

Background: The tetraspanin CD63 is known to regulate protein trafficking, leukocyte recruitment, and adhesion processes.
Results: Silencing of CD63 disrupts complex formation between β1 integrin and VEGFR2, resulting in impaired downstream signaling.
Conclusion: CD63 supports VEGFR2 activation and signaling in vitro and in vivo.
Significance: A novel role for the tetraspanin CD63 in the convergence between integrin and growth factor signaling in angiogenesis.
Keywords: Angiogenesis, Endothelial Cell, Receptor Tyrosine Kinase, Tetraspanins, Vascular Endothelial Growth Factor (VEGF), CD63, Angiogenesis
Abstract
CD63 is a member of the transmembrane-4 glycoprotein superfamily (tetraspanins) implicated in the regulation of membrane protein trafficking, leukocyte recruitment, and adhesion processes. We have investigated the involvement of CD63 in endothelial cell (EC) signaling downstream of β1 integrin and VEGF. We report that silencing of CD63 in primary ECs arrested capillary sprouting and tube formation in vitro because of impaired adhesion and migration of ECs. Mechanistically, CD63 associated with both β1 integrin and the main VEGF receptor on ECs, VEGFR2. Our data suggest that CD63 serves to bridge between β1 integrin and VEGFR2 because CD63 silencing disrupted VEGFR2-β1 integrin complex formation identified using proximity ligation assays. Signaling downstream of β1 integrin and VEGFR2 was attenuated in CD63-silenced cells, although their cell surface expression levels remained unaffected. CD63 was furthermore required for efficient internalization of VEGFR2 in response to VEGF. Importantly, systemic delivery of VEGF failed to potently induce VEGFR2 phosphorylation and downstream signaling in CD63-deficient mouse lungs. Taken together, our findings demonstrate a previously unrecognized role for CD63 in coordinated integrin and receptor tyrosine kinase signaling in vitro and in vivo.
Introduction
Angiogenesis is a physiological/pathological process that results in the formation of new blood vessels from the pre-existing vasculature. It consists of several sequential steps including extracellular matrix (ECM)3 degradation, sprout initiation, proliferation and migration of endothelial cells (ECs), sprout anastomosis, lumen formation, and stabilization (1). The sprouting of ECs is tightly regulated and demands a balanced integration of signals from growth factors and adhesion molecules (2). Among multiple proangiogenic factors, VEGF and its main receptor, VEGFR2, are essential in angiogenesis. Activation of VEGFR2 on ECs initiates several signaling cascades that regulate EC biology during development, in health, and in disease (3).
During the angiogenic process, ECs establish dynamic interactions with their surrounding ECM through integrins, a large family of heterodimeric cell surface proteins that link ECM proteins to the actin cytoskeleton in different adhesive structures (4). Integrin-mediated cell attachment to the ECM results in the recruitment of adaptor proteins and the activation of signaling mediators including the dual kinase complex formed by the focal adhesion kinase (FAK) and c-Src (5), critical mediators of integrin-dependent adhesion turnover and cell migration (6). Importantly, integrin ligation is required for several growth factor-induced biological processes. Thus, signaling pathways induced as a consequence of integrin activation are also employed by growth factor receptors, providing opportunities for cross-talk (7).
CD63 is a member of the transmembrane-4 glycoprotein superfamily, known as tetraspanins. The tetraspanins contain four transmembrane domains, two extracellular loops flanked by short intracellular N and C termini, and a highly conserved sequence motif within the larger extracellular domain (8). Tetraspanins interact among themselves and with other transmembrane proteins to form membrane domains denoted tetraspanin-enriched microdomains (TEMs) (9). At least in part through the TEMs, tetraspanins regulate a variety of cellular processes including cell fusion, intracellular trafficking, cell adhesion, motility, invasion, and cell differentiation (10).
CD63 was originally identified as a protein present in the surface of activated platelets and in early stage human melanoma cells (11). CD63 gene targeting in mice results in a relatively mild phenotype with increased gastrointestinal and kidney diuresis (12). Moreover, leukocyte attachment to cd63−/− ECs is reduced because of altered trafficking of endothelial P-selectin, resulting in decreased leukocyte rolling and extravasation (13). A number of other cell types have also been shown to depend on CD63 for efficient adhesion, spread, and migration including platelets, monocytes, neutrophils, dendritic, intestinal, and melanoma cells (14–19). In addition, CD63 has been implicated in the regulation of cell survival and polarization in breast epithelial cells, where it was found to interact with the tissue inhibitor of metalloproteinase-1 (20). Interestingly, CD63 is one of several lysosomal proteins affected in Hermansky-Pudlak syndrome with adaptor protein complex-3 deficiency, a rare autosomal recessive disorder characterized by defective intracellular vesicle formation and a platelet storage pool deficiency (21).
CD63 is among the most highly expressed tetraspanins in ECs (22). Still, the role of CD63 in endothelial biology and angiogenesis remains to be addressed. We show that CD63 is co-localized with the type I integral lysosomal membrane protein (LAMP-1) but also present on the EC surface. By silencing CD63 expression in primary ECs, we demonstrate that CD63 associated with both β1 integrin and VEGFR2 in the plasma membrane and was required for VEGFR2-β1 integrin complex formation. Loss of CD63 expression disrupted downstream signaling pathways both in endothelial cells in culture and in intact tissues. As a consequence, CD63-deficient ECs failed to engage in sprouting angiogenesis and organize into tube structures.
MATERIALS AND METHODS
Animals
C57BL/6 WT mice were purchased from B&K Scanbur. cd63−/− mice (12) were kindly provided by Prof. Paul Saftig (Christian-Albrechts University, Kiel, Germany) and subsequently propagated at the local animal facility under laminar air flow conditions with a 12-h light/dark cycle at a temperature of 22–25 °C. All animal work was approved by the Uppsala University board of animal experimentation. VEGF (5 μg/mouse) in 100 μl of PBS was injected in the tail vein of wild type and cd63−/− mice, and after 1 min of circulation, the lungs were harvested, snap frozen, and processed for immunoblotting as described below.
Cell Culture
Human umbilical vein ECs (HUVECs; ATCC) were cultured on gelatin-coated dishes in endothelial cell basal medium MV 2 (EBM-2, C-22221; PromoCell) with supplemental pack C-39221, containing 5% FCS, epidermal growth factor (5 ng/ml), VEGF (0.5 ng/ml), FGF2 (10 ng/ml), long R3 insulin growth factor-1 (20 ng/ml), hydrocortisone (0.2 μg/ml), and ascorbic acid (1 μg/ml). HUVECs at passages 3–6 were used. For experimental purposes, ECs were serum-starved overnight and plated in EBM-2 medium, 1% FCS without growth factor supplement and treated with VEGF or FGF2 for the indicated time points.
siRNA Knockdown of CD63 in HUVECs
HUVECs were transfected with StealthTM siRNA duplex oligoribonucleotides against human CD63 (si-CD631, UAUGGUCUGACUCAGGACAAGCUGU; si-CD632, AAGACAAUAGUUCUCCUUGCAGGCC) or with control siRNA (12935-300; Invitrogen) using the transfection reagent Lipofectamine RNAiMAX (Invitrogen).
In Vitro Tube Formation Assay
Collagen type I (Vitrogen; Cohesion Technologies, Palo Alto, CA) was mixed with 0.1 m NaOH and 10× Ham's F-12 medium (PromoCell) (8:1:1), and components were added to final concentrations indicated: 0.02 m HEPES, 0.1% (w/v) bicarbonate, 2 mm Glutamax-I (Invitrogen). The collagen mix polymerized at 37 °C overnight. Serum-starved cells were seeded out at 67,600 cells/cm2 and left for 2 h at 37 °C. A second layer of collagen was added and left to polymerize for 1 h. Thereafter, cultures were treated with VEGF or FGF2, each at 50 ng/ml (Peprotech EC) in EBM-2 medium in 1% FCS for up to 24 h. The cells were washed three times with ice-cold PBS and fixed in 4% paraformaldehyde (PFA) in PBS for 10 min at 4 °C followed by immunostaining with Alexa 555-conjugated phalloidin (Molecular Probes, Invitrogen). To quantify effects of siRNA on tubular morphogenesis, random images were taken using a LSM 510 META confocal laser-scanning inverted microscope (Carl Zeiss International, Oberkochen, Germany). Tube area was quantified using ImageJ (National Institutes of Health, Bethesda, MD) and represented as a percentage of phalloidin-positive area/total area.
