

Background: The Tat OYI vaccine might reduce HIV-1-infected cells due to neutralizing antibodies targeting extracellular Tat.
Results: MIMOOX is a peptide designed to mimic a three-dimensional epitope of Tat OYI-inducing neutralizing antibodies against Tat variants.
Conclusion: There is a highly conserved surface on Tat variants that the Tat OYI vaccine helps to recognize.
Significance: The Tat OYI vaccine could be an alternative to antiretroviral therapy.
Keywords: Cellular Immune Response, Epitope Mapping, HIV-1, Protein Design, Vaccine Development, HIV-1, Tat, Vaccine
Abstract
Extracellular Tat is suspected to protect HIV-1-infected cells from cellular immunity. Seropositive patients are unable to produce neutralizing antibodies against Tat, and Tat is still secreted under antiviral treatment. In mice, the Tat OYI vaccine candidate generates neutralizing antibodies such as the mAb 7G12. A peptide called MIMOOX was designed from fragments of Tat OYI identified as the possible binding site for mAb 7G12. MIMOOX was chemically synthesized, and its structure was stabilized with a disulfide bridge. Circular dichroism spectra showed that MIMOOX had mainly β turns but no α helix as Tat OYI. MIMOOX was recognized by mAb 7G12 in ELISA only in reduced conditions. Moreover, a competitive recognition assay with mAb 7G12 between MIMOOX and Tat variants showed that MIMOOX mimics a highly conserved surface in Tat variants. Rat immunizations with MIMOOX induce antibodies recognizing Tat variants from the main HIV-1 subtypes and confirm the Tat OYI vaccine approach.
Introduction
Tat is a potential target for the development of a vaccine (1). Tat is a short protein (average 101 residues) expressed in the early stages of infection that was first described as a trans-activator of HIV-1 genes (2). However, another interesting property of Tat is its extracellular role and its high level of secretion from HIV-1-infected cells, suggesting that this viral protein plays a major role in the capacity of HIV-1 to evade immune response in infected patients (3). Extracellular Tat crosses the membrane of a diversity of uninfected bystander cells such as cytotoxic T lymphocytes to trigger apoptosis (4). Moreover, it was reported that antiretroviral treatments (ART)2 do not block the secretion of Tat even for HIV-1-infected patients with an undetectable viremia, which may explain why cellular immunity is ineffective against HIV-1-infected cells (5).
Serological studies show that only half of the HIV-infected patients have antibodies against Tat either in Africa (6) or Europe (5). A serological survey for one year shows that Tat antibodies can disappear and/or appear with blood samplings carried out every three months in a same patient (5). Up to 40% diversity is observed between Tat variant amino acid sequences from the five main HIV-1 subtypes or clades (1). Human anti-Tat antibodies recognized mainly variable regions of the protein and do not have a cross-clade capacity (5, 6). Moreover, anti-Tat antibodies appear to have no impact on disease progression since patients with or without anti-Tat antibodies progress to AIDS without ART (7). For patients under ART with or without an undetectable viremia, antibodies against Tat have no effect on the level of CD4 or CD8 T cells (5).
Tat is divided into six different regions: region I (residues 1–21) is a proline-rich region and has a conserved Trp11; region II (residues 22–37) has seven well conserved cysteines; region III (residues 38–48) has a conserved Phe38 and the conserved sequence 43LGISYG; region IV (residues 49–59) is rich in basic residues and has the rather well conserved sequence 49RKKRRQRRRPP; region V (residues 60–72) is the glutamine-rich region and has the highest rate of sequence variation; and region VI constitutes the C terminus of Tat (1). The basic region (IV) appear to be the most important functional region of Tat because it is essential to cross the cell membrane and to bind on the trans-activating region an hairpin structure at the 5′-end of the viral mRNA, but it is not recognized by sera from HIV-1-infected patients due to sequence homology with human proteins such as protamine (1). However, regions III and V are important because they play a role of hinge for region IV (8). The presence of a disulfide bridge in region II was proposed (9), but the transactivation activity with full Tat protein requires free cysteines (1). There is a controversy regarding Tat structure with studies showing that Tat has a folding (10–14) and studies showing that Tat is a naturally unfolded protein (15–16). Tat NMR studies on full size Tat variants with the biological activity show that Trp11 and Phe38 constitute an hydrophobic core with other aliphatic residues from other regions, whereas most of regions II, III, IV, and VI are exposed to the solvent with β turns constituting the main secondary structure (1). In the blood of HIV-1-infected patients, Tat variants are naturally folded proteins (17).
Different approaches to obtain a vaccine against Tat were developed but were unable to prevent viral rebound after ART cessation in phase II clinical trial (18–27). We plan to test in a phase II clinical trial a vaccine using the Tat OYI variant (1). The base of this approach was the study of an HIV-1-infected cohort of 23 patients who resisted to HIV-1 infection in Gabon (28). HIV-1 OYI was isolated from one of these Gabonese patients, and Tat OYI has specific mutations never observed in other Tat variants (29). Experiments on rabbits immunized with Tat OYI show a production of antibodies recognizing not only Tat OYI but also Tat variants from the five major HIV-1 subtypes (30). Furthermore, a Tat OYI vaccine protected macaques from heterologous mucosal SHIV challenge with SHIV-infected cells no longer detectable two months after SHIV challenge (31). Tat OYI lost its capacity to induce cross-clade antibodies once it is denatured, showing that a three-dimensional epitope is probably involved to induce cross-clade antibodies (30). The rational of the Tat OYI vaccine is that specific mutations would transform a highly conserved surface in Tat variants in a three-dimensional epitope triggering the production of neutralizing antibodies recognizing all Tat variants. Mouse immunization with Tat OYI made it possible to obtain a cross-clade neutralizing mAb, called 7G12, thereby confirming the rational of the Tat OYI vaccine (32). mAb 7G12 cannot recognize unfolded Tat OYI (32). It is worth noting that all other mAbs obtained after immunizing mice with distinct Tat variants or Tat-derived peptides are not able to recognize and neutralize different Tat variants (33–37).
