

Background: 6-Aminophenanthridine (6AP) is an inhibitor of the protein folding activity of the ribosome (PFAR).
Results: The protein substrates and 6AP bind at common sites on rRNA; mutations at those sites abolish binding and inhibit PFAR.
Conclusion: 6AP competitively obstructs the protein-binding sites and