Spheroid-based Angiogenesis Assay
EC spheroids were generated by culturing HUVECs overnight in medium containing 0.25% carboxymethylcellulose in round-bottomed 96-well plates. The spheroids were embedded in collagen gels in 24-well plates. Culture medium containing human VEGF (50 ng/ml) or human FGF2 (50 ng/ml) was added on the gel. After 24 h, the gels were washed three times in ice-cold PBS and fixed in 4% PFA in PBS for 10 min at 4 °C followed by immunostaining with Alexa 555-conjugated phalloidin (Molecular Probes, Invitrogen). Alternatively, spheroids were immunostained with CD63 (Serotec), cleaved caspase-3 (Cell Signaling), or VE-cadherin (Santa Cruz Biotechnology) following the immunofluorescence protocol described below. To quantify effects of siRNA transfection on sprouting angiogenesis, images spanning the entire wells were taken using a LSM 510 META confocal microscope, and sprout length was quantified with ImageJ (National Institutes of Health).
Chemotaxis Assay
The chemotaxis assay was performed using a modified Boyden chamber with 8-μm micropore polycarbonate filters coated with collagen (Neuro Probe Inc., Gaithersburg, MD). HUVECs transfected with the corresponding siRNA were starved overnight in EBM-2 medium, 1% FCS, trypsinized, and resuspended at 4 × 105 cells/ml in RPMI medium with 0.25% BSA and Trasylol at 1,000 kallikrein inhibition units/ml. The cell suspension was added in the upper chamber and 10 ng/ml VEGF or 20 ng/ml FGF2 in the lower chamber. After 5 h at 37 °C, cells that had migrated through the filter were fixed in methanol, stained with Giemsa, and counted and analyzed using ImageJ (National Institutes of Health). The data are presented as the number of cells that migrated through the filter/field.
Adhesion Assay
To assess cell adhesion and spreading, siRNA-transfected HUVECs were suspended in EBM-2 medium with 0.25% BSA and seeded onto plates coated with collagen (160 μg/ml), fibronectin (50 μg/ml), laminin (20 μg/ml), and vitronectin (5 μg/ml) for different time periods. The cells were fixed in 4% PFA for 10 min, and the nuclei were visualized using 1 μg/ml Hoechst 33342 (Molecular Probes, Invitrogen). Images were taken with a LSM 510 META confocal laser-scanning inverted microscope (Carl Zeiss International). Cell adhesion was calculated as the number of cells attached/field.
Alamar Blue Cell Viability Assay
After plating in 24-well plates, HUVECs were allowed to attach for 24 h, transfected with control and CD63 siRNAs, and cultured in EBM-2 medium containing 0.5% FCS. Before the end of treatment (48 and 72 h), the cells were incubated with Alamar Blue (Invitrogen) for 5 h, and the plates were read in a microplate reader at 590 nm. Relative cell viability was calculated and expressed as Alamar Blue counts.
Crystal Violet Assay
Twenty-four hours after transfection, the cells were seeded into 96-well culture plates (5.0 × 103 cells/well) in triplicate in fresh EBM-2 medium containing 0.5% FCS. After 24 and 48 h of incubation at 37 °C, the cells were stained with 0.1% crystal violet, 20% methanol for 10 min. Then the dye was eluted in 0.1 m sodium citrate, 75% ethanol, and the absorbance at 540 nm was determined in an ELISA reader.
Immunofluorescence
Transfected HUVECs on glass coverslips were cultured for 48 h. Following starvation, the cells were treated with VEGF (25 ng/ml), washed three times with ice-cold PBS, and fixed in 4% PFA in PBS for 10 min at 4 °C. The cells were permeabilized in 0.2% Triton X-100 in PBS for 5 min at room temperature and then washed three times in PBS. Fixed and permeabilized cells were blocked in 3% BSA in PBS (blocking solution) for 1 h before incubation with primary antibodies diluted in blocking solution overnight at 4 °C. The following primary antibodies were used: mouse anti-human CD63 (Serotec), Tyr(P)-397 FAK (Cell Signaling), Tyr(P)-416 Src (Invitrogen), mouse anti-human paxillin (Millipore), and goat anti-human VE-cadherin (Santa Cruz Biotechnology). The cells were then washed three times in PBS and incubated for 45 min at room temperature with the appropriate fluorescently conjugated secondary antibodies (Alexa 488 or 555; Molecular Probes). Finally, the cells were washed three times in PBS. Actin stress fibers were stained with Alexa 488-conjugated phalloidin, and the nuclei were visualized using 1 μg/ml Hoechst 33342 (Molecular Probes, Invitrogen). Coverslips were mounted onto microscope slides using Fluoromount-G (Southern Biotech). Immunofluorescent staining was analyzed using a Nikon Eclipse E100 microscope or an LSM 510 META confocal laser-scanning inverted microscope.
Proximity Ligation Assay (PLA)
ECs were seeded into collagen-coated 8-well chamber slides at a density of 60,000 cells/well. After 24 h, the cells were left in full medium or serum-starved overnight (EBM-2, 1% FCS) and stimulated with 50 ng/ml VEGF in EBM-2 medium, 1% FCS at 37 °C. The cells were fixed in 4% PFA and subjected to PLA. Briefly, slides were blocked and incubated with primary antibodies; goat anti-human VEGFR2 (R & D Systems), mouse anti-human CD63 (Serotec), rabbit anti-human CD63 (Santa Cruz Biotechnology), mouse anti-human β1 integrin (Millipore), mouse anti-human β3 integrin (Becton Dickinson), mouse IgG1 (Becton Dickinson), normal goat IgG (Vector Laboratories), and rabbit IgG control (Santa Cruz Biotechnology). Secondary antibodies (anti-mouse, anti-rabbit, and anti-goat) conjugated to unique oligonucleotide fragments (Olink Bioscience, Uppsala, Sweden) were added. Ligation and circularization of the DNA was followed by a rolling circle amplification step, and reactions were detected by using a complementary Cy3-labeled DNA linker. Slides were mounted using Vectashield (Vector Laboratories, Burlingame, CA) and evaluated using an LSM 510 META confocal. The PLA signal was quantified as a mean intensity of fluorescence per cell using ImageJ.
Immunoblotting
HUVECs were lysed in Nonidet P-40 lysis buffer (1% Nonidet P-40, 150 mm NaCl, 10% glycerol, 20 mm HEPES, 1 mm phenylmethylsulfonyl fluoride, 2.5 mm EDTA, 100 μm Na3VO4, and 1% aprotinin). Mouse lungs were lysed in radioimmune precipitation assay buffer containing phosphatase and protease inhibitors (ProteinSimple).