The purpose of the present study was to identify the Tat OYI three-dimensional epitope recognized by mAb 7G12. From fragments of a Tat OYI sequence gathered on a same spot of Tat OYI surface and suspected to be the possible binding site of mAb 7G12. A molecular modeling of a peptide of 56 residues called MIMOOX3 was designed to mimic the Tat OYI three-dimensional epitope recognized by mAb 7G12. MIMOOX was synthesized and successfully purified. Circular dichroism spectra show that MIMOOX has mainly β turn secondary structures and no α helix as Tat OYI. In contrast to Tat OYI, MIMOOX fails to stimulate reporter gene expression in Hela P4 cells. mAb 7G12 is able to recognize MIMOOX in ELISA. MIMOOX has a disulfide bridge that does not exist in Tat variants. mAb 7G12 cannot recognize MIMOOX without the disulfide bridge. Rat immunizations with MIMOOX induce antibodies able to recognize Tat OYI and Tat variants from the five main HIV-1 subtypes. This study confirms the rational of the Tat OYI vaccination with the identification of a highly conserved surface in Tat variants essential for its activity, which is recognized by the mAb 7G12.
EXPERIMENTAL PROCEDURES
Tat OYI and MIMOOX Molecular Modeling
Molecular modeling was carried out with the Insight II package, including Biopolymer, Discover, and Homology (Accelrys, San Diego, CA). The Tat OYI model was obtained from molecular modeling using sequence homologies with the Tat ELI NMR structure (13). The consistent valence force field was used to determine the internal energies, using van der Waals energy to monitor each step of the modeling. The pH was set up at 7. Energy minimization was performed with steepest descent and conjugate gradient algorithms with a maximum derivative of 0.001 kcal/Å in the final steps. Dynamic steps were performed at 300 K for 1.1 ps.
Tat Variants and MIMO Synthesis
MIMO, Tat OYI peptides, and Tat variants representative of the main HIV-1 subtypes in the world (UG11RP (A), OYI(B), 96BW(C), ELI(D), CRF_AE(AE)) were synthesized in solid phase Fmoc chemistry and purified as described previously (14). Mass and purity were analyzed by mass spectrometry using an Ettan matrix-assisted laser desorption ionization time-of-flight apparatus (Amersham Biosciences). Purity of the protein was up to 95%. After lyophilization, biological activity of Tat variants was checked by transactivation assays with HeLa P4 cells as described previously (13). MIMO cysteine residues (Cys22, Cys54/Cys55) were oxidized to obtain a disulfide bridge in 100 mm phosphate buffer at pH 8.3 for 6 h and 30 min. A kinetic was done to determine optimum MIMO oxidation. MIMO-oxidized, named MIMOOX, was desalted in HPLC, and the main peak was recovered. MIMO and MIMOOX outlines were compared with HPLC analysis, and the oxidation (disulfide bridge formation) was controlled by alkylation assays using 85 mm iodoacetamide (Sigma). Alkylated proteins were compared in mass spectrometry before and after oxidation.
CD
CD spectra were measured with a 100-μm path length from 260–178 nm at 10–70 °C on a JASCO J-810 spectropolarimeter. Data were collected at 0.5-nm intervals using a step auto response procedure (JASCO). CD spectra were presented as Δϵ per amide. Protein concentration was 1 mg/ml in 20 mm phosphate buffer, pH 4.5, for MIMOOX and Tat OYI. The CD data were analyzed with VARSELEC software to determine the secondary structure content according to the method of Manavalan and Johnson (38) using a set of 32 reference proteins and an average of 4960 calculations.
Transactivation Activity
Transactivation was performed with HIV LTR-transfected HeLa cells (HeLa-P4) and analyzed by monitoring the production of β-gal as described previously (13). Briefly, Hela-P4 cells were cultivated in DMEM with glutamine supplemented with 10% (v/v) heat-inactivated fetal calf serum and 50 units/ml neomycin. 2 × 105 cells/well were incubated in 24-well plates (Falcon) in 400 μl of DMEM supplemented with 0.01% (w/v) protamine (Sigma-Aldrich) and 0.1% (w/v) BSA (Sigma). Lyophilized Tat OYI, MIMOOX, and MIMO were diluted in 100 mm phosphate buffer, pH 4.5 (5 μm final concentration) Volumes were completed to with 20 mm Hepes buffer, 120 mm NaCl, pH 7.3 (0.5 μm final concentration). After 18 h at 37 °C, β-gal production was measured with a β-gal ELISA kit (Roche Diagnostics). Background against BSA was removed. The positive control was the amount of β-gal in presence of Tat OYI C22 and the negative control was the amount of β-gal without Tat OYI C22.
Dot Blot
MIMO, MIMOOX, denatured MIMO with urea, denatured MIMOOX with urea, and DTT were spotted (50 ng) on nitrocellulose membranes. MIMO was denatured by heating at 90 °C for 45 min in the presence of 3 m urea. MIMOOX was denatured by heating at 90 °C for 45 min in presence of 3 m urea and 20 mm DTT. Nitrocellulose membranes were incubated for 1 h with PBS supplemented with 5% milk and then overnight with mAbs 7G12 (1 mg/ml) or 6E7 (0.1 mg/ml) at 1/500 and 1/5 in PBS with 0.5% milk at 4 °C, respectively. After washing steps, HRP-conjugated anti-mouse (1/1000, GE Healthcare) were added and incubated for 1 h at room temperature. After washing steps, spots were revealed with tetramethylbenzidine (BIOFAX) as substrate.
Production of MIMOOX Antibodies
Five Wistar rats were immunized by the intramuscular route in the lower back with 25 μg of MIMOOX bound on the surface of immunopotentiating reconstituted influenza virosomes (39) with the last free cysteine in C-terminal. Rat immune systems were boosted with the same preparation 4 and 8 weeks later. Rats were euthanized and terminally bled on week 10. Serum was obtained after centrifugation at 4500 rpm during 30 min at 4 °C and tested for MIMOOX and Tat variant cross-recognition in ELISA.
Antibody Affinity Determination by ELISA
Plates (96-wells) were coated with MIMOOX (25 ng) and Tat (50 ng) or denatured MIMOOX (25 ng) in 100 μl of 100 mm phosphate buffer, pH 4. MIMOOX (0.25 μg/ml) was denatured by heating at 90 °C for 45 min in presence of 3 m urea, 20 mm DTT and then diluted 10-fold with 100 mm cold phosphate buffer, pH 4.5. To control urea/DTT effect, native MIMOOX was coated in presence to 0.3 m urea, 2 mm DTT. Nonspecific signal was controlled in coating native or denatured BSA with the same protocol. Following blocking with 2% skim milk and washing steps, plates were incubated with 100 μl of week 10 rat serum (or serum before rat immunizations) at dilution 1/20 or with 100 μl of diluted mouse mAbs 7G12 (1 mg/ml) or with 6E7 (0.1 mg/ml) for 1 h at room temperature. These two mAbs were previously characterized as a conformational and linear anti-Tat antibody, respectively (5). After washing steps, 100 μl of HRP-conjugated anti-rat or anti-mouse (1/1000, GE Healthcare) were added, and the plate was incubated for 1 h at room temperature. After washing steps, 100 μl of ABTS substrate (Roche Diagnostics) were added. The absorbance was measured at wavelength 405 nm 1 h later. An ELISA-based method was used as described previously (40) for measurement association rate constants of mAb 7G12 with MIMOOX in solution.
