (Fig. 1B). Targeting free fatty acids, precursors to ACs, in the metabolomics data revealed a global downward trend, albeit insignificant (supplemental Fig. S2B). A heat map, generated from correlations between metabolite levels across the four dose levels by computing the pairwise Pearson's correlation, displayed a high correlation among ACs and among DC fatty acids highlighting the concerted changes within lipid classes (supplemental Fig. S2C). Parameters used to identify individual metabolites can be found in supplemental Table S1. Because muscle tissue is the major consumer of fatty acids during fasting, we hypothesized the decrease in plasma ACs and DCs was a reflection of altered muscle metabolism. To test this, we treated muscle cells with XN and used bioenergetic and metabolomic approaches to uncover its effects.
XN Increases OCR and at High Concentrations Decreases OCR through ROS
Electrons are transferred along the mitochondrial electron transport chain (ETC) where they ultimately split molecular oxygen (O2) and water is formed. The energy gained from this process is used to transfer protons and generate ΔΨ. Mitochondria are responsible for 70–90% of total cellular O2 consumption, and as such changes in cellular O2 consumption largely reflect changes in mitochondrial O2 consumption. At 5 μm, XN increased the OCR in C2C12 mouse skeletal muscle cells by roughly 30% compared with controls (Fig. 2A). XN (5 μm) also led to an increase in the ECAR, a measure of glycolytic ATP production (Fig. 2B). Together, these findings suggest a compensatory increase in energy generation in response to XN treatment.
FIGURE 2.

XN has a hormetic effect on respiration and glycolytic energy production. A, 5 μm XN acutely increases OCR in C2C12 mouse myocytes by 25% (p = 0. 0037, Student's two-tailed t test, n = 10). B, 5 μm XN increased the ECAR by 20% (p = 0.0041, 3 h post treatment, Student's two-tailed t test, n = 10). C, shown is the dose-dependent effect of XN on myocyte OCR. D, shown is the dose-dependent effect of XN on myocyte ECAR 2 h post treatment. E, shown is the effect of antioxidant pretreatment on the 70 μm XN induced decrease in OCR; An, antioxidant. In C–E, data are represented as the mean ± S.E., n = 4.
Interestingly, at concentrations above 8 μm, XN inhibited OCR and ECAR almost completely (Fig. 2, C and D). When these cells were treated with XN at concentrations between 5 and 8 μm, inhibition of OCR was moderate, and ∼50% inhibition was achieved with 6.5 μm (supplemental Fig. S3A). ECAR was still completely inhibited after 3 h at XN concentrations above 5 μm; however, the rate of ECAR decline was slower at lower concentrations, and 2 h post XN treatment, a noticeable dose-dependent inhibition with an IC50 close to 6.5 μm could be observed (supplemental Fig. S3B). Although some studies suggest flavonoids inhibit electron transport by directly binding to ETC complexes, it has also been shown that XN promotes mitochondrial superoxide formation and, separately, that ROS can inhibit ETC activity (17, 28). To determine if ROS played a role in the XN-induced OCR decrease, myocytes were pretreated with antioxidants. The superoxide dismutase (SOD)/peroxynitrite dismutase mimetic MnTMPyP and the SOD/catalase mimetic EUK 134 both prevented the acute decrease in OCR caused by 70 μm XN and allowed for a substantial increase (Fig. 2E). However, the well characterized antioxidant NAC (a relatively poor scavenger of superoxide) did not prevent the decrease in OCR, implicating superoxide or peroxynitrite as possible culprits (Fig. 2E). Neither EUK 134 (50 μm) nor MnTMPyP (50 μm) affected OCR, but a relatively low (compared with other studies) concentration of NAC (0.5 mm) was used because higher concentrations greatly decreased respiration (supplemental Fig. S4). These results suggest XN inhibits the ETC and OCR through the production of ROS.
XN Increases Uncoupled Respiration
The majority of O2 consumed inside cells is coupled to ATP synthesis. To test whether the increase in OCR due to XN was coupled to ATP production, we pharmacologically inhibited ATP synthase with oligomycin A before XN treatment. In the presence of oligomycin A, XN increased OCR 2-fold, indicative of mitochondrial uncoupling (Fig. 3A). This effect was even more pronounced at 70 μm XN but only with antioxidant pretreatment (Fig. 3B).
FIGURE 3.

XN acutely increases uncoupled respiration in muscle and liver cells. A, shown is the effect of 5 μm XN on the rate of uncoupled respiration in myocytes; n = 5. O = oligomyxin, X = xanthohumol. B, shown is the effect of 70 μm XN with antioxidant pretreatment on the rate of uncoupled respiration in myocytes; n = 5. M = MnTMPyP. C, shown is the dose-dependent effect of XN on hepatocyte (HepG2 cells) OCR; n = 4. D, shown is the effect of antioxidant pretreatment on the XN-induced decrease in OCR in hepatocytes; n = 4, M = MnTMPyP. E, shown is the effect of 70 μm XN with antioxidant pretreatment on the rate of uncoupled respiration in hepatocytes; n = 5.
Mitochondria are present in almost every cell type and provide a ubiquitous target for energy expenditure. As such, non-selective mitochondrial uncouplers increase respiration in most cell types limited largely by their ability to reach their target. We treated HepG2 hepatoma cells as well as brown and white preadipocytes with XN to assess the effect on OCR in different cell lines. XN increased OCR in HepG2 cells but only when pretreated with an antioxidant, perhaps due to a heightened sensitivity to ROS in this cell line compared with C2C12 myocytes (Fig. 3, C and D). But similar to the myocytes, the increase in respiration in HepG2 cells was not coupled to ATP synthesis (Fig. 3E). XN also increased uncoupled respiration in white and brown preadipocytes (Fig. 4A). And again, at higher concentrations, XN inhibited respiration through ROS (Fig. 4, B and C).
FIGURE 4.

XN acutely increases uncoupled respiration in preadipocytes. A, shown is the effect of 8 μm XN on the rate of uncoupled respiration (70 min after XN) in white (WA) and brown preadipocytes (BA). B, shown is the effect of 70 μm XN with antioxidant pretreatment on the rate of uncoupled respiration in white preadipocytes. C, shown is the effect of 70 μm XN with antioxidant pretreatment on rate of uncoupled respiration in brown preadipocytes. Data are represented as the mean ± S.E., n = 5, and p values are from a Student's two-tailed t test.