Samples separated in NuPage 4–12% Bis-Tris gels using MOPS buffer (Invitrogen) and transferred to Hybond-C extra membranes (GE Healthcare). The membranes were blocked in 5% nonfat dry milk in TBS + 0.1% Tween (blocking solution) for 1 h, followed by incubation with primary antibodies overnight. The following antibodies were used: goat anti-human VEGFR2 (R & D Systems), rabbit anti-human Tyr(P)-951 VEGFR2, rabbit anti-human Tyr(P)-1175 VEGFR2, rabbit anti-human Thr(P)-202/Tyr(P)-204 Erk mouse anti-human Erk1/2, rabbit anti-human Ser(P)-473 Akt, rabbit anti-human Akt (all from Cell Signaling, Danvers, MA), rabbit anti-human Tyr(P)-783 PLCγ (BIOSOURCE, Invitrogen), rabbit anti-human PLCγ (Cell Signaling), rabbit anti-human FAK (Santa Cruz Biotechnology), rabbit anti-human Tyr(P)-577 and Tyr(P)-861 FAK (Cell Signaling), mouse anti-human β1 integrin (Millipore), mouse anti-human β2-microglobulin and goat anti-human actin (Santa Cruz Biotechnology). After three washes with TBS + 0.1% Tween 20, the membranes were incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies. Immune complexes were visualized by enhanced chemiluminescence (Amersham Biosciences).
Flow Sorting Analysis (FACS)
The cells were resuspended in FACS buffer (1% FCS in PBS) and labeled with primary antibodies for 45 min at 4 °C. The following antibodies were used: mouse anti-human β1 integrin (Millipore), goat anti-human VEGFR2 (R & D Systems), mouse IgG (Becton Dickinson), and normal goat IgG (Vector Laboratories). The cells were subsequently washed and incubated with appropriate fluorescently conjugated secondary antibodies (Alexa 488; Molecular Probes) for 30 min at 4 °C. DAPI was used for discrimination of dead cells, and samples were analyzed on an LSRII cytometer (BD Biosciences). All antibodies and corresponding isotype controls were used at a concentration of 2 μg/ml. To check for membrane levels of VEGFR2, the cells were serum-starved overnight and stimulated with a pulse of VEGF (25 ng/ml) for 15 min. The cells were washed and harvested immediately or at 1 and 3 h after the pulse. A goat anti-human VEGFR2 antibody was used for labeling as described above.
Activation-dependent ligand Huts-21 was used to determine the activation status of integrins. To analyze Huts-21 binding, serum-starved HUVECs were cultured in 1 mm MnCl2 followed by the addition of Huts-21-PE (BD Biosciences) in adherent conditions for 30 min. Total β1 integrin levels were assessed using the β1 integrin-allophycocyanin antibody. The wells were washed in PBS, detached, vortexed, and washed two more times in PBS. FACS was performed using FACSCalibur, and the data were analyzed using FACS DIVA 6.0 Software (BD Biosciences). Only intact cells were analyzed, excluding cell debris.
RNA Isolation and Quantitative RT-PCR
For quantitative PCR, ECs were left in full growth medium (EBM-2 with supplemental pack) for 48 h, followed by RNA extraction. One microgram of DNase-treated total RNA was used for cDNA synthesis using dT18 and murine moloney leukemia virus reverse transcriptase (USB Corp., Cleveland, OH). Real time PCR was performed on cDNA using 2× SYBR Green PCR Master Mix (Applied Biosystems) and run in triplicate on an ABI Prism 7700 sequence detection system instrument (Applied Biosystems) with an initial 10 min at 95 °C, followed by 45 cycles at 95 °C for 15 s and 60 °C for 60 s. The calculated threshold cycle value for each transcript was normalized against the corresponding hypoxanthine-guanine phosphoribosyltransferase (HPRT) calculated threshold cycle value. The normalized values were related to control siRNA and presented as fold change. The following oligonucleotides (Invitrogen) were used for real time PCR analysis (forward, reverse primers; 5′-3′): HPRT, CTTTGCTGACCTGCTGGATT and TCCCGTGTTGACTGGTCATT; and CD63, TGGAAGGAGGAATGAAATGTG and GCAATCAGTCCCACTGCAC.
Statistical Analysis
The data were analyzed using GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA). The bars represent mean values ± S.E. For comparison of two groups, Student's unpaired t test was used. A p value < 0.05 was considered as statistically significant.
RESULTS
CD63 Is Localized in Late Endosomes/lysosomes and on the Plasma Membrane in Primary ECs
To determine the role of CD63 in endothelial cell biology, we first investigated its subcellular distribution in ECs by immunofluorescence and flow cytometric analyses. CD63 has previously been shown to be a component of Weibel-Palade bodies (23), specialized secretory organelles in ECs. We found that the majority of CD63 immunostaining co-localized with the lysosomal protein LAMP-1 in late endocytic organelles, in agreement with previous reports on the subcellular localization of CD63 (Fig. 1a) (10). Moreover, FACS analyses of nonpermeabilized ECs showed that CD63 was localized on the cell surface of quiescent ECs (Fig. 1b). Two siRNAs efficiently and specifically silenced CD63 mRNA and protein expression in ECs (Fig. 1c) without causing morphological alterations in general or specifically of the late LAMP-1-positive endosomal/lysosomal compartment (Fig. 1d), in accordance with the literature (12).
FIGURE 1.
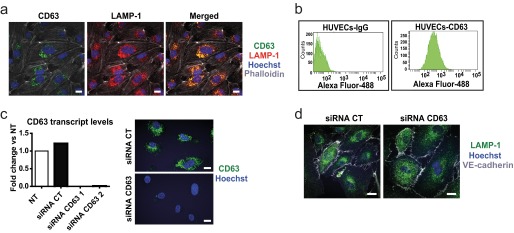
Expression of CD63 in endothelial cells in LAMP-1 vesicles and on the cell surface. a, representative maximum intensity projection confocal images of permeabilized HUVECs immunostained for CD63 and LAMP-1. b, flow cytometric analysis of CD63 expression in nonpermeabilized HUVECs labeled with mouse IgG (left panel) or a monoclonal antibody against CD63 (right panel). c, CD63 expression in siRNA-transfected HUVECs (transcript levels to the left and immunofluorescent staining to the right) of HUVECs transfected with control (CT) siRNA and two different CD63 siRNAs, at 48 h after transfection. The data show fold induction versus not treated (NT). d, immunofluorescence for LAMP-1 and VE-cadherin (as a membrane marker) in siRNA-transfected HUVECs. Scale bars, 20 μm.
CD63 Ablation Results in Impaired Sprouting Angiogenesis and Tube Formation
To investigate the potential role of CD63 in angiogenesis, we analyzed endothelial sprouting and network formation in three-dimensional collagen gels after CD63 silencing. As shown in Fig. 2 (a and b), CD63 silencing impaired endothelial sprouting from EC spheroids and EC tube formation in three-dimensional collagen gels in response to VEGFA and FGF2. Unlike control siRNA-treated ECs, CD63-silenced ECs remained as isolated cells or cell clusters. Higher magnification images showed deficient and disorganized sprout formation by CD63-silenced cells already at 12 h after growth factor addition (Fig. 2c). Immunohistochemistry for cleaved caspase-3 revealed a similar extent of apoptosis in the presence and absence of CD63 (Fig. 2d). Together, these data showed that CD63 is required for endothelial tube formation and sprouting angiogenesis and that the dysregulated angiogenic sprouting is not due to endothelial cell apoptosis.
FIGURE 2.
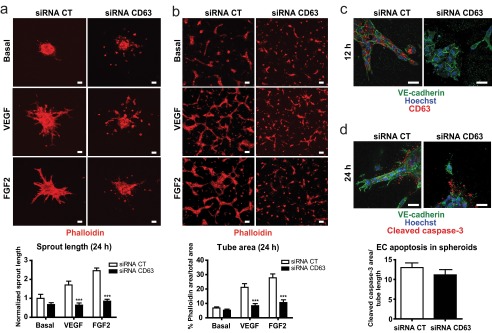
CD63 is required for sprouting angiogenesis and tube formation. a, HUVECs spheroids from siRNA-transfected cells placed in three-dimensional collagen gels and treated with VEGF or FGF2 (both 50 ng/ml) for 24 h. Quantification shows sprout length normalized to basal levels of siRNA control (CT; below). b, siRNA-transfected HUVECs seeded on three-dimensional collagen, treated with VEGF or FGF2 for 24 h, and immunostained with phalloidin-Alexa 555. Quantification shows area of capillary-like structures defined as a percentage of phalloidin area/total area. c, high magnification images of sprouts in siRNA-transfected HUVECs spheroids at 12 h of growth factor treatment. d, apoptotic cells in siRNA-transfected HUVECs spheroids at 24 h detected by immunostaining for cleaved caspase-3. ***, p < 0.001. The data are shown as the means ± S.E. Scale bars, 40 μm.