Competitive Recognition Assays between MIMOOX and Tat Variants
Plates (96 wells) were coated with 25 ng of MIMOOX (or Tat variants) in 100 μl of 100 mm phosphate buffer, pH 4.5. Following blocking with 5% skim milk and washing steps, plates were incubated with 100 μl of mAb7G12 in an equimolar amount relative to coated protein mixed with different dilutions of native or denatured Tat OYI (or MIMOOX) for 20 min at room temperature. Because DTT could not be used, MIMO denatured by heating at 90 °C for 45 min in the presence of 3 m urea and diluted 10-fold was used as “denatured MIMOOX.” Assays with native proteins were performed in the presence of 0.3 m urea. Nonspecific signal was controlled on coating BSA. The other steps of the protocol were done as described above, but the absorbance was measured 30 min later.
Statistical Analysis
Statistical differences were analyzed with a Mann-Whitney test. A p value < 0.05 was considered significant.
RESULTS
Molecular Modeling of Tat OYI
From the atomic coordinates of the Tat ELI two-dimensional NMR structure (13), a Tat OYI model was built using the Insight II software. The Tat ELI coordinates were directly used to build the model when a strict sequence homology of three residues or more was observed, whereas only the Cα coordinate was used in case of a partial homology. Due to sequence homology between the two sequences, no loop research was necessary to build the Tat OYI model. Once atomic coordinates for all Tat OYI atoms were attributed, the Tat OYI model was optimized with energy minimization algorithms associated to a dynamic at 300 K. An analysis of the dynamic trajectory determined the lowest energy wheel corresponding to the most probable folding. A final step of minimization made possible to obtain a Tat OYI model (Fig. 1A) with a van der Waals energy similar to the Tat ELI two-dimensional NMR structure.
FIGURE 1.

Tat OYI and MIMOOX molecular modeling. Tat OYI (A, C) is divided into six regions: region I (residues 1–21) is depicted in red, region II (cysteine-rich region, residues 22–37) in orange, region III (residues 38–48) in yellow, region IV (basic region, residues 49–59) in green, region V (residues 60–72) in light blue, region VI (residues 73–101) in blue. MIMOOX (B and C) comprises the N-terminal region (red), followed by the inverted sequence of the C terminus (blue) and then region III (yellow), and a part of the arginine-rich region (green). Glycines serve as a linker between the three Tat regions are in purple. Two of the three cysteines create a disulfide bridge that is essential to have a three-dimensional epitope in the MIMO equivalent to the three-dimensional epitope in Tat OYI.
Molecular Modeling of MIMOOX
A highly conserved surface on Tat variants involves highly conserved amino acids on Tat variants. The most conserved sequence in Tat variants corresponds to segments 44 and 50 located in region III. Fig. 1 shows that region III in yellow is surrounded by short sequences of region I in red, region III in green, and region VI in blue. The amino acid glutamate at position 100 (presents only in Tat OYI) could play an important role because it displays a charge in this conserved surface that might transform this surface in an epitope only for Tat OYI (1). Therefore, three β turns involving sequences (1MEPV4, 44GISYGRKK51, and three residues of the C terminus (99PED101)) gathered on the same surface of Tat OYI (Fig. 1A) were identified as the possible binding site of mAb 7G12. To validate this hypothesis, a linear peptide called MIMO was designed to reproduce a surface similar to the Tat OYI surface where the following sequences are gathered: 1MEPV4, 44GISYGRKK51, and 99PED101). It was necessary to use other Tat OYI amino acid residues that are not exposed but play a structural role to obtain the appropriate structure. Therefore, three peptides corresponding to the Tat OYI sequences 1–21, 38–54, and 91–101 were used to reproduce the Tat OYI three-dimensional epitope (Fig. 1, B and C). MIMO has 56 residues, including four glycines and three cysteines, that are not found in Tat OYI sequence. There is no disulfide bridge in Tat variants, and the highly conserved cysteines, which are found exclusively in the cysteine-rich region from positions 22 to 37, must be free for Tat transactivation activity (1). The role of the disulfide bridge in MIMO is to block the conformation of the three Tat OYI sequences as it is in Tat OYI. The role of the four glycines is to make the disulfide bridge possible . Energy minimization was performed on MIMO without the disulfide bridge, and no change was observed in the conformation. It was still possible to create the disulfide bridge after energy minimization, which was the goal of modeling. MIMO with the disulfide bridge was called MIMOOX.
Synthesis and Characterization of MIMO and MIMOOX
After chemical synthesis, purity and mass were controlled by HPLC (Fig. 2A) and mass spectrometry (Fig. 2B). To verify oxidation, MIMO and MIMOOX were compared by HPLC analysis (Fig. 2A). A delay of 1 min was observed between the peak of MIMO before and after oxidation, suggesting an increase of hydrophobicity due to the disulfide bond. MIMO (before oxidation) and MIMOOX were alkylated, and their mass was controlled in mass spectrometry (Fig. 2B). Alkylated MIMO mass showed an increase of 171 Da, corresponding to a covalent binding following a nucleophile chemical reaction of the free thiol groups of the three cysteines with iodoacetamide (Fig. 1B). After oxidation, a mass increase of 57 Da only was observed, corresponding to one alkylation. These results confirmed the establishment of a disulfide bond between the cysteine at position 22 and the C-terminal cysteines. CD spectra of MIMOOX and Tat OYI were similar (Fig. 2C). According to the method of Manavalan and Johnson (38), mostly β turn structures were deduced from CD spectra analysis, but no α helix was found. The transactivation activity of MIMOOX and MIMO was tested with Tat OYI with a cysteine in position 22 as a positive control and without Tat as a negative control. A basal level of transactivation similar to the negative control was observed with MIMOOX and MIMO (data not shown).
FIGURE 2.