XN Electrophilicity Is Not Necessary for Effects on Respiration
Uncoupling protein 3 (UCP3) and adenine nucleotide translocase are both mitochondrial proteins that contribute to a basal level of proton leak and mitochondrial uncoupling in muscle (29). Both possess exposed thiol groups that can affect enzyme activity upon modification (1). UCP3 activity is in part regulated by glutathionylation and adenine nucleotide translocase by thiol oxidation (30, 31). Furthermore, the electrophilic lipid peroxidation product, 4-hydroxy-2(E)-nonenal, can uncouple respiration, likely through UCPs and adenine nucleotide translocase (32). Because XN also has an electrophilic group capable of thiol alkylation, we hypothesized that XN may act in a manner similar to 4-hydroxy-2(E)-nonenal. To test this idea, we utilized the hydrogenated product of XN, TXN, which lacks the electrophilic α,β-unsaturated ketone functionality of XN (Fig. 5A). Treatment of myocytes with TXN resulted in an increase in OCR at low concentrations and a decrease at higher concentrations, similar to the hormetic dose response observed with XN (Fig. 5B). However, TXN was more effective at increasing respiration compared with XN, resulting in a similar increase in OCR but at less than half the concentration. 8PNG is an estrogenic in vivo metabolite of XN that had little effect on myocyte OCR, even at 12 μm (Fig. 5C).
FIGURE 5.

Effects on respiration are not dependent on electrophilicity. A, shown are structural differences between XN and a dehydrogenation product, TXN. B, shown is the dose-dependent effect of TXN on myocyte OCR. C, shown is the dose-dependent effect of 8PNG on myocyte OCR. Data are represented as the mean ± S.E., n = 4.
Time Course Metabolomics Reveals a Catabolic Phenotype and Induction of an ASR
XN (5 μm) uncoupled respiration and activated glycolysis in myocytes for several hours, indicating a highly catabolic state inside the cell (Fig. 2, A and B). To capture the associated, acute metabolic changes as well as long term changes in metabolism, we performed untargeted, time course metabolomics on myocytes treated with XN (5 μm). To provide an accurate correlation between the myocyte metabolome and the previous respiration measurements, experimental conditions for the two separate assays were kept similar. Serum-free medium is required for the OCR/ECAR assays and was, therefore, also used for the metabolomics experiments. However, serum-free medium was replaced with 10% FBS medium at 4 h post treatment to maintain cell viability for later time points (Fig. 6A). Cells were extracted at eight different time points after XN addition, from 10 min to 48 h (Fig. 6A). Results were not normalized because cell viability and cellular protein content were unaffected by concentrations less than 25 μm XN (supplemental Fig. S5, A and B). Significantly differing metabolites (Student's t test, control versus 5 μm XN, >2-fold change, p < 0.01, for any time point) are displayed at their most significant time point (time point with greatest -fold change with p < 0.01) in Fig. 6B.
FIGURE 6.

Untargeted, time course metabolomics of muscle cell response to XN. A, shown is the experimental design and relative cellular XN levels of the time course metabolomics assay. B, shown are metabolites with p < 0.01 from Student's two-tailed t test and -fold change > 2 at any time point. The time point displayed here is different for each metabolite and is the time point with the greatest -fold change with a p value < 0.01.
The largest change observed in XN-treated cells was a 10.5-fold increase in GSH (Fig. 6B). GSH represents the main endogenous antioxidant inside cells, and when ROS increases, GSH is consumed, and oxidized glutathione (GSSG) is produced (33). GSSG is recycled enzymatically back to GSH in a NADPH-dependent manner to maintain the GSH/GSSG ratio. After XN addition, we observed an acute increase in GSH as well as a decrease in GSSG, suggesting acute activation of GSSG recycling systems (Fig. 7A). At 16 h, GSH and GSSG were elevated 10.5- and 1.7-fold respectively, indicating up-regulated GSH synthesis separate from recycling (Fig. 7B). The GSH/GSSG ratio was elevated in XN-treated myocytes during the entire experiment (Fig. 7C). In contrast, cysteine glutathione disulfide (cySSG) was decreased during the entire experiment (Fig. 7C). cySSG is a product of ROS, cysteine, and GSH but may also serve as a GSH reservoir in times of need (34, 35). We observed a highly significant, inverse correlation between cySSG and GSH, but only in XN-treated cells, supporting this notion (Fig. 7D).
FIGURE 7.
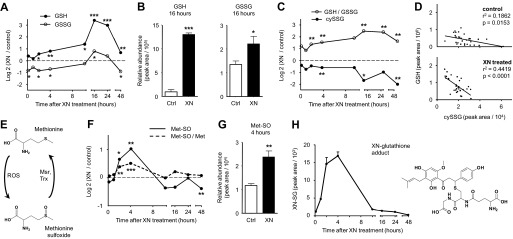
XN induces oxidative and electrophilic stress and an adaptive response. A, shown is the time course response of reduced (GSH) and oxidized (GSSG) glutathione after 5 μm XN treatment. B, shown are GSH and GSSG levels 16 h after 5 μm XN treatment. C, shown is the time course response of GSH/GSSG ratio and cysteine-glutathione disulfide (cySSG) after XN treatment. D, shown is the correlation between GSH and cySSG levels in XN-treated and control myocytes. All time points are included. E, methionine can be chemically oxidized to methionine sulfoxide by ROS but is enzymatically reduced back to methionine by methionine sulfoxide reductase (Msr) in a thioredoxin (Trx)-dependent manner. F, shown is the time course response of methionine sulfoxide with and without normalization to free methionine (Met) after 5 μm XN treatment. G, shown are methionine sulfoxide (Met-SO) levels 4 h after 5 μm XN treatment. H, temporal changes in cellular levels of a XN-glutathione conjugate (XN-SG) after 5 μm XN treatment are shown. In B and G data represented are the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001, from Student's two-tailed t test.