CD63 Ablation Impairs EC Adhesion and Migration toward VEGF and FGF2
To identify the possible mechanisms by which CD63 regulates EC sprouting in vitro, we first examined the effect of CD63 knockdown on HUVECs adhesion to different ECM proteins. As shown in Fig. 3a, CD63 silencing reduced cell adhesion at 30 min after seeding to all the ECM protein tested including collagen, fibronectin, laminin, and vitronectin. There was a consequent reduction in the capacity of CD63-silenced ECs to migrate through collagen-coated filters toward VEGF and FGF2 in a modified Boyden chamber (Fig. 3b). Cell survival and proliferation were not affected by CD63 silencing at the time point (48 h) when adhesion and migration were assayed. However at later time points, 72 h after siRNA transfection, CD63 deficiency also impaired endothelial survival and proliferation (Fig. 3, c and d).
FIGURE 3.
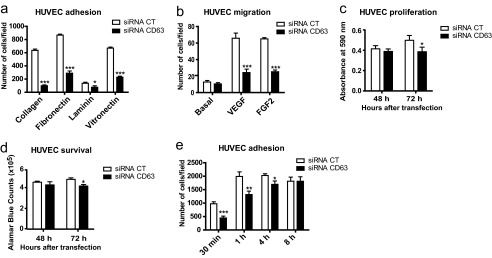
CD63 silencing interferes with endothelial cell adhesion. a, siRNA-transfected HUVECs seeded on 96-well plates coated with collagen, fibronectin, laminin, or vitronectin were allowed to adhere for 30 min. Quantification shows number of cells attached/field in random images. b, migration of siRNA-transfected HUVECs in a modified Boyden chamber toward VEGF (10 ng/ml) and FGF2 (20 ng/ml) for 5 h. Quantification shows the number of migrated cells/field. c, siRNA-transfected HUVECs were cultured for 48 or 72 h in 0.5% FCS, and the cells were stained with crystal violet. Quantification shows absorbance at 590 nm. d, cell viability was measured using an Alamar Blue assay in siRNA-transfected HUVECs in 0.5% FCS at 48 and 72 h after transfection. Quantification shows absorbance at 590 nm. e, siRNA-transfected HUVECs in 96-well plates coated with collagen were allowed to adhere for 30 min and 1, 4, and 8 h in BSA-containing medium. Quantification shows the number of cells attached/field on images spanning the entire well. CT, control. *, p < 0.05; **, p < 0.01; ***, p < 0.001. The data are mean values ± S.E.
Time course studies of cell adhesion on collagen showed an impaired adhesive capacity of CD63-silenced cells at 30 min and 1 h after seeding in medium devoid of growth factors. However, adhesion to collagen rebound to 90% of the control after 4 h and was completely restored after 8 h in CD63-silenced cells (Fig. 3e). Thus, EC adhesion and spreading was delayed but not arrested in the absence of CD63. We conclude that CD63 regulates several cell functions relevant to initiation and progression of angiogenesis such as adhesion and migration of vascular endothelial cells.
CD63 Associates with β1 Integrin and Affects Integrin Downstream Signaling
CD63 silencing caused impaired adhesion to all the ECM proteins tested (Fig. 3a). Because β1 is the most common subunit among the ECM-binding integrins and because tetraspanins are known to engage in complex formation with integrins, we asked whether CD63 and β1 integrin exist in complex. We used the PLA (Fig. 4a) to detect complex formation between CD63 and β1 integrin in situ in quiescent, primary ECs. The PLA employs oligonucleotide-ligated antibodies, which when brought in close proximity because of the binding to epitopes on proteins, e.g., engaged in complex formation, initiate a rolling circle amplification detected using Cy3-labeled probes, resulting in the visualization of complexes as fluorescent spots (24).
FIGURE 4.
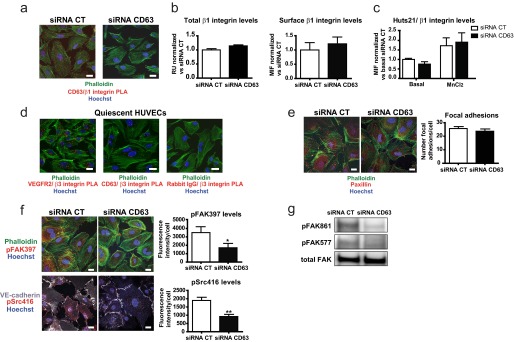
CD63 silencing interferes with integrin-signaling. a, detection of CD63-β1 integrin complex formation in siRNA-transfected HUVECs using in situ PLA. The images show representative confocal maximal projections of surface PLA signals. b, immunoblot analysis of total β1 integrin protein levels in siRNA-transfected HUVECs in relation to β2-microglobulin expression; diagram shows quantification of immunoblotted β1 integrin/β2 microglobulin bands in relative units (RU); fold change in CD63-siRNA treated cells versus control (CT) siRNA cells (left panel). Flow cytometric analysis of surface β1 integrin expression in siRNA-transfected HUVECs. Quantification shows the mean intensity of fluorescence (MIF) of β1 integrin compared with IgG control; normalized data shows fold change in CD63-siRNA treated cells versus control (CT) siRNA cells (right panel). c, siRNA-transfected HUVECs were labeled with HUTS-21-PE and total β1 integrin-APC, and binding to intact cells was analyzed by flow cytometry in the presence or absence of MnCl2. Quantification shows MIF of Huts-21 compared with IgG control and related to surface β1 integrin levels; normalized data show fold change in CD63-siRNA treated cells versus basal levels of control (CT) siRNA cells. d, in situ PLA detection of VEGFR2-β3 integrin and CD63-β3 integrin complexes in nonpermeabilized HUVECs. Images show representative confocal maximal projections of surface PLA signals. e, immunofluorescence for paxillin in siRNA-transfected HUVECs. Quantification shows the number of focal adhesions/cell. f, immunofluorescent staining for pFAK397 and pSrc416 in siRNA-transfected HUVECs cultured without growth factors and serum. Quantification shows intensity of fluorescence/cell. g, immunoblot analysis of FAK, pFAK861 and pFAK577 in whole cell lysates from HUVECs treated as in e. *, p < 0.05; **, p < 0.01. The data are mean values ± S.E. Bars, 20 μm.
CD63 silencing attenuated the PLA signals, demonstrating specificity of the reactions (Fig. 4a). There was no effect of CD63 silencing, neither on total expression levels nor on the cell surface expression levels of β1 integrin (Fig. 4b). To determine whether CD63 regulated integrin activation, we used the Huts-21 antibody, known to specifically detect the active conformation of β1 integrin. The Huts-21 antibody was equally reactive with β1 integrin in the presence and absence of CD63 expression (Fig. 4c), both when cells were in a quiescent state and when integrins had been activated by the addition of Mn2+ to the culture medium.
Because perturbation of adhesion on vitronectin (Fig. 3a) could be likely mediated by the αvβ3 integrin and because CD63 has been shown to associate with the β3 integrin in platelets (25), we also investigated complex formation between CD63 and β3 integrin in HUVECs. As shown in Fig. 4d, only sparse CD63-β3 integrin complexes were detected in quiescent HUVECs, as compared with the more frequent VEGFR2-β3 integrin (Fig. 4d) and CD63-β1 integrin complexes (Fig. 4a).