Control of MIMOOX folding. A, oxidation of MIMOOX was verified in HPLC method between T = 0 h (dashed line) and T = 6 h (continuous line). B, mass spectrometry after alkylation with iodoacetamide of MIMO (black), MIMOOX in oxidation conditions at T = 0 h (dashed lines) and at T = 6 h (gray). In C, CD spectra of MIMOOX and Tat OYI were measured from 260 to 178 nm with a 50-mm path length in 20 mm phosphate buffer (pH 4.5). The same percentage of secondary structures of MIMOOX and Tat OYI was determined from the CD data analysis with no α helix, 23% extended structure, 30% β turns, and 47% of other structures. The absence of the α helix in MIMOOX is in accordance with a similar structure of the conserved surface of Tat variants that contain only β turn secondary structure. Furthermore, MIMOOX having 56 residues, 30% β turns correspond to four β turns. This is compatible with the presence of at least β turns in the MIMOOX structure. mAU, milliabsorbance units.
MIMOOX Is Recognized by the mAb 7G12
An ELISA-based method was used to measure antigen/antibody association rate constants in solution. Antigen and antibody were mixed and aliquots were withdrawn at different time intervals to determine the amount of free antibodies. The disappearance of free antibodies reflected the time course of the association reaction. Affinity curves (Fig. 3A) showed that MIMOOX is recognized by mAb 7G12 with an affinity constant obtained for MIMOOX (KD = 15.8 ± 0.5 nm) twice lower compared with Tat OYI (KD = 7 ± 0.4 nm). In ELISA, this recognition was not longer observed with a reducing reagent (Fig. 3B). As a control, another monoclonal antibody called mAb 6E7 was used (Fig. 3B). The mAb 6E7 recognized a linear epitope located in the N terminus of Tat OYI whatever its folding (32). A Dot Blot assay was performed to test mAbs 7G12 and 6E7 recognition against denatured MIMO, native MIMOOX, and denatured MIMOOX (Fig. 3C). The mAb 7G12 did not recognize denatured MIMO or denatured MIMOOX, whereas mAb 6E7 was able to recognize denatured MIMOOX. These results showed that mAb 7G12 recognizes a conformation of MIMOOX and not a sequence corresponding to a linear epitope as mAb 6E7.
FIGURE 3.
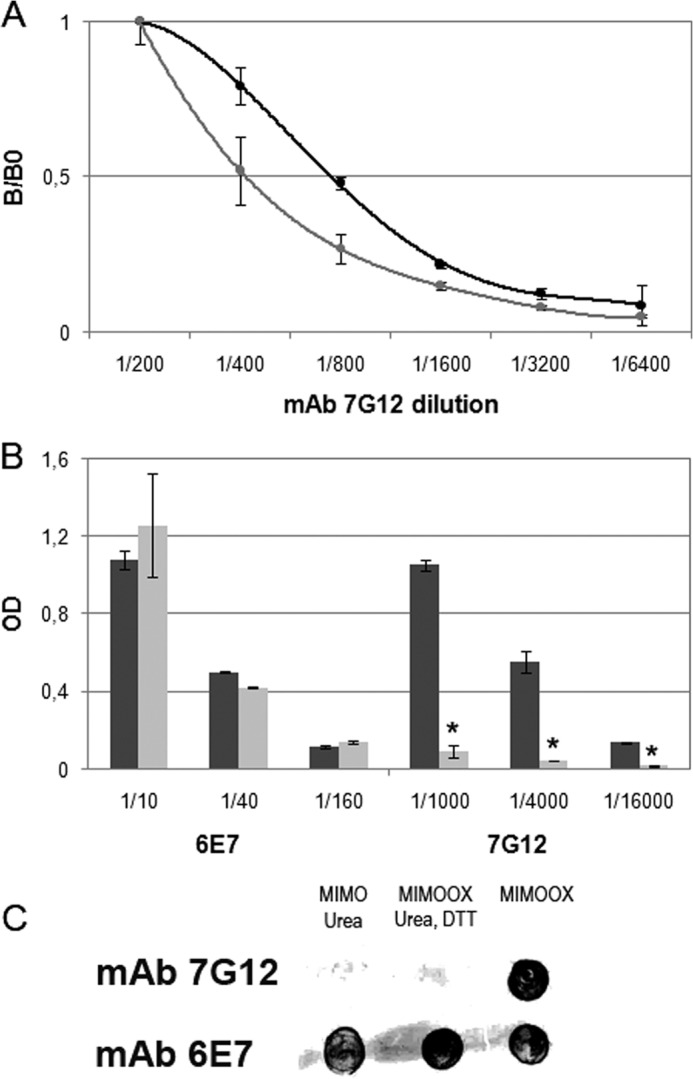
MIMOOX was recognized by the conformational mAb 7G12 able to cross-neutralize Tat variants. In A, the affinity curves (mean ± S.D., n ≥ 3) of mAb 7G12 with Tat OYI (black) and MIMOOX (gray) were measured in ELISA. In B, the affinity curves (mean ± S.D., n ≥ 3) of mAbs 7G12 and 6E7 with native (dark gray) or denatured (light gray) MIMOOX were measured in ELISA. Different dilutions of mAbs were used. In bar graphs, statistical significant differences (p < 0.05) between the affinity of mAbs 7G12 with native and denatured MIMOOX were indicated by an asterisk. In C, the recognition of native or denatured MIMOOX by mAbs 7G12 and 6E7 in dot blot assay was represented (n = 3). Lane 1, denatured MIMOOX; lane 2, denatured MIMOOX; lane 3, native MIMOOX.
MIMOOX Competes with Tat Variants, Including Tat OYI on mAb 7G12
Competitive recognition assays with mAb 7G12 between coated Tat OYI and soluble MIMOOX (or coated MIMOOX and soluble Tat OYI) were carried out in ELISA (Fig. 4, A and B). mAb 7G12 was used in equimolar amounts regarding coated proteins. Increasing MIMOOX concentration in solution progressively decreased mAb binding for coated Tat OYI (2 pmol) (Fig. 4A). For an equimolar ratio of 2 pmol (corresponding to 12.5 ng of MIMOOX), a decrease of 31% of the signal was observed. This decrease was not observed with denatured MIMOOX (Fig. 4A). A higher decrease was observed when MIMOOX was coated (25 ng, 4pmol) (Fig. 4B). In this case, 65% of the signal was removed for 50 ng of Tat OYI in solution corresponding to 4 pmol. A similar experiment was carried out with Tat Ug 11 RP, Tat 96BW, Tat ELI, and Tat CM240 representative of HIV-1 subtypes A, C, D, and E. Only one concentration was used for coated and soluble proteins corresponding to an equimolar amount of 2 pmol. A decrease (mean of 29.8 ± 2.1%) was observed for Tat variants signal when MIMOOX was in solution (Fig. 4C). A higher decrease (mean of 56.7 ± 8.7%) was observed with Tat variants in solution and coated MIMOOX (Fig. 4D). These results were similar to the competition assay between MIMOOX and Tat OYI observed previously and were in agreement with similar Tat variant KD values (32). The decrease of signal was not observed with denatured MIMOOX or denatured Tat variants in solution (Fig. 4, C and D). These experiments showed a structural homogeneity of MIMOOX with Tat variants and confirmed that a conserved surface recognized by mAb 7G12 existed among Tat variants.