Methionine is highly sensitive to oxidation by ROS. Its mild oxidation product, methionine sulfoxide (Met-SO), is enzymatically recycled back to methionine by methionine sulfoxide reductase (Msr) in a thioredoxin (Trx)-dependent manner. Therefore, Met-SO may serve as a sensitive marker for acute induction of ROS and their effective removal (Fig. 7E) (36). XN induced a transient increase in Met-SO with maximal levels at the 4-h time point (Fig. 7, F and G). The increase was less apparent when normalized to free methionine but still significant (Student's t test, p < 0.001) (Fig. 7F).
The electrophilicity of XN and the observed changes in glutathione metabolism prompted us to search for a XN-glutathione adduct (XN-SG). XN-SG was detected and identified by mass, isotope distribution, and fragmentation pattern where the [GS]− ion was the dominant fragment (supplemental Fig. S6). Furthermore, the time course profile is similar to that of free XN (Figs. 6A and 7H).
Aside from changes in metabolites associated with oxidative and electrophilic stress, we uncovered alterations in metabolites reflective of energy stress and subsequent adaptation. Ophthalmic acid (OP) is a GSH analog and is identified as a potential marker of oxidative stress in liver, increased when GSH is lower (37). In our case, OP and norophthalmic acid were both decreased when GSH was increased but not until 16 h after XN exposure (Fig. 8A). In fact, both OP and norophthalmic acid were unchanged or increased up to the 10 h time point and had similar time course profiles to several uncharacterized di- and tripeptides (Fig. 8, A and B). Six peptides (tentatively identified by MS as three tripeptides and three dipeptides) in total were uncovered as significantly differing metabolites (Fig. 8, A and B). Interestingly, all six increased the first 4 h after XN treatment but were sharply decreased at 24 h (Fig. 8, A and B).
FIGURE 8.

XN affects protein degradation, cofactor, and energy metabolism. A, shown is the time course response of three tripeptides after XN treatment. All three tripeptides were significantly increased (p < 0.05) 4 h post XN treatment, and all were decreased (p < 0.01) at 24 h. B, shown is the time course response of three dipeptides after XN treatment. All three tripeptides were significantly decreased (p < 0.01) 24 h post XN treatment. C, shown is the time course response of pyridoxine and thiamine after XN treatment. D, shown is the correlation between thiamine and pyridoxine levels in XN-treated and control myocytes. All time points are included. E, shown is the time course response of folic acid, nicotinamide, and pantothenic acid after XN treatment. F, shown is the time course response of AMP and ADP after XN treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001, from Student's two-tailed t test.
We observed an acute decrease in the active B vitamin precursors thiamine and pyridoxine after XN treatment (Fig. 8C). Thiamine and pyridoxine levels were positively correlated in treated and untreated samples (Fig. 8D). For most time points, both of these B vitamins were lower in XN-treated cells, and accordingly, the treated samples occupy the lower left portion of the plot (Fig. 8D). Folic acid and nicotinamide were also lower in XN-treated myocytes, whereas pantothenic acid was unchanged (Fig. 8E).
It may also be worth mentioning that adenosine phosphates were altered by XN. ADP was elevated 4.7-fold 16 h after XN treatment (Fig. 8F). AMP levels were mildly altered (Fig. 8F).
The remaining findings, depicted in Fig. 9, are the result of a targeted data mining effort specifically focusing on metabolites central to energy metabolism. Metabolites targeted include those from glycolysis, the tricarboxylic acid (TCA) cycle, phosphocreatine metabolism, amino acids, and lipids (supplemental Fig. S7). The parameters used to identify individual metabolites can be found in supplemental Table S2. Overall, these changes appear to reflect a state of acute energy stress and hint toward induction of protein degradation (Fig. 9; supplemental Fig. S7). These changes and their implications are discussed in detail below.
FIGURE 9.
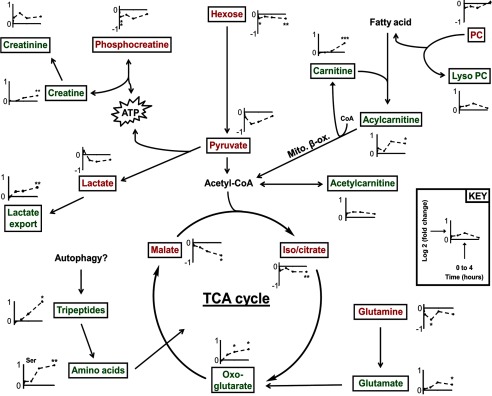
XN induces a transient catabolic phenotype. Because XN uncoupled respiration, several pathways and molecules central to muscle energy metabolism (e.g. glycolysis, TCA cycle, phosphocreatine) were targeted during data analysis. Time course profiles of the detected metabolites for the first 4 h post XN treatment (10 min, 1 h, 2 h, 4 h) are shown here. Green lettering, metabolite trending up after XN treatment. Red lettering, metabolite trending down after XN treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001, from Student's two-tailed t test.
DISCUSSION
The most commonly used plasma markers of dysfunctional β-oxidation in type 2 diabetes are ACs, thought to be byproducts of mitochondrial dysfunction and fatty acid overload in skeletal muscle (19, 38). During fasting, fatty acids are released into the bloodstream for uptake and mitochondrial β-oxidation by peripheral tissues (39). Skeletal muscle constitutes about 40% of total body mass and is the major consumer of fatty acids during fasting (40). The presence of dysfunctional or too few mitochondria in skeletal muscle will result in high rates of incomplete fatty acid oxidation and elevated levels of ACs in plasma (19, 41). Decreasing levels of these species will either alleviate mitochondrial dysfunction or their reduction serves as a marker of prior alleviation of such dysfunction. We found that XN-treated Zucker rats had globally decreased levels in plasma ACs, but individual ACs failed to reach statistical significance with a Student's t test (Fig. 1B). However, Fisher's grouping analysis of the ACs and separately the medium chain ACs did result in statistical significance for both groupings.