To study the potential impact of CD63 on focal adhesions, we compared the localization and phosphorylation of the focal adhesion constituent paxillin. As shown in Fig. 4e, CD63-deficient cells formed paxillin-containing focal adhesions associated with actin-positive stress fibers to the same extent as control siRNA-treated cells.
The loss of CD63-β1 integrin complex formation did, however, affect downstream integrin-dependent signaling. As shown in Fig. 4f, silencing of CD63 resulted in decreased levels of FAK tyrosine phosphorylated at Tyr-397, Tyr-577, and Tyr-861 (Fig. 4, f and g). There was also decreased downstream activation of c-Src, as judged from immunostaining with antibodies reactive to Tyr(P)-416 Src, the activating tyrosine residue in the c-Src kinase domain (Fig. 4f). Collectively, these results suggest that the loss of CD63 results in an impaired integrin signaling without alterations in β1 integrin expression levels, conformational state, and formation of focal adhesions.
CD63 Regulates Complex Formation between VEGFR2 and β1 Integrin and Modulates VEGFR2 Signaling and Internalization
β1 integrin has been shown to exist in complex with VEGFR2 and affect VEGFR2 signal transduction (26). In accordance, VEGFR2-β1 integrin complexes were identified by in situ PLA, on the EC surface (Fig. 5a). Importantly, VEGFR2 also associated with CD63 in HUVECs (Fig. 5b), and the formation of these complexes gradually disappeared after treatment of cells with VEGF, probably because of internalization of the receptor. Of note, VEGFR2-β1 integrin complex formation was dependent on CD63 expression because silencing of CD63 resulted in a significant decrease in the number of VEGFR2-β1 integrin complexes (Fig. 5c).
FIGURE 5.

VEGFR2-β1 integrin-complex formation is dependent on CD63. a, in situ PLA detection of VEGFR2-β1 integrin complexes in nonpermeabilized, siRNA-transfected HUVECs. The images show representative confocal maximal projections of surface PLA signals. b, In situ PLA detection of VEGFR2-CD63 complexes in serum-starved, nonpermeabilized HUVECs, treated with VEGF as indicated. The images show representative confocal maximal projections of surface PLA signals. c, PLA detection of VEGFR2-β1 integrin complexes in nonpermeabilized, siRNA-transfected HUVECs. Quantification shows area of PLA signals/cells. CT, control. *, p < 0.05. The data are mean values ± S.E. Bars, 20 μm.
Because integrin-dependent adhesion signaling converges with growth factor signaling (5), we examined the potential effect of loss of β1 integrin and CD63 complex formation on VEGFR2 signaling in HUVECs. As shown in Fig. 6a, a short pulse of VEGF induced tyrosine phosphorylation of VEGFR2 at the autophosphorylation sites Tyr-1175 and Tyr-951. CD63 silencing led to markedly decreased VEGFR2 tyrosine phosphorylation. Importantly, the total VEGFR2 (Fig. 6a) and β1 integrin (Fig. 6b) expression levels were not affected by CD63 deficiency. Immunoblotting for downstream signal transducers and their tyrosine-phosphorylated counterparts showed activation of PLCγ, Erk1/2, and Akt in response to VEGF stimulation in control siRNA-treated HUVECs, which was attenuated after silencing of CD63 (Fig. 6b). Other VEGF-regulated pathways involving cytoskeletal rearrangement such as FAK and Src were also impaired in CD63-deficient cells (Fig. 6, c and d).
FIGURE 6.

CD63 silencing attenuates VEGFR2 signaling. a, immunoblot analysis of VEGFR2, Tyr(P)-1175 VEGFR2, and Tyr(P)-951 VEGFR2 in whole cell lysates from serum-starved siRNA-transfected HUVECs treated with VEGF as indicated. Quantifications to the right show ratios of phosphorylated protein versus total protein bands. b, immunoblot analysis of pErk1/2, Erk1/2, Ser(P)-473 Akt, Akt, Tyr(P)-783 PLCγ, PLCγ, β1 integrin, and β2-microglobulin (β2m) in whole cell lysates from siRNA-transfected, serum-starved HUVECs, treated with VEGF as indicated. Quantifications show the ratio of phosphorylated protein versus total protein bands normalized to basal levels of siRNA control. The total levels of β1 integrin were calculated as ratios of β1 integrin versus β2m and normalized to basal levels of siRNA control. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the corresponding time points for siRNA control. c, immunofluorescent staining for pFAK397 and pSrc416 in siRNA-transfected, serum-starved HUVECs treated with VEGF for 10 min. Quantification shows the intensity of fluorescence/cell. d, immunoblot analysis of FAK, pFAK577, and pFAK861 in whole cell lysates from siRNA-transfected HUVECs. The cells were serum-starved for 12 h and stimulated with VEGF for different time periods as indicated and processed for immunoblotting, at 48 h after transfection. Quantification shows the ratio of pFAK861 protein signal versus actin (left panel) and pFAK577 protein signal versus total FAK (right panel). e, flow cytometry analysis of surface VEGFR2 in siRNA-transfected HUVECs serum-starved for 12 h, at 48 h after transfection. Quantification shows MIF of VEGFR2 compared with IgG control; normalized data show fold change in CD63-siRNA treated cells versus control siRNA cells. f, flow cytometry analysis of surface VEGFR2 in siRNA-transfected, serum-starved HUVECs treated with VEGF for 15 min, washed, and further incubated for 30 min and 1 h (indicated as ap for after pulse). Quantification shows a percentage of VEGFR2 that remained in the membrane measured as MIF of VEGFR2 compared with IgG control. The data were normalized to the corresponding basal levels in siRNA control and siRNA CD63-treated cells (right panel). g, immunoblot analysis of pErk1/2, Erk1/2, pAkt, and Akt in whole lysates from siRNA-transfected HUVECs for 36 h, serum-starved for 12 h, and stimulated with FGF2 at different time points. Quantifications show the ratio of phosphorylated protein versus total protein bands normalized to basal levels of siRNA control (CT). *, p < 0.05; **, p < 0.01. The data are mean values ± S.E. Bars, 20 μm.
Because VEGF signaling through VEGFR2 depends on the internalization and trafficking of the receptor (3), we analyzed the surface levels of VEGFR2 at different time points after a 15-min pulse of VEGF. In unstimulated ECs, silencing of CD63 resulted in increased surface levels of VEGFR2 (Fig. 6e). Moreover, CD63-deficient cells showed impaired internalization of VEGFR2 after 15 min of VEGF treatment, whereas trafficking back to the cell membrane was not affected (Fig. 6f).
Because the sprouting defects described in Fig. 2 for CD63-depleted ECs were not unique for VEGF, we also analyzed the effects of CD63 silencing on FGF2 signaling. Indeed, activation of Erk1/2 and Akt in response to FGF2 was attenuated in CD63-silenced HUVECs (Fig. 6g).
To validate the role for CD63 in VEGF-induced VEGFR2 signaling in vivo, we studied activation of VEGFR2 and its downstream signaling pathways in WT and cd63−/− mice after systemic delivery of VEGF through tail vein injection. Lungs were harvested at 1 min after VEGF injection, and tissue lysates were analyzed by immunoblotting (Fig. 7). VEGF induced very rapid phosphorylation of VEGFR2 at Tyr-949 and Tyr-1173 (corresponding to Tyr-951 and Tyr-1175 in the human VEGFR2) in WT lungs. In contrast, the phosphorylation of VEGFR2 on both tyrosines was clearly decreased in lungs from cd63−/− mice. A corresponding decrease was seen in phosphorylation of PLCγ in CD63-deficient mice. Whereas pErk1/2 was induced by VEGF in the WT mouse lung, there was no induction above the basal level of pErk1/2 in response to VEGF administration, in cd63−/− lungs. The increased basal level of pErk1/2 was most likely due to the elevated expression of Erk1/2 in the absence of CD63. These findings indicate a compensatory increase and constitutive activation of pathways regulating cell proliferation, as a consequence of impaired growth factor signal transduction in endothelial cells. Collectively, these results show an important role for CD63 in the formation of VEGFR2-β1 complexes and thereby in activation of VEGFR2 and downstream signaling.