FIGURE 4.

Competitive ELISA (mean ± S. D.; n ≥ 3) with mAb 7G12 between MIMOOX in solution and coated Tat OYI (A) or Tat in solution and coated MIMOOX (B) are shown. Native (continuous line) and denatured (dashed line) form of proteins in solution were shown. In C, competitive ELISA (mean ± S.D., n ≥ 3) with mAb 7G12 between different coated Tat variants and native (dark gray) or denatured (light gray) MIMOOX or coated MIMOOX and native (dark gray) or denatured (light gray) Tat from different clades (D). Signal without competitor represented the 100% (black) for each Tat tested. These experiments were done in triplicate. In bar graphs, statistical significant differences (p < 0.05) between the condition without competitor and with native competitor were indicated by an asterisk.
Rat Immunization with MIMOOX Induces Cross-recognition Antibodies
To assess whether MIMOOX could generate antibodies able to recognize heterologous and homologous Tat variants, rats were immunized with MIMOOX linked on the surface of an immunopotentiating reconstituted influenza virosome (39, 41). The polyclonal anti-MIMOOX serum obtained at week 10 was tested in ELISA against five Tat variants representative of the main HIV-1 subtypes as described previously. Fig. 5 shows that the anti-MIMOOX serum recognizes the five Tat variants. Tat OYI was recognized as well as MIMOOX. Serum dilution shows that Tat Eli is better recognized compared with the other Tat variants. A similar result was observed with previously published anti-Tat OYI polyclonal sera (31).
FIGURE 5.
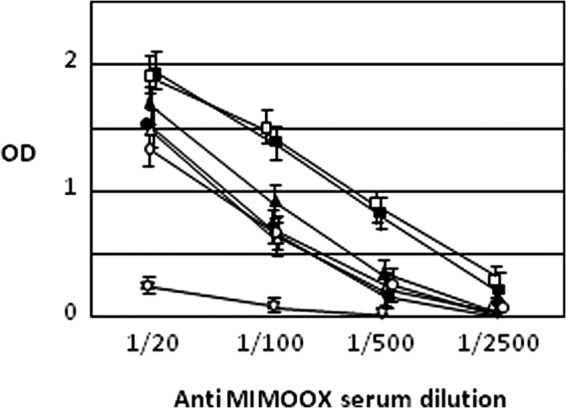
MIMOOX and Tat variants recognition by anti-MIMOOX serum dilution. ELISA (n = 2) was performed with four dilutions of the anti-MIMOOX rat serum with coated MIMOOX (black squares), Tat OYI (white squares), Tat UG11RP (white triangles), Tat 96BW (white circles), Tat Eli (black triangles), and Tat CM240 (black circles). ELISA was performed also with a negative control (white star) made of a pool of three peptides corresponding respectively to regions 1, 3, and 6 of Tat OYI. The anti-MIMOOX serum recognizes the five Tat variants and MIMOOX. Tat OYI was recognized as well as MIMOOX. Serum dilution shows that Tat Eli is better recognized compared with the other Tat variants. A similar result was observed with a previously published Tat OYI immunization (14).
DISCUSSION
An effective therapeutic vaccine against AIDS should maintain an undetectable viremia without ART, but none have been reported so far with or without Tat protein (42). SHIV Challenge carried out on macaques is certainly the best animal model to validate potential vaccine on human. To determine whether a vaccine can be effective against HIV-1 infection in humans and due to the high genetic diversity of HIV-1, a SHIV challenge in macaques should be heterologous, which means that the SHIV used should be a genetically distinct virus compared with the recombinant HIV-1 strains, proteins, or peptides used in the vaccine (43). It is therefore important to outline that the first successful protection against heterologous SHIV challenges was obtained with Tat OYI (31). A second successful heterologous SHIV challenge was reported with a Tat variant closely related to Tat OYI mixed with other active principles such as multimeric HIV-1 gp160 and SIV Gag-Pol particles (44). Interestingly, it was shown recently, using a biopanning strategy, that only anti-Tat neutralizing antibody were present in completely or partially protected macaques (45).
The rational of the Tat OYI vaccine is that Tat OYI would transform a highly conserved surface in Tat variants in a three-dimensional epitope, triggering the production of neutralizing antibodies recognizing all Tat variants (1). mAb 7G12 was characterized previously as an antibody recognizing different Tat variants showing that a highly conserved surface exists in all Tat variants (32). This common surface required a conserved folding in Tat variants that plays an important role for Tat activity (1). The most conserved sequence in Tat variants corresponds to the segment between positions ∼44 and 50 located in region III. This sequence constitutes a type 2 β turn that is observed in two-dimensional NMR studies of Tat variants alone (10–13) or an x-ray diffraction study of Tat in a complex with the human P-TEFb protein (14). The amino acid glutamate at position 100 (presents only in Tat OYI) plays an important role because it displays a charge in this conserved surface that might transform this surface in an epitope only for Tat OYI (1).
As mentioned, the Tat OYI vaccine approach suggests that a common folding exists among Tat variants, but there is a controversy regarding the existence of a folding in Tat variants (1). A two-dimensional NMR study of a peptide corresponding to a first exon of Tat(1–72) showed that no structure could be identified in this peptide and concluded that Tat variants were natively unfolded proteins (15). A potential Tat activity was not tested with this peptide, and no viable HIV-1 strain consisting of only the first exon of Tat has ever been observed in vivo. Furthermore, the sequence used for this study does not correspond to a viable HIV-1 strain, as the peptide contained a supplemental 20 residues at the N terminus that are unrelated to Tat (1). This study was in contradiction with three former two-dimensional NMR studies that we published with Tat variants having the transactivation activity in a cellular assay and showing a conserved folding among Tat variants (11–13). However, structural heterogeneities in Tat variants are observed, which are localized mainly in region V, which can adopt an α helix or a β turn conformation as a function of mutations (1). Furthermore, the broad peaks in NMR spectra indicate that Tat is a flexible protein that can become a random coil as a function of the buffer or pH used to solubilize Tat (17). The study with the Fab against the N terminus of Tat bound to a Tat variant and concluding that Tat was intrinsically disordered was carried out in a Tat-denaturing buffer (16). It is important to note that HIV-1-infected patients have antibodies that recognize only structured Tat variants, which indicates that Tat variants are naturally folded proteins in their blood (17). This study confirms also that there is a conserved folding because the mAb 7G12 is no longer able to recognize MIMOOX with a reducing reagent (Fig. 3B).