The most significant change in our metabolomics data, however, appears to be the decrease in DC fatty acids, also a marker of dysfunctional lipid metabolism although typically measured in the urine for diagnosis of inherited β-oxidation disorders (42) (Fig. 1A). DC fatty acids are endogenous products of cellular fatty acid metabolism, first ω-hydroxylated by endoplasmic reticulum-associated enzymes then further oxidized in the peroxisome (43). DC fatty acids are normally present at relatively low levels, but their production increases when fatty acid supply is excessive or mitochondrial β-oxidation is dysfunctional (44). DC fatty acids are able to undergo β-oxidation in the peroxisome or mitochondria or they can be excreted in the urine due increased hydrophilicity (43). The decrease in DC fatty acids and other lipid species with XN treatment strongly hints toward amelioration of mitochondrial dysfunction in skeletal muscle (Fig. 1C). Also, given the small degree of intergroup variation observed in DC fatty acids relative to ACs, fasting levels of DC fatty acids may serve as a more consistent indicator for mitochondrial dysfunction, especially when group size is small.
Hydroxylated chalcones have previously been implicated as mitochondrial uncouplers (45). Prenylated derivatives such as XN may be more effective uncouplers as they are highly nonpolar and easily diffuse across lipid bilayers. This notion taken together with our metabolomics data prompted us to investigate the effects of XN on respiration and energy metabolism in muscle cells. We found that XN acutely increased uncoupled respiration in muscle cells, and at 5 μm this effect was significant (Figs. 2A and 3A). The effective concentration of XN in cell culture (2–5 μm) was similar to levels observed in Zucker rats (1–3 μm in liver, <1 μm in plasma) (18, 46). Interestingly, XN at 8 μm and above inhibited respiration in myocytes (Fig. 2, C and D). Pharmacological intervention with antioxidants MnTMPyP and EUK 134, but not NAC, completely prevented the decrease in OCR induced by 70 μm XN, hinting toward superoxide- or peroxynitrite-mediated inhibition of respiration (Fig. 2E). XN also increased uncoupled respiration in HepG2 hepatoma cells, but at higher concentrations, and only with antioxidant (MnTMPyP) pretreatment (Fig. 3, D and E). XN had a similar effect in white and brown preadipocytes as in myocytes; low levels increased uncoupled respiration, and high levels inhibited respiration in a ROS-dependent manner (Fig. 4). It was previously hypothesized that XN inhibits respiration in cancer cell lines by directly binding the Fe centers of the ETC (17). Our results suggest the inhibitory effects of XN on respiration may simply stem from ROS production.
To test if the electrophilicity of XN played a role in the OCR increase or decrease, we treated myocytes with TXN, a hydrogenation product of XN that is not electrophilic (Fig. 5A). TXN had a similar dose-response curve to that of XN but was equally effective at increasing OCR at less than half the concentration of XN (Fig. 5B). This increased efficacy likely results from a lack in protein thiol interactions due to the absence of electrophilicity. From these results we conclude that the α,β-unsaturated ketone functionality of XN is not required for the acute effects on respiration. 8PNG is a potent phytoestrogen and an in vivo metabolite of XN. However, 8PNG failed to have any effect on OCR in myocytes (Fig. 5C).
Post translational signaling events such as protein phosphorylation, glutathionylation, and thiol modification aid in the acute regulation of metabolism and respiration and help facilitate the stress response (47–50). To monitor all the modifications to proteins involved in energy metabolism would require an amazing effort. Metabolomics allows for a relatively easy correlation to cellular phenotype due to the lack of modifications, such as those made to proteins after translation (51). In muscle cells, XN (5 μm) acutely uncoupled respiration and increased ECAR, reflecting an elevated state of energy production. To uncover the associated changes in metabolism, we performed a time course metabolomics experiment on myocytes treated with XN (Fig. 6A). Unexpectedly, glutathione recycling was immediately increased, and synthesis was greatly elevated 16 h after XN exposure, when GSH was increased 10.5-fold (Fig. 7, A and B). The oxidation product of GSH, cySSG, has previously been hypothesized to act as a reservoir for GSH (35). We observed a highly significant (p < 0.0001) correlation between cySSG and GSH but only in XN-treated cells (Fig. 7C). These results support the role of cySSG as a GSH reservoir during times of stress. Activation of the ASR requires Nrf2, which is critical for glutathione recycling and targeted degradation of damaged proteins (52). Because XN can prevent inactivation of Nrf2 via Keap1 adduction, these changes may be a reflection of increased Nrf2 activity. We were able to detect the XN-SG adduct, a marker of electrophilic stress (Fig. 7H).
ROS can also modify the sensitive Keap1 thiols allowing for Nrf2 release and activation. Our respiration experiments indicate that high concentrations of XN inhibit respiration through ROS. Metabolomics revealed that XN (5 μm) transiently increased Met-SO, a product of methionine and ROS (Fig. 7E). Met-SO was elevated 2-fold at 4 h but normalized by 10 h (Fig. 7, F and G). To try and account for changes due to protein degradation, we normalized Met-SO to free methionine levels and found a slightly blunted but still significant (p < 0.0001) increase at 4 h (Fig. 7F). These findings, however, do not account for selective degradation of proteins with high levels of Met-SO.
Ophthalmic acid is a GSH analog lacking the thiol and is a potential marker of oxidative stress (37). Although OP and its analog norophthalmic acid were both found to be decreased long term when GSH was greatly elevated, their overall time course profiles were strikingly similar to those of several uncharacterized peptides, increasing roughly 2-fold over the first 4 h then dropping 3–4-fold below control levels 24 h post XN treatment (Fig. 8, A and B). In the context of this experiment, we believe these peptides are products of incomplete protein degradation, potentially both proteasomal and lysosomal. In general, peptides were initially increased (presumably to support the enhanced respiration) followed by an even greater decrease (presumably due to up-regulated protein synthesis and organelle biogenesis) (Fig. 8, A and B). In addition, given the consistency in the profiles of these peptides over time, we propose that their monitoring by LC-MS/MS may provide a small molecule-based method for the determination and tracking of cellular autophagy. Because the proteases in the cytosol and lysosomes have different amino acid specificities, we hypothesize differential di- and tripeptides may arise from the two processes. This idea is being explored.