FIGURE 7.
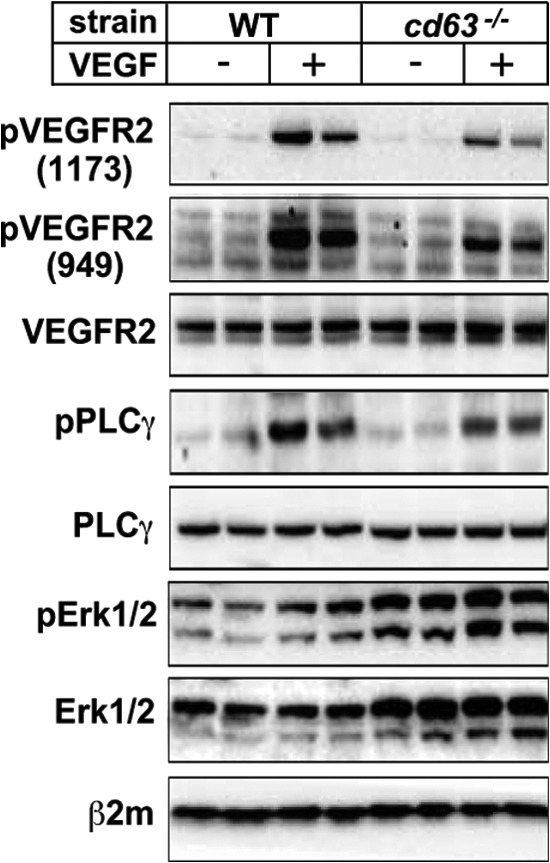
CD63 deficiency attenuates phosphorylation of VEGFR2 in vivo. WT and cd63−/− mice were injected in the tail vein with VEGF, and the lungs were harvested 1 min later. Blotting was performed on total lung lysates using antibodies against VEGFR2 phosphorylation sites Tyr(P)-1173 and Tyr(P)-949, VEGFR2, pPLCγ, PLCγ, pErk1/2, Erk1/2, and β2-microglobulin (β2m). The data are shown for two mice injected with PBS and two mice injected with VEGF per strain.
DISCUSSION
Studies addressing the role of CD63 on ECs have so far been focused on leukocyte recruitment during the initiation of inflammation (13). Here we show that the convergence between integrin and growth factor signaling in ECs depends on CD63. VEGFR2-β1 integrin complex formation involves cell surface-localized CD63, and silencing of CD63 disrupts complex formation and signaling downstream of both β1 integrin and VEGFR2. Whereas β1 integrin cell surface expression and state of activation was not affected by removal of CD63, downstream signaling was impaired, leading to defective adhesion and migration (see schematic outline in Fig. 8). VEGFR2 phosphorylation and therefore activity and consequent internalization were markedly impaired in CD63-deficient cells, leading to suppressed downstream signaling. In accordance, VEGF responsiveness in various EC in vitro assays was attenuated. The effect described here for CD63-depleted ECs is not unique for VEGF because FGF2-dependent responses were also impaired.
FIGURE 8.

Schematic figure illustrating downstream VEGFR2 signaling pathways in ECs in the presence and absence of the tetraspanin CD63. CD63 is essential for VEGFR2-β1 integrin complex formation, promoting potent activation, and signaling downstream of both β1 integrin and VEGFR2 in VEGF-treated ECs (left panel). ECs thereby undergo adhesion, migration, survival, and proliferation, essential in angiogenesis. In contrast, CD63 deficiency suppresses VEGFR2-β1 integrin complex formation, leading to impaired activation and downstream signaling and ultimately, reduced angiogenesis (right panel). The effects described here for CD63-depleted ECs are not unique for VEGF, because FGF2-dependent responses were also impaired (see text and Figs. 2a and 6g).
Even though CD63 is a well established marker for late endosomes/lysosomes, our data indicate that CD63 is localized also in the plasma membrane of resting ECs. In agreement, other studies have reported the presence of CD63 on the cell surface of several types of human cancer cells (27, 28). We propose that the membrane pool of CD63 might be responsible for all of the aspects of endothelial cell biology studied here.
To sprout toward angiogenic stimuli, ECs must coordinate their adhesion to the ECM. Integrins have a key role in anchoring ECs to matrix proteins, allowing flexible responses to changes in the microenvironment (4). There are numerous studies reporting the association of tetraspanins with integrins in different cell types (29). The main integrins found associated to tetraspanins contain the β1 subunit, an essential component in angiogenesis (30). CD63 associates with β1 integrin in human melanoma and osteosarcoma cells (31, 32), α3β1 integrin in lymphocytes and melanoma cells (33–35), α4β1 in T lymphoblasts (36), and α6β1 in different cell lines (29). We report the formation of cell surface complexes between CD63 and β1 integrin in HUVECs. The loss of CD63 resulted in changes neither in the total or cell surface expression levels β1 integrin nor in its conformational state, in agreement with earlier reports (27). CD63 has been reported to interact with β3 integrin in platelets (25); in contrast, only sparse CD63-β3 integrin complexes were detected in HUVECs, suggesting that the integrin partner for CD63 may be cell type-specific.
Integrin-induced adhesion signaling involves activation of c-Src and FAK in focal adhesions. CD63 ablation led to decreased phosphorylation of c-Src at the activating tyrosine Tyr-416, as well as decreased FAK phosphorylation at Tyr-397, Tyr-577, and Tyr-861, of which the latter is phosphorylated by c-Src and required for maximal FAK catalytic activity (37). The contribution of CD63 to adhesion-dependent signaling could rely on the ability of tetraspanins to recruit specific signaling molecules into integrin complexes (38). For example, the CD63 cytoplasmic tail interacts with c-Src in platelets (39).
Whether the activation of c-Src in ECs requires its localization in CD63-enriched microdomains needs further investigation. We found no changes in focal adhesion formation in HUVECs after CD63 silencing, indicating that the impaired activation of c-Src and FAK did not affect the formation of these structures per se. This is congruent with the fact that focal adhesions form even in the absence of FAK or Src family proteins (40). The reduced integrin signaling via c-Src and FAK in CD63-ablated cells was accompanied by reduced acute EC adhesion and migration. However, with time, the adhesion defect subsided, and under conditions where growth factor signaling was analyzed, CD63-silenced ECs showed adhesion comparable to control siRNA-treated cells. Therefore, we do not regard the decrease in VEGFR2 activation and signaling in the absence of CD63 as a consequence of impaired adhesion and spreading of cells as such, but as a result of the interrupted complex formation with CD63 and β1 integrin.
Using in situ PLA, we identified different constellations of cell surface complexes in quiescent ECs: CD63-VEGFR2, VEGFR2-β1 integrin, and CD63-β1 integrin. In the absence of CD63, there was a clear reduction in the number of VEGFR2-β1 integrin complexes. These data suggest the existence of trimeric VEGFR2-CD63-β1 integrin complexes. The phosphorylation levels of VEGFR2 and downstream signaling molecules were markedly decreased in VEGF-stimulated CD63 siRNA-treated cells, indicating an important role for CD63-containing complexes in VEGFR2 activation. How might this occur? Tetraspanins provide a framework to organize proteins on the cell membrane (41). Thus, CD63 might be important for maintaining properly structured TEMs in ECs, thereby facilitating the formation and stability of signaling complexes involving VEGFR2 and β1 integrin. VEGFR2-β1 integrin complexes have previously been implicated in the phosphorylation of VEGFR2 at Tyr-1214 when HUVECs were treated with matrix-bound VEGF (23). Our data show that disruption of VEGFR2-β1 integrin complexes attenuated phosphorylation of VEGFR2 at both Tyr-1175 and Tyr-951, accompanied by marked reduction in all analyzed downstream pathways, implying a more global role for integrated VEGFR2 and β1 integrin signaling. In accordance with the in vitro results, the phosphorylation of VEGFR2 and PLCγ in cd63−/− lungs after systemic VEGF delivery was markedly decreased. On the other hand, phosphorylation of Erk1/2 did not increase in response to VEGF, at least in part because of the elevated expression levels of Erk1/2 and increased constitutive phosphorylation in the absence of CD63. Modulation of Erk1/2 expression levels in the CD63-deficient mice may indicate a compensatory effect because of the loss of signaling regulating endothelial cell proliferation. Because CD63 is widely expressed (29), we cannot exclude that the effects observed for VEGF signaling to some extent is influenced by the loss of CD63 in other cell types. Specific deletion of CD63 in ECs is required to further address the role of CD63 in EC physiology and pathology.