Competitive assay between Tat variants and MIMOOX regarding mAb 7G12 demonstrates the presence of a highly conserved surface in Tat variants involving 15 amino acid residues from sequences in region I, III, IV, and VI (Fig. 1). This study confirms the Tat OYI vaccine approach relying upon the presence of this highly conserved surface in Tat variants that is normally not recognized by the human immune system but becomes a three-dimensional epitope in Tat OYI (1). MIMOOX was a useful tool to identify this highly conserved surface in Tat variants. MIMOOX could also be a valuable surrogate antigen in immunoassays to screen neutralizing antibodies in serum patients vaccinated with Tat OYI. However, MIMOOX cannot replace Tat OYI yet in a vaccine candidate because it has an affinity twice lower compared with Tat OYI for mAb 7G12. Point mutation experiments could help to obtain a real mimotope of Tat OYI using the scaffold of MIMOOX. This step requires a long development. A serum obtained after immunization cannot be tested directly for neutralization because animal sera modify the production of β-galactosidase in the neutralization test performed with HIV LTR-transfected HeLa cells. Therefore, it will be necessary to produce monoclonal antibodies against these MIMOOX derivatives to test their capacity to be a mimotope able to replace Tat OYI in a vaccine.
Clinical trials with Tat OYI were begun in France in February 2013. The protocol is entitled “Evaluation on seropositive patients of a synthetic vaccine targeting the HIV Tat protein” with the acronym: “EVA TAT.” The protocol was declared in the European Clinical trial database (EudraCT) under ID no. 2012-000374-36. The protocol received favorable advice from an ethics committee (CPP SudMed 2) on November 9th, 2012 and an authorization from the French drug agency (Agence Nationale de Sécurité du Médicament) on January 14th, 2013. It is a phase I/II clinical trial to test the Tat OYI vaccine. It is a randomized double-blinded clinical trial with anonymous syringes containing either the vaccine or placebo. Phase I/IIa will be carried out on four groups of 12 patients with an undetectable viremia (inferior to 40 copies/ml) and a level of CD4 cells superior to 350/mm3 for at least one year. Three groups (1, 2, and 3) will have three injections at M0, M1, and M2 of a solution containing 10, 33, and 100 μg, respectively, of the Tat OYI vaccine. Group 4 will have three injections of a placebo with no active principle. It will be proposed to all patients to stop their antiviral treatment for two months from M5 to M7. The treatment interruption will make possible to evaluate the optimal vaccine dose to evaluate the efficacy of the vaccine in a phase IIb (27). This phase IIb will be carried out on 80 patients divided in two groups (vaccine/placebo) who did not participate to the phase I/IIa. The efficacy of this therapeutic vaccine could be considered if 30% of patients from the vaccine group can maintain their viremia undetectable (inf 40 copies/ml) after interruption of ART for two months. Result of phase IIa should be available in January 2014.
Synthetic Tat OYI and MIMOOX could also be considered as a new step in vaccine development because their diameters, 100 and 50 nm, respectively, are largely inferior to commercially available vaccines with active principles that have diameters in the range of μm. These nanovaccines that contain a three-dimensional epitope triggering neutralizing antibodies will be safer because they will trigger an immune response with a lower diversity of polyclonal antibodies. This should reduce significantly the risk of autoimmune response of classical vaccine approach using an adjuvant such as Alum to increase the diversity of polyclonal antibodies. The coast of production of these nano vaccines is largely compensated by the easiness to obtain germ free preparation with a double filtration with 0.2-μm filters. Furthermore, the Tat OYI vaccine is stable in solution for at least one month at 4 °C (data not shown). This remarkable stability is essential for mass vaccination.
Acknowledgments
We thank Daniel Laffite, Claude Villars, and Therese Schembri for technical assistance. We thank Sylvain Fleury for fruitful discussion regarding the use of a virosome for rat immunization with MIMOOX.
This work was supported by BIOSANTECH (Sofia Antipolis, France) and MIMETICS (Lausanne, Switzerland).
The main abbreviations used are MIMO and MIMOOX, which correspond to a same peptide of 56 residues with the sequence described in the legend to Fig. 1. MIMOOX is the oxidized form of MIMO, which means that there is a disulfide in MIMOOX but not in MIMO.
- ART
- antiretroviral treatment(s).