ADP was increased ∼4-fold 16 h post treatment (Fig. 8F). As an adaptation to exercise, the mitochondrial affinity for cytosolic ADP in muscle decreases as the myofibers become more oxidative (53). It is tempting to speculate that the long term increase we observed in ADP may be related to this switch.
Also of interest, we observed a decrease in several precursors to active B vitamins, cofactors necessary for the catabolism of macronutrients. Thiamine and pyridoxine are B vitamin precursors to the biologically active cofactors, thiamine pyrophosphate and pyridoxal phosphate. Thiamine pyrophosphate and pyridoxal phosphate are cofactors for many enzymatic reactions and are required for the degradation of fatty acids, sugars, and amino acids. We observed an acute decrease in these precursors with XN treatment, but their levels were correlated regardless of treatment, suggesting constitutive, coordinate regulation (Fig. 8D). Similar to the other B vitamins, nicotinamide and folic acid were lower in XN-treated myocytes over the first 4 h (Fig. 8E). We hypothesize that these changes are a reflection of up-regulated, active B vitamin production for use as cofactors in catabolic reactions.
An acute increase in OCR and ECAR, indicating a catabolic state within the cell, was accompanied by a variety of alterations in energy metabolism. Creatine kinase catalyzes the transfer of a high energy phosphate from phosphocreatine to ADP, resulting in ATP and creatine. The maximal rate of ATP synthesis through the creatine kinase system is much higher than oxidative phosphorylation, and as such, phosphocreatine serves as an energy reservoir that supports acute energy depletion (54). After XN treatment, time course metabolomics revealed activation of the acute compensatory creatine kinase system, indicated by a decrease in phosphocreatine at 10 min. Creatine did not increase until 4 h post treatment. However, the spontaneous intramolecular condensation product of creatine, creatinine, increased immediately (p > 0.05). Lactate production from pyruvate also produces ATP and is another mechanism for muscle to quickly recover from energy depletion. Although lactate and pyruvate were both decreased for the first 4 h (p > 0.05), we observed a sustained increase in ECAR, a measure of cellular lactate export and glycolytic activity (Fig. 2B). Furthermore, intracellular hexose (our method cannot distinguish glucose from other hexoses) was decreased short term, perhaps due to enhanced glucose utilization.
Under conditions of energy depletion, cells maintain energy status by selectively degrading macronutrient complexes including proteins and organelle membranes, a process known as autophagy. In XN-treated myocytes, metabolomics revealed a transient increase in several uncharacterized di- and tripeptides as well as the majority of amino acids, indicative of protein degradation and amino acid mobilization for entry into the TCA cycle. Free glutamine is present in muscle at high concentrations and is a major amino acid feeding the TCA cycle (55, 56). Glutamine is first converted to glutamate, which can then produce the TCA cycle intermediate oxoglutarate. Interestingly, glutamine was the only amino acid that significantly decreased in the first 4 h, perhaps due to a higher rate of utilization than production (supplemental Fig. S7).
To undergo β-oxidation, fatty acyl CoA species are converted to ACs for transport into the mitochondria. Plasma ACs increase during exercise due to an elevation in fatty acid supported respiration in muscle (57). Again in agreement with a state of acutely increased respiration, we observed modest increases in both acetylcarnitine and long chain ACs in the first 4 h post treatment. Possibly due to increased demand and synthesis, carnitine was elevated after 4 h. Perhaps most importantly, these results highlight the dynamic nature of the cellular stress response and suggest metabolomics may be an effective strategy for studying this response.
Together, our cell culture experiments suggest XN has three general, acute effects: mitochondrial uncoupling, ROS generation, and electrophilic adduction (Scheme 1). Although the ROS production and electrophilic adduction are previously documented, the uncoupling activity of XN has not been. Separately, these mechanisms are capable of activating an ASR, and their simultaneous occurrence, as observed with XN treatment, may ensure this induction. For example, Nrf2 is activated by both ROS and electrophile adduction, whereas PGC1α is activated by a decreased energy status. AMP-activated protein kinase is a central regulator of cell metabolism, acting in part through PGC1α, and is itself activated through multiple mechanisms including decreased energy status and ROS. Energy expenditure could also be enhanced and ROS emission decreased through increased UCP3 expression, which can be mediated by both PGC1α- and AMP-activated protein kinase in muscle. These proteins together constitute a driving force of the ASR and result in enhanced ROS scavenging capacity, autophagy and mitophagy, and increased mitochondrial biogenesis and oxidative capacity. We propose that the beneficial effects observed in XN-treated Zucker rats stem at least in part from these three general mechanisms and subsequent activation of adaptive cellular processes (Scheme 1).
SCHEME 1.

Proposed downstream cellular responses to acute effects of XN. Three general effects of XN include electrophilic adduction, ROS generation, and mitochondrial uncoupling. Uncoupling induces energy expenditure and mitophagy. A decreased energy status activates proteins central to the adaptive stress response including AMP-activated protein kinase and PGC1α. Separately, ROS generation and electrophilic adduction can activate AMP-activated protein kinase (AMPK) as well as Nrf2. Activation of these proteins leads to an array of adaptive cellular processes thought to enhance mitochondrial and cellular function.
Although XN treatment reduced Zucker rat body weight gain in a previous study by our group, it is important to note this change was only significant in males (supplemental Fig. S1). Because protonophoric mitochondrial uncouplers act directly, this result does not appear to fall in line with this proposed action of XN. However, it is a well documented phenomenon that females have an enhanced response to a high fat feeding by increasing UCP expression in muscle and brown adipose tissue (58, 59). Importantly, enhanced UCP expression could lower mitochondrial membrane potential and decrease the effectiveness of weakly acidic mitochondrial uncouplers, where activity is driven in part by membrane potential and pH (60–62). We propose that enhanced UCP expression in the female Zucker rats may have blunted the effects of XN on body weight. However, the decrease in plasma markers of dysfunctional lipid metabolism described in this study were not found to be sex-specific, suggesting these changes may not be related to the protonophoric properties of XN. Instead, we hypothesize they are a reflection of an adaptive cellular response to electrophilic and oxidative stress induced by XN (Scheme 1).