To what extent the effect of CD63 ablation on VEGFR2 activation was dependent on a loss in potential TEM clustering or more specifically on loss in VEGFR2 association with β1 integrin is difficult to distinguish. The existence of a tight knit molecular cross-talk between receptor tyrosine kinases and integrins is well established, often involving Src family kinases (42, 43). For instance, c-Src activity is crucial for the integrin-induced phosphorylation of EGFR and VEGFR3 (44, 45). In a reciprocal reaction, receptor tyrosine kinases may affect integrin biology. Thus, VEGF receptor-mediated activation of FAK via c-Src allows formation of FAK-αvβ5 integrin signaling complexes (46). Furthermore, VEGF induces α3 integrin tyrosine phosphorylation via c-Src, regulating adhesion and migration of endothelial cells (47).
Our data implied a role for CD63 in trafficking of VEGFR2, which may be direct or indirect. The cell surface levels of VEGFR2 and the fraction of VEGFR2 that remained cell surface-expressed in the presence of VEGF were increased in the absence of CD63. In accordance, CD63 has been implicated in the trafficking of the chemokine receptor CXCR4, (48), and of P-selectin (13). Moreover, adaptor protein complex-3 deficiency in patients with Hermansky-Pudlak syndrome is associated with increased surface expression of CD63 (21).
The reduced VEGFR2 internalization in the absence of CD63 may be a consequence of the impaired VEGFR2 tyrosine phosphorylation and therefore kinase activation. It is well known that activated growth factor receptors are rapidly internalized to attenuate signaling. Moreover, endocytosis of tyrosine kinase receptors is required for induction of certain pathways, because substrates are differently distributed between endosomes and the plasma membrane (49). Thus, the impaired signal transduction downstream VEGFR2 observed in CD63-silenced cells may in part be a consequence of a deficient internalization of the receptor, which in turn may further have impaired downstream signaling.
Even though several tetraspanins have been implicated as key players in vascular functions (50, 51), their knock-out phenotypes are usually subtle or undetectable. Here, we show that endothelial cells in cd63−/− mouse lungs failed to respond efficiently to VEGF stimulation, resulting in suppressed VEGFR2 activation and signaling. Still, CD63 knock-out mice survive embryonic development without overt signs of disturbed vascular development (12), possibly because of redundancy or functional compensation among different tetraspanin members or between different angiogenic pathways in a global CD63 knock-out. Clearly therefore, further studies of the consequence of conditional deletion of CD63 in ECs are warranted.
Acknowledgments
We thank Dr. Kathrin Zeller and Prof. Staffan Johansson (Uppsala University) for expert advice and Prof. Paul Saftig (Christian-Albrechts-Universität Kielfor) the kind gift of cd63−/− mice.
This work was supported by grants from the Swedish Cancer Foundation, the Swedish Science Council, and the Knut and Alice Wallenberg Foundation (to L. C.-W.).
- ECM
- extracellular matrix
- EC
- endothelial cell
- FAK
- focal adhesion kinase
- TEM
- tetraspanin-enriched microdomain
- HUVEC
- human umbilical vein endothelial cell
- PFA
- paraformaldehyde
- PLA
- proximity ligation assay.
REFERENCES
- 1. Potente M., Gerhardt H., Carmeliet P. (2011) Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 [DOI] [PubMed] [Google Scholar]
- 2. Eilken H. M., Adams R. H. (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 22, 617–625 [DOI] [PubMed] [Google Scholar]
- 3. Koch S., Tugues S., Li X., Gualandi L., Claesson-Welsh L. (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 437, 169–183 [DOI] [PubMed] [Google Scholar]
- 4. Margadant C., Monsuur H. N., Norman J. C., Sonnenberg A. (2011) Mechanisms of integrin activation and trafficking. Curr. Opin. Cell Biol. 23, 607–614 [DOI] [PubMed] [Google Scholar]
- 5. Mitra S. K., Schlaepfer D. D. (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 [DOI] [PubMed] [Google Scholar]
- 6. Ramjaun A. R., Hodivala-Dilke K. (2009) The role of cell adhesion pathways in angiogenesis. Int. J. Biochem. Cell Biol. 41, 521–530 [DOI] [PubMed] [Google Scholar]
- 7. Ross R. S. (2004) Molecular and mechanical synergy. Cross-talk between integrins and growth factor receptors. Cardiovasc. Res. 63, 381–390 [DOI] [PubMed] [Google Scholar]
- 8. Hemler M. E. (2005) Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 [DOI] [PubMed] [Google Scholar]
- 9. Yunta M., Lazo P. A. (2003) Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell. Signal. 15, 559–564 [DOI] [PubMed] [Google Scholar]
- 10. Yáñez-Mó M., Barreiro O., Gordon-Alonso M., Sala-Valdés M., Sánchez-Madrid F. (2009) Tetraspanin-enriched microdomains. A functional unit in cell plasma membranes. Trends Cell Biol. 19, 434–446 [DOI] [PubMed] [Google Scholar]
- 11. Hotta H., Ross A. H., Huebner K., Isobe M., Wendeborn S., Chao M. V., Ricciardi R. P., Tsujimoto Y., Croce C. M., Koprowski H. (1988) Molecular cloning and characterization of an antigen associated with early stages of melanoma tumor progression. Cancer Res. 48, 2955–2962 [PubMed] [Google Scholar]
- 12. Schröder J., Lüllmann-Rauch R., Himmerkus N., Pleines I., Nieswandt B., Orinska Z., Koch-Nolte F., Schröder B., Bleich M., Saftig P. (2009) Deficiency of the tetraspanin CD63 associated with kidney pathology but normal lysosomal function. Mol. Cell. Biol. 29, 1083–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doyle E. L., Ridger V., Ferraro F., Turmaine M., Saftig P., Cutler D. F. (2011) CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 118, 4265–4273 [DOI] [PubMed] [Google Scholar]
- 14. Koyama Y., Suzuki M., Yoshida T. (1998) CD63, a member of tetraspan transmembrane protein family, induces cellular spreading by reaction with monoclonal antibody on substrata. Biochem. Biophys. Res. Commun. 246, 841–846 [DOI] [PubMed] [Google Scholar]
- 15. Toothill V. J., Van Mourik J. A., Niewenhuis H. K., Metzelaar M. J., Pearson J. D. (1990) Characterization of the enhanced adhesion of neutrophil leukocytes to thrombin-stimulated endothelial cells. J. Immunol. 145, 283–291 [PubMed] [Google Scholar]
- 16. Skubitz K. M., Campbell K. D., Iida J., Skubitz A. P. (1996) CD63 associates with tyrosine kinase activity and CD11/CD18, and transmits an activation signal in neutrophils. J. Immunol. 157, 3617–3626 [PubMed] [Google Scholar]
- 17. Mantegazza A. R., Barrio M. M., Moutel S., Bover L., Weck M., Brossart P., Teillaud J. L., Mordoh J. (2004) CD63 tetraspanin slows down cell migration and translocates to the endosomal-lysosomal-MIICs route after extracellular stimuli in human immature dendritic cells. Blood 104, 1183–1190 [DOI] [PubMed] [Google Scholar]
- 18. Radford K. J., Thorne R. F., Hersey P. (1997) Regulation of tumor cell motility and migration by CD63 in a human melanoma cell line. J. Immunol. 158, 3353–3358 [PubMed] [Google Scholar]