REFERENCES
- 1. Campbell G. R., Loret E. P. (2009) What does the structure-function relationship of the HIV-1 Tat protein teach us about developing an AIDS vaccine? Retrovirology 6, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong-Staal F., Gallo R. C. (1985) Human T-lymphotropic retroviruses. Nature 317, 395–403 [DOI] [PubMed] [Google Scholar]
- 3. Debaisieux S., Rayne F., Yezid H., Beaumelle B. (2012) The ins and outs of HIV-1 Tat. Traffic 13, 355–363 [DOI] [PubMed] [Google Scholar]
- 4. Westendorp M. O., Frank R., Ochsenbauer C., Stricker K., Dhein J., Walczak H., Debatin K. M., Krammer P. H. (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500 [DOI] [PubMed] [Google Scholar]
- 5. Mediouni S., Darque A., Baillat G., Ravaux I., Dhiver C., Tissot-Dupont H., Mokhtari M., Moreau H., Tamalet C., Brunet C., Paul P., Dignat-George F., Stein A., Brouqui P., Spector S. A., Campbell G. R., Loret E. P. (2012a) Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect. Disord. Drug Targets 12, 81–86 [DOI] [PubMed] [Google Scholar]
- 6. Campbell G. R., Senkaali D., Watkins J., Esquieu D., Opi S., Yirrell D. L., Kaleebu P., Loret E. P. (2007) Tat mutations in an African cohort that do not prevent transactivation but change its immunogenic properties. Vaccine 25, 8441–8447 [DOI] [PubMed] [Google Scholar]
- 7. Senkaali D., Kebba A., Shafer L. A., Campbell G. R., Loret E. P., Van Der Paal L., Grosskurth H., Yirrell D., Kaleebu P. (2008) Tat-specific binding IgG and disease progression in HIV type 1-infected Ugandans. AIDS Res. Hum. Retroviruses 24, 587–594 [DOI] [PubMed] [Google Scholar]
- 8. Loret E. P., Georgel P., Johnson W. C., Jr., Ho P. S. (1992) Circular dichroism and molecular modeling yield a structure for the complex of human immunodeficiency virus type 1 trans-activation response RNA and the binding region of Tat, the trans-acting transcriptional activator. Proc. Natl. Acad. Sci. U.S.A. 89, 9734–9738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koken S. E., Greijer A. E., Verhoef K., van Wamel J., Bukrinskaya A. G., Berkhout B. (1994) Intracellular analysis of in vitro modified HIV Tat protein. J. Biol. Chem. 269, 8366–8375 [PubMed] [Google Scholar]
- 10. Bayer P., Kraft M., Ejchart A., Westendorp M., Frank R., Rösch P. (1995) Structural studies of HIV-1 Tat protein. J. Mol. Biol. 247, 529–535 [DOI] [PubMed] [Google Scholar]
- 11. Péloponèse J. M., Jr., Grégoire C., Opi S., Esquieu D., Sturgis J., Lebrun E., Meurs E., Collette Y., Olive D., Aubertin A. M., Witvrow M., Pannecouque C., De Clercq E., Bailly C., Lebreton J., Loret E. P. (2000) 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C. R. Acad. Sci. III 323, 883–894 [DOI] [PubMed] [Google Scholar]
- 12. Grégoire C., Péloponèse J. M., Jr., Esquieu D., Opi S., Campbell G., Solomiac M., Lebrun E., Lebreton J., Loret E. P. (2001) Homonuclear 1H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers 62, 324–335 [DOI] [PubMed] [Google Scholar]
- 13. Watkins J. D., Campbell G. R., Halimi H., Loret E. P. (2008) Homonuclear 1H NMR and circular dichroism study of the HIV-1 Tat Eli variant. Retrovirology 5, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tahirov T. H., Babayeva N. D., Varzavand K., Cooper J. J., Sedore S. C., Price D. H. (2010) Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465, 747–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shojania S., O'Neil J. D. (2006) HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1–72) by NMR spectroscopy. J. Biol. Chem. 281, 8347–8356 [DOI] [PubMed] [Google Scholar]
- 16. Serrière J., Dugua J. M., Bossus M., Verrier B., Haser R., Gouet P., Guillon C. (2011) Fab-induced folding of antigenic N-terminal peptides from intrinsically disordered HIV-1 Tat revealed by x-ray crystallography. J. Mol. Biol. 405, 33–42 [DOI] [PubMed] [Google Scholar]
- 17. Mediouni S., Baillat G., Darque A., Ravaux I., Loret E. (2011) HIV-1 infected patients have antibodies recognizing folded Tat. Infect. Disord. Drug Targets 11, 57–63 [DOI] [PubMed] [Google Scholar]
- 18. Caselli E., Betti M., Grossi M. P., Balboni P. G., Rossi C., Boarini C., Cafaro A., Barbanti-Brodano G., Ensoli B., Caputo A. (1999) DNA immunization with HIV-1 tat mutated in the trans activation domain induces humoral and cellular immune responses against wild-type Tat. J. Immunol. 162, 5631–5638 [PubMed] [Google Scholar]
- 19. Ensoli B., Fiorelli V., Ensoli F., Lazzarin A., Visintini R., Narciso P., Di Carlo A., Monini P., Magnani M., Garaci E. (2008) The therapeutic phase I trial of the recombinant native HIV-1 Tat protein. AIDS 22, 2207–2209 [DOI] [PubMed] [Google Scholar]
- 20. Longo O., Tripiciano A., Fiorelli V., Bellino S., Scoglio A., Collacchi B., Alvarez M. J., Francavilla V., Arancio A., Paniccia G., Lazzarin A., Tambussi G., Din C. T., Visintini R., Narciso P., Antinori A., D'Offizi G., Giulianelli M., Carta M., Di Carlo A., Palamara G., Giuliani M., Laguardia M. E., Monini P., Magnani M., Ensoli F., Ensoli B. (2009) Phase I therapeutic trial of the HIV-1 Tat protein and long term follow-up. Vaccine 27, 3306–3312 [DOI] [PubMed] [Google Scholar]
- 21. Ensoli B., Fiorelli V., Ensoli F., Lazzarin A., Visintini R., Narciso P., Di Carlo A., Tripiciano A., Longo O., Bellino S., Francavilla V., Paniccia G., Arancio A., Scoglio A., Collacchi B., Ruiz Alvarez M. J., Tambussi G., Tassan Din C., Palamara G., Latini A., Antinori A., D'Offizi G., Giuliani M., Giulianelli M., Carta M., Monini P., Magnani M., Garaci E. (2009) The preventive phase I trial with the HIV-1 Tat-based vaccine. Vaccine 28, 371–378 [DOI] [PubMed] [Google Scholar]
- 22. Ensoli B., Bellino S., Tripiciano A., Longo O., Francavilla V., Marcotullio S., Cafaro A., Picconi O., Paniccia G., Scoglio A., Arancio A., Ariola C., Ruiz Alvarez M. J., Campagna M., Scaramuzzi D., Iori C., Esposito R., Mussini C., Ghinelli F., Sighinolfi L., Palamara G., Latini A., Angarano G., Ladisa N., Soscia F., Mercurio V. S., Lazzarin A., Tambussi G., Visintini R., Mazzotta F., Di Pietro M., Galli M., Rusconi S., Carosi G., Torti C., Di Perri G., Bonora S., Ensoli F., Garaci E. (2010) Therapeutic immunization with HIV-1 Tat reduces immune activation and loss of regulatory T-cells and improves immune function in subjects on HAART. PLoS One 5, e13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guillon C., Mayol K., Terrat C., Compagnon C., Primard C., Charles M. H., Delair T., Munier S., Verrier B. (2007) Formulation of HIV-1 Tat and p24 antigens by PLA nanoparticles or MF59 impacts the breadth, but not the magnitude, of serum and faecal antibody responses in rabbits. Vaccine 25, 7491–7501 [DOI] [PubMed] [Google Scholar]