Although XN clearly uncouples respiration in cell culture, our experiments do not rule out other mechanisms that may be responsible for the effects of XN in Zucker rats. Alternatively, we propose that XN may exert its beneficial effects through increased bile acid production. Plasma bile acids induce energy expenditure by activating the G-protein-coupled receptor, TGR5, which promotes intracellular thyroid hormone activation and, subsequently, UCP expression in both muscle and brown adipose tissue (63, 64). Although no reports describe a direct increase in plasma bile acids with XN treatment, at least two hint strongly toward it. In 2005, Nozawa (65) reported a 7-fold induction in the mRNA of CYP7A1 in the livers of mice fed XN. CYP7A1 hydroxylates cholesterol, and this is the rate-limiting step in bile acid synthesis. In 2012, Hirata et al. (66) saw an increase of bile acids in the feces of XN-treated mice. Neither study reported bile acid measurements in plasma. However, the large diurnal and interindividual variation in plasma bile acid concentrations makes comparative measurements difficult (67). In agreement with this, targeted data mining of bile acids revealed great variation between XN-treated and control Zucker rats, and no significant changes were observed (data not shown).
CONCLUSIONS
In vivo, metabolomics revealed that XN treatment markedly and globally reduced plasma markers of ROS and dysfunctional lipid oxidation in the muscle tissue of Zucker rats. In vitro we show that XN uncouples respiration in several cell types, and metabolomics revealed an adaptive stress response in myocytes. The degree to which these mechanisms occur and contribute to the anti-obesity effects of XN in vivo is currently under investigation.
Supplementary Material
Acknowledgments
We thank Dr. Chrissa Kioussi (Oregon State University-OSU College of Pharmacy) for the C2C12 myocytes and Jeff Morre of the OSU mass spectrometry facility. The brown preadipocytes were a kind gift from Dr. Bruce Spiegelman (Harvard Medical School Department of Cell Biology). For help with the Seahorse XF Analyzer, we acknowledge Justin X. Zhang and Jeffrey A. Greenwood, Director, Cell Imaging and Analysis Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University Grant P30 ES00210, NIEHS, National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grants R21AT005294, S10RR027878, and P30ES000210.

This article contains supplemental Experimental Procedures and references, Figs. S1–S7, and Tables S1 and S2.
- ROS
- reactive oxygen species
- PGC-1α
- peroxisome proliferator-activated receptor-coactivator 1α
- ASR
- adaptive stress response
- XN
- xanthohumol
- DC
- dicarboxylic
- AC
- acylcarnitine
- HP
- hydroperoxy
- TXN
- tetrahydroxanthohumol
- 8PNG
- 8-prenylnaringenin
- OCR
- oxygen consumption rate
- ECAR
- extracellular acidification rate
- TM
- treatment medium
- NAC
- N-acetylcysteine
- ETC
- electron transport chain
- UCP3
- uncoupling protein 3
- cySSG
- cysteine glutathione disulfide
- OP
- ophthalmic acid
- MnTMPyP
- (manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin).
REFERENCES
- 1. Harper M. E., Green K., Brand M. D. (2008) The efficiency of cellular energy transduction and its implications for obesity. Annu. Rev. Nutr. 28, 13–33 [DOI] [PubMed] [Google Scholar]
- 2. Amara C. E., Shankland E. G., Jubrias S. A., Marcinek D. J., Kushmerick M. J., Conley K. E. (2007) Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 1057–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rose G., Crocco P., De Rango F., Montesanto A., Passarino G. (2011) Further support to the uncoupling-to-survive theory. The genetic variation of human UCP genes is associated with longevity. PLoS ONE 6, e29650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anderson E. J., Yamazaki H., Neufer P. D. (2007) Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem. 282, 31257–31266 [DOI] [PubMed] [Google Scholar]
- 5. Rohas L. M., St-Pierre J., Uldry M., Jäger S., Handschin C., Spiegelman B. M. (2007) A fundamental system of cellular energy homeostasis regulated by PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 7933–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camacho A., Rodriguez-Cuenca S., Blount M., Prieur X., Barbarroja N., Fuller M., Hardingham G. E., Vidal-Puig A. (2012) Ablation of PGC1 β prevents mTOR dependent endoplasmic reticulum stress response. Exp. Neurol. 237, 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calabrese E. J. (2009) Getting the dose-response wrong. Why hormesis became marginalized and the threshold model accepted. Arch. Toxicol. 83, 227–247 [DOI] [PubMed] [Google Scholar]
- 8. Lee H. R., Cho J. M., Shin D. H., Yong C. S., Choi H. G., Wakabayashi N., Kwak M. K. (2008) Adaptive response to GSH depletion and resistance to l-buthionine-(S,R)-sulfoximine. Involvement of Nrf2 activation. Mol. Cell. Biochem. 318, 23–31 [DOI] [PubMed] [Google Scholar]
- 9. Buckley B. J., Whorton A. R. (2000) Adaptive responses to peroxynitrite. increased glutathione levels and cystine uptake in vascular cells. Am. J. Physiol. Cell Physiol. 279, C1168–C1176 [DOI] [PubMed] [Google Scholar]
- 10. Luo Y., Eggler A. L., Liu D., Liu G., Mesecar A. D., van Breemen R. B. (2007) Sites of alkylation of human Keap1 by natural chemoprevention agents. J. Am. Soc. Mass Spectrom. 18, 2226–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Findeisen H. M., Gizard F., Zhao Y., Qing H., Jones K. L., Cohn D., Heywood E. B., Bruemmer D. (2011) Glutathione depletion prevents diet-induced obesity and enhances insulin sensitivity. Obesity 19, 2429–2432 [DOI] [PubMed] [Google Scholar]