- 19. Lee M., Hadi M., Halldén G., Aponte G. W. (2005) Peptide YY and neuropeptide Y induce villin expression, reduce adhesion, and enhance migration in small intestinal cells through the regulation of CD63, matrix metalloproteinase-3, and Cdc42 activity. J. Biol. Chem. 280, 125–136 [DOI] [PubMed] [Google Scholar]
- 20. Jung K. K., Liu X. W., Chirco R., Fridman R., Kim H. R. (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 25, 3934–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dell'Angelica E. C., Shotelersuk V., Aguilar R. C., Gahl W. A., Bonifacino J. S. (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β 3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 [DOI] [PubMed] [Google Scholar]
- 22. Bailey R. L., Herbert J. M., Khan K., Heath V. L., Bicknell R., Tomlinson M. G. (2011) The emerging role of tetraspanin microdomains on endothelial cells. Biochem. Soc. Trans. 39, 1667–1673 [DOI] [PubMed] [Google Scholar]
- 23. Vischer U. M., Wagner D. D. (1993) Cd63 Is A Component of Weibel-Palade Bodies of Human Endothelial-Cells. Blood 82, 1184–1191 [PubMed] [Google Scholar]
- 24. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation in situ of individual endogenous protein complexes by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 25. Israels S. J., McMillan-Ward E. M., Easton J., Robertson C., McNicol A. (2001) CD63 associates with the αIIb β3 integrin-CD9 complex on the surface of activated platelets. Thromb. Haemost. 85, 134–141 [PubMed] [Google Scholar]
- 26. Chen T. T., Luque A., Lee S., Anderson S. M., Segura T., Iruela-Arispe M. L. (2010) Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J. Cell Biol. 188, 595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Latysheva N., Muratov G., Rajesh S., Padgett M., Hotchin N. A., Overduin M., Berditchevski F. (2006) Syntenin-1 is a new component of tetraspanin-enriched microdomains. Mechanisms and consequences of the interaction of syntenin-1 with CD63. Mol. Cell. Biol. 26, 7707–7718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lekishvili T., Fromm E., Mujoomdar M., Berditchevski F. (2008) The tumour-associated antigen L6 (L6-Ag) is recruited to the tetraspanin-enriched microdomains. Implication for tumour cell motility. J. Cell Sci. 121, 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berditchevski F. (2001) Complexes of tetraspanins with integrins. More than meets the eye. J. Cell Sci. 114, 4143–4151 [DOI] [PubMed] [Google Scholar]
- 30. Eliceiri B. P., Cheresh D. A. (2001) Adhesion events in angiogenesis. Curr. Opin. Cell Biol. 13, 563–568 [DOI] [PubMed] [Google Scholar]
- 31. Radford K. J., Thorne R. F., Hersey P. (1996) CD63 associates with transmembrane 4 superfamily members, CD9 and CD81, and with β1 integrins in human melanoma. Biochem. Biophys. Res. Commun. 222, 13–18 [DOI] [PubMed] [Google Scholar]
- 32. Iizuka S., Kudo Y., Yoshida M., Tsunematsu T., Yoshiko Y., Uchida T., Ogawa I., Miyauchi M., Takata T. (2011) Ameloblastin regulates osteogenic differentiation by inhibiting Src kinase via cross talk between integrin β1 and CD63. Mol. Cell. Biol. 31, 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berditchevski F., Bazzoni G., Hemler M. E. (1995) Specific association of Cd63 with the Vla-3 and Vla-6 integrins. J. Biol. Chem. 270, 17784–17790 [DOI] [PubMed] [Google Scholar]
- 34. Berditchevski F., Zutter M. M., Hemler M. E. (1996) Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins). Mol. Biol. Cell 7, 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berditchevski F., Tolias K. F., Wong K., Carpenter C. L., Hemler M. E. (1997) A novel link between integrins, transmembrane-4 superfamily proteins (CD63 and CD81), and phosphatidylinositol 4-kinase. J. Biol. Chem. 272, 2595–2598 [DOI] [PubMed] [Google Scholar]
- 36. Mannion B. A., Berditchevski F., Kraeft S. K., Chen L. B., Hemler M. E. (1996) Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63, and CD53 specifically associate with integrin α4β1 (CD49d/CD29). J. Immunol. 157, 2039–2047 [PubMed] [Google Scholar]
- 37. Calalb M. B., Polte T. R., Hanks S. K. (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity. A role for Src family kinases. Mol. Cell. Biol. 15, 954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hemler M. E. (1998) Integrin associated proteins. Curr. Opin. Cell Biol. 10, 578–585 [DOI] [PubMed] [Google Scholar]
- 39. Heijnen H. F., Van Lier M., Waaijenborg S., Ohno-Iwashita Y., Waheed A. A., Inomata M., Gorter G., Möbius W., Akkerman J. W., Slot J. W. (2003) Concentration of rafts in platelet filopodia correlates with recruitment of c-Src and CD63 to these domains. J. Thromb. Haemost. 1, 1161–1173 [DOI] [PubMed] [Google Scholar]
- 40. Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T., Aizawa S. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from Fak-deficient mice. Nature 377, 539–544 [DOI] [PubMed] [Google Scholar]
- 41. Levy S., Shoham T. (2005) Protein-protein interactions in the tetraspanin web. Physiology 20, 218–224 [DOI] [PubMed] [Google Scholar]
- 42. Sundberg C., Rubin K. (1996) Stimulation of β1 integrins on fibroblasts induces PDGF independent tyrosine phosphorylation of PDGF beta-receptors. J. Cell Biol. 132, 741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang R., Kobayashi R., Bishop J. M. (1996) Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. U.S.A. 93, 8425–8430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X., Groopman J. E., Wang J. F. (2005) Extracellular matrix regulates endothelial functions through interaction of VEGFR-3 and integrin α5β1. J. Cell Physiol. 202, 205–214 [DOI] [PubMed] [Google Scholar]
- 45. Moro L., Venturino M., Bozzo C., Silengo L., Altruda F., Beguinot L., Tarone G., Defilippi P. (1998) Integrins induce activation of EGF receptor. Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17, 6622–6632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eliceiri B. P., Puente X. S., Hood J. D., Stupack D. G., Schlaepfer D. D., Huang X. Z., Sheppard D., Cheresh D. A. (2002) Src-mediated coupling of focal adhesion kinase to integrin αvβ5 in vascular endothelial growth factor signaling. J. Cell Biol. 157, 149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahabeleshwar G. H., Feng W., Reddy K., Plow E. F., Byzova T. V. (2007) Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circ. Res. 101, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pols M. S., Klumperman J. (2009) Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 315, 1584–1592 [DOI] [PubMed] [Google Scholar]
- 49. Hupalowska A., Miaczynska M. (2012) The new faces of endocytosis in signaling. Traffic 13, 9–18 [DOI] [PubMed] [Google Scholar]
- 50. Takeda Y., Kazarov A. R., Butterfield C. E., Hopkins B. D., Benjamin L. E., Kaipainen A., Hemler M. E. (2007) Deletion of tetraspanin Cd151 results in decreased pathologic angiogenesis in vivo and in vitro. Blood 109, 1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamisasanuki T., Tokushige S., Terasaki H., Khai N. C., Wang Y., Sakamoto T., Kosai K. (2011) Targeting CD9 produces stimulus-independent antiangiogenic effects predominantly in activated endothelial cells during angiogenesis. A novel antiangiogenic therapy. Biochem. Biophys. Res. Commun. 413, 128–135 [DOI] [PubMed] [Google Scholar]