- 24. Mayol K., Munier S., Beck A., Verrier B., Guillon C. (2007) Design and characterization of an HIV-1 Tat mutant: inactivation of viral and cellular functions but not antigenicity. Vaccine 25, 6047–6060 [DOI] [PubMed] [Google Scholar]
- 25. Lecoq A., Moine G., Bellanger L., Drevet P., Thai R., Lajeunesse E., Ménez A., Léonetti M. (2008) Increasing the humoral immunogenic properties of the HIV-1 Tat protein using a ligand-stabilizing strategy. Vaccine 26, 2615–2626 [DOI] [PubMed] [Google Scholar]
- 26. Liao W., Chen Q., Cao J., Tan G., Zhu Z., Zhang H., Chai Y., Pan W. (2012) A designed Tat immunogen generates enhanced anti-Tat C-terminal antibodies. Vaccine 14, 2453–2461 [DOI] [PubMed] [Google Scholar]
- 27. Goldstein G., Damiano E., Donikyan M., Pasha M., Beckwith E., Chicca J. (2012) HIV-1 Tat B-cell epitope vaccination was ineffectual in preventing viral rebound after ART cessation: HIV rebound with current ART appears to be due to infection with new endogenous founder virus and not to resurgence of pre-existing Tat-dependent viremia. Hum. Vaccin. Immunother. 8, 1425–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huet T., Dazza M. C., Brun-Vézinet F., Roelants G. E., Wain-Hobson S. (1989) A highly defective HIV-1 strain isolated from a healthy Gabonese individual presenting an atypical western blot. AIDS 3, 707–715 [DOI] [PubMed] [Google Scholar]
- 29. Gregoire C. J., Loret E. P. (1996) Conformational heterogeneity in two regions of TAT results in structural variations of this protein as a function of HIV-1 isolates. J. Biol. Chem. 271, 22641–22646 [DOI] [PubMed] [Google Scholar]
- 30. Opi S., Péloponèse J. M., Jr., Esquieu D., Campbell G., de Mareuil J., Walburger A., Solomiac M., Grégoire C., Bouveret E., Yirrell D. L., Loret E. P. (2002) Tat HIV-1 primary and tertiary structures critical to immune response against non-homologous variants. J. Biol. Chem. 277, 35915–35919 [DOI] [PubMed] [Google Scholar]
- 31. Watkins J. D., Lancelot S., Campbell G. R., Esquieu D., de Mareuil J., Opi S., Annappa S., Salles J. P., Loret E. P. (2006) Reservoir cells no longer detectable after a heterologous SHIV challenge with the synthetic HIV-1 Tat OYI vaccine. Retrovirology 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mediouni S., Watkins J. D., Pierres M., Bole A., Loret E. P., Baillat G. (2012b) A monoclonal antibody directed against a conformational epitope of the HIV-1 trans-activator (Tat) protein neutralizes cross-clade. J. Biol. Chem. 287, 11942–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steinaa L., Sørensen A. M., Nielsen J. O., Hansen J. E. (1994) Antibody to HIV-1 Tat protein inhibits the replication of virus in culture. Arch. Virol. 139, 263–271 [DOI] [PubMed] [Google Scholar]
- 34. Re M. C., Furlini G., Vignoli M., Ramazzotti E., Roderigo G., De Rosa V., Zauli G., Lolli S., Capitani S., La Placa M. (1995) Effect of antibody to HIV-1 Tat protein on viral replication in vitro and progression of HIV-1 disease in vivo. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10, 408–416 [DOI] [PubMed] [Google Scholar]
- 35. Valvatne H., Szilvay A. M., Helland D. E. (1996) A monoclonal antibody defines a novel HIV type 1 Tat domain involved in trans-cellular trans-activation. AIDS Res. Hum. Retroviruses 12, 611–619 [DOI] [PubMed] [Google Scholar]
- 36. Rodman T. C., Lutton J. D., Jiang S., Al-Kouatly H. B., Winston R. (2001) Circulating natural IgM antibodies and their corresponding human cord blood cell-derived MAbs specifically combat the Tat protein of HIV. Exp. Hematol. 29, 1004–1009 [DOI] [PubMed] [Google Scholar]
- 37. Moreau E., Hoebeke J., Zagury D., Muller S., Desgranges C. (2004) Generation and characterization of neutralizing human monoclonal antibodies against human immunodeficiency virus type 1 Tat antigen. J. Virol. 78, 3792–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manavalan P., Johnson W. C., Jr. (1987) Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 167, 76–85 [DOI] [PubMed] [Google Scholar]
- 39. Pfeiffer B., Peduzzi E., Moehle K., Zurbriggen R., Glück R., Pluschke G., Robinson J. A. (2003) A virosome-mimotope approach to synthetic vaccine design and optimization, synthesis, conformation, and immune recognition of a potential malaria-vaccine candidate. Angew Chem. Int. Ed. Engl. 42, 2368–2371 [DOI] [PubMed] [Google Scholar]
- 40. Hardy F., Djavadi-Ohaniance L., Goldberg M. E. (1997) Measurement of antibody/antigen association rate constants in solution by a method based on the enzyme-linked immunosorbent assay. J. Immunol. Methods 200, 155–159 [DOI] [PubMed] [Google Scholar]
- 41. Bomsel M., Tudor D., Drillet A. S., Alfsen A., Ganor Y., Roger M. G., Mouz N., Amacker M., Chalifour A., Diomede L., Devillier G., Cong Z., Wei Q., Gao H., Qin C., Yang G. B., Zurbriggen R., Lopalco L., Fleury S. (2011) Immunization with HIV-1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity 34, 269–280 [DOI] [PubMed] [Google Scholar]
- 42. Reynell L., Trkola A. (2012) HIV vaccines, an attainable goal? Swiss Med Wkly. 142, w13535. [DOI] [PubMed] [Google Scholar]
- 43. Feinberg M. B., Moore J. P. (2002) AIDS vaccine models, challenging challenge viruses. Nat. Med. 8, 207–210 [DOI] [PubMed] [Google Scholar]
- 44. Lakhashe S. K., Velu V., Sciaranghella G., Siddappa N. B., Dipasquale J. M., Hemashettar G., Yoon J. K., Rasmussen R. A., Yang F., Lee S. J., Montefiori D. C., Novembre F. J., Villinger F., Amara R. R., Kahn M., Hu S. L., Li S., Li Z., Frankel F. R., Robert-Guroff M., Johnson W. E., Lieberman J., Ruprecht R. M. (2011) Prime-boost vaccination with heterologous live vectors encoding SIV gag and multimeric HIV-1 gp160 protein, efficacy against repeated mucosal R5 clade C SHIV challenges. Vaccine 29, 5611–5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bachler B. C., Humbert M., Palikuqi B., Siddappa N. B., Lakhashe S. K., Rasmussen R. A., Ruprecht R. M. (2013) Novel biopanning strategy to identify epitopes associated with vaccine protection. J. Virol. 87, 4403–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]