- 12. Zarse K., Schmeisser S., Groth M., Priebe S., Beuster G., Kuhlow D., Guthke R., Platzer M., Kahn C. R., Ristow M. (2012) Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B. J., Skiba B., Ooms L. M., Stepto N., Wu B., Mitchell C. A., Tonks N. K., Watt M. J., Febbraio M. A., Crack P. J., Andrikopoulos S., Tiganis T. (2009) Reactive oxygen species enhance insulin sensitivity. Cell Metab. 10, 260–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mesquita A., Weinberger M., Silva A., Sampaio-Marques B., Almeida B., Leão C., Costa V., Rodrigues F., Burhans W. C., Ludovico P. (2010) Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. U.S.A. 107, 15123–15128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dietz B. M., Kang Y. H., Liu G., Eggler A. L., Yao P., Chadwick L. R., Pauli G. F., Farnsworth N. R., Mesecar A. D., van Breemen R. B., Bolton J. L. (2005) Xanthohumol isolated from Humulus lupulus Inhibits menadione-induced DNA damage through induction of quinone reductase. Chem. Res. Toxicol. 18, 1296–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Festa M., Capasso A., D'Acunto C. W., Masullo M., Rossi A. G., Pizza C., Piacente S. (2011) Xanthohumol induces apoptosis in human malignant glioblastoma cells by increasing reactive oxygen species and activating MAPK pathways. J. Nat. Prod. 74, 2505–2513 [DOI] [PubMed] [Google Scholar]
- 17. Strathmann J., Klimo K., Sauer S. W., Okun J. G., Prehn J. H., Gerhäuser C. (2010) Xanthohumol-induced transient superoxide anion radical formation triggers cancer cells into apoptosis via a mitochondria-mediated mechanism. FASEB J. 24, 2938–2950 [DOI] [PubMed] [Google Scholar]
- 18. Legette L. L., Moreno Luna A. Y., Reed R. L., Miranda C. L., Bobe G., Proteau R. R., Stevens J. F. (2013) Xanthohumol lowers body weight and fasting plasma glucose in obese male Zucker fa/fa rats. Phytochemistry 91, 236–241 [DOI] [PubMed] [Google Scholar]
- 19. Mihalik S. J., Goodpaster B. H., Kelley D. E., Chace D. H., Vockley J., Toledo F. G., DeLany J. P. (2010) Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 18, 1695–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wada F., Usami M. (1977) Studies on fatty acid omega-oxidation. Antiketogenic effect and gluconeogenicity of dicarboxylic acids. Biochim. Biophys. Acta 487, 361–368 [PubMed] [Google Scholar]
- 21. Stevens J. F., Ivancic M., Hsu V. L., Deinzer M. L. (1997) Prenylflavonoids from Humulus lupulus. Phytochemistry 44, 1575–1585 [Google Scholar]
- 22. Miranda C. L., Aponso G. L., Stevens J. F., Deinzer M. L., Buhler D. R. (2000) Prenylated chalcones and flavanones as inducers of quinone reductase in mouse Hepa 1c1c7 cells. Cancer Lett. 149, 21–29 [DOI] [PubMed] [Google Scholar]
- 23. Stevens J. F., Taylor A. W., Clawson J. E., Deinzer M. L. (1999) Fate of xanthohumol and related prenylflavonoids from hops to beer. J. Agric. Food Chem. 47, 2421–2428 [DOI] [PubMed] [Google Scholar]
- 24. Kirkwood J. S., Lebold K. M., Miranda C. L., Wright C. L., Miller G. W., Tanguay R. L., Barton C. L., Traber M. G., Stevens J. F. (2012) Vitamin C deficiency activates the purine nucleotide cycle in zebrafish. J. Biol. Chem. 287, 3833–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B. M. (2006) Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 3, 333–341 [DOI] [PubMed] [Google Scholar]
- 26. Agresti A. (2012) Categorical Data Analysis, 3rd Ed., pp. 274–282, John Wiley & Sons, Inc., New York [Google Scholar]
- 27. Food and Drug Administration (2005) Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, p. 7, Rockville, MD [Google Scholar]
- 28. Brown G. C., Borutaite V. (2004) Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite, and S-nitrosothiols. Biochim. Biophys. Acta 1658, 44–49 [DOI] [PubMed] [Google Scholar]
- 29. Brand M. D., Pakay J. L., Ocloo A., Kokoszka J., Wallace D. C., Brookes P. S., Cornwall E. J. (2005) The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McStay G. P., Clarke S. J., Halestrap A. P. (2002) Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 367, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mailloux R. J., Seifert E. L., Bouillaud F., Aguer C., Collins S., Harper M. E. (2011) Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 286, 21865–21875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Echtay K. S., Esteves T. C., Pakay J. L., Jekabsons M. B., Lambert A. J., Portero-Otín M., Pamplona R., Vidal-Puig A. J., Wang S., Roebuck S. J., Brand M. D. (2003) A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 22, 4103–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hughes R. E. (1964) Reduction of dehydroasorbic acid by animal tissues. Nature 203, 1068–1069 [DOI] [PubMed] [Google Scholar]
- 34. Ashfaq S., Abramson J. L., Jones D. P., Rhodes S. D., Weintraub W. S., Hooper W. C., Vaccarino V., Alexander R. W., Harrison D. G., Quyyumi A. A. (2008) Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension 52, 80–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berkeley L. I., Cohen J. F., Crankshaw D. L., Shirota F. N., Nagasawa H. T. (2003) Hepatoprotection by l-cysteine-glutathione-mixed disulfide, a sulfhydryl-modified prodrug of glutathione. J. Biochem. Mol. Toxicol. 17, 95–97 [DOI] [PubMed] [Google Scholar]
- 36. Lee B. C., Gladyshev V. N. (2011) The biological significance of methionine sulfoxide stereochemistry. Free Radic. Biol. Med. 50, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soga T., Baran R., Suematsu M., Ueno Y., Ikeda S., Sakurakawa T., Kakazu Y., Ishikawa T., Robert M., Nishioka T., Tomita M. (2006) Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 281, 16768–16776 [DOI] [PubMed] [Google Scholar]
- 38. Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk G. D., Muoio D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 39. Cahill G. F., Jr., Herrera M. G., Morgan A. P., Soeldner J. S., Steinke J., Levy P. L., Reichard G. A., Jr., Kipnis D. M. (1966) Hormone-fuel interrelationships during fasting. J. Clin. Invest. 45, 1751–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Masoro E. J. (1967) Skeletal muscle lipids. 3. Analysis of the functioning of skeletal muscle lipids during fasting. J. Biol. Chem. 242, 1111–1114 [PubMed] [Google Scholar]
- 41. Bell J. A., Reed M. A., Consitt L. A., Martin O. J., Haynie K. R., Hulver M. W., Muoio D. M., Dohm G. L. (2010) Lipid partitioning, incomplete fatty acid oxidation, and insulin signal transduction in primary human muscle cells. Effects of severe obesity, fatty acid incubation, and fatty acid translocase/CD36 overexpression. J. Clin. Endocrinol. Metab. 95, 3400–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tserng K. Y., Griffin R. L., Kerr D. S. (1996) Distinction of dicarboxylic aciduria due to medium-chain triglyceride feeding from that due to abnormal fatty acid oxidation and fasting in children. Metabolism 45, 162–167 [DOI] [PubMed] [Google Scholar]
- 43. Grego A. V., Mingrone G. (1995) Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition. An update. Clin. Nutr. 14, 143–148 [DOI] [PubMed] [Google Scholar]
- 44. Mortensen P. B. (1992) Formation and degradation of dicarboxylic acids in relation to alterations in fatty acid oxidation in rats. Biochim. Biophys. Acta 1124, 71–79 [DOI] [PubMed] [Google Scholar]
- 45. Guzy J., Vasková-Kubálková J., Rozmer Z., Fodor K., Mareková M., Poskrobová M., Perjési P. (2010) Activation of oxidative stress response by hydroxyl substituted chalcones and cyclic chalcone analogues in mitochondria. FEBS Lett. 584, 567–570 [DOI] [PubMed] [Google Scholar]
- 46. Legette L., Ma L., Reed R. L., Miranda C. L., Christensen J. M., Rodriguez-Proteau R., Stevens J. F. (2012) Pharmacokinetics of xanthohumol and metabolites in rats after oral and intravenous administration. Mol. Nutr. Food Res. 56, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcia J., Han D., Sancheti H., Yap L. P., Kaplowitz N., Cadenas E. (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zmijewski J. W., Banerjee S., Bae H., Friggeri A., Lazarowski E. R., Abraham E. (2010) Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 285, 33154–33164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mailloux R. J., Bériault R., Lemire J., Singh R., Chénier D. R., Hamel R. D., Appanna V. D. (2007) The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2, e690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patti G. J., Yanes O., Siuzdak G. (2012) Innovation. Metabolomics. The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Harvey C. J., Thimmulappa R. K., Singh A., Blake D. J., Ling G., Wakabayashi N., Fujii J., Myers A., Biswal S. (2009) Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol. Med. 46, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zoll J., Koulmann N., Bahi L., Ventura-Clapier R., Bigard A. X. (2003) Quantitative and qualitative adaptation of skeletal muscle mitochondria to increased physical activity. J. Cell Physiol. 194, 186–193 [DOI] [PubMed] [Google Scholar]
- 54. Wallimann T., Wyss M., Brdiczka D., Nicolay K., Eppenberger H. M. (1992) Intracellular compartmentation, structure, and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands. The “phosphocreatine circuit” for cellular energy homeostasis. Biochem. J. 281, 21–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bowtell J. L., Bruce M. (2002) Glutamine. An anaplerotic precursor. Nutrition 18, 222–224 [DOI] [PubMed] [Google Scholar]
- 56. Bergström J., Fürst P., Norée L. O., Vinnars E. (1974) Intracellular free amino acid concentration in human muscle tissue. J. Appl. Physiol. 36, 693–697 [DOI] [PubMed] [Google Scholar]
- 57. Lehmann R., Zhao X., Weigert C., Simon P., Fehrenbach E., Fritsche J., Machann J., Schick F., Wang J., Hoene M., Schleicher E. D., Häring H. U., Xu G., Niess A. M. (2010) Medium chain acylcarnitines dominate the metabolite pattern in humans under moderate intensity exercise and support lipid oxidation. PLoS ONE 5, e11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li L., Hossain M. A., Sadat S., Hager L., Liu L., Tam L., Schroer S., Huogen L., Fantus I. G., Connelly P. W., Woo M., Ng D. S. (2011) Lecithin cholesterol acyltransferase null mice are protected from diet-induced obesity and insulin resistance in a gender-specific manner through multiple pathways. J. Biol. Chem. 286, 17809–17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodríguez A. M., Quevedo-Coli S., Roca P., Palou A. (2001) Sex-dependent dietary obesity, induction of UCPs, and leptin expression in rat adipose tissues. Obes. Res. 9, 579–588 [DOI] [PubMed] [Google Scholar]
- 60. Mao W., Yu X. X., Zhong A., Li W., Brush J., Sherwood S. W., Adams S. H., Pan G. (1999) UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 443, 326–330 [DOI] [PubMed] [Google Scholar]
- 61. Marti A., Larrarte E., Novo F. J., Garcia M., Martinez J. A. (2001) UCP2 muscle gene transfer modifies mitochondrial membrane potential. Int. J. Obes. Relat. Metab. Disord. 25, 68–74 [DOI] [PubMed] [Google Scholar]
- 62. Ozaki S., Kano K., Shirai O. (2008) Electrochemical elucidation on the mechanism of uncoupling caused by hydrophobic weak acids. Phys. Chem. Chem. Phys. 10, 4449–4455 [DOI] [PubMed] [Google Scholar]
- 63. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 [DOI] [PubMed] [Google Scholar]
- 64. Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., Pellicciari R., Auwerx J., Schoonjans K. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nozawa H. (2005) Xanthohumol, the chalcone from beer hops (Humulus lupulus L.), is the ligand for farnesoid X receptor and ameliorates lipid and glucose metabolism in KK-A(y) mice. Biochem. Biophys. Res. Commun. 336, 754–761 [DOI] [PubMed] [Google Scholar]
- 66. Hirata H., Yimin, Segawa S., Ozaki M., Kobayashi N., Shigyo T., Chiba H. (2012) Xanthohumol prevents atherosclerosis by reducing arterial cholesterol content via CETP and apolipoprotein E in CETP-transgenic mice. PLoS ONE 7, e49415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Steiner C., Othman A., Saely C. H., Rein P., Drexel H., von Eckardstein A., Rentsch K. M. (2011) Bile acid metabolites in serum. Intraindividual variation and associations with coronary heart disease, metabolic syndrome, and diabetes mellitus. PLoS ONE 6, e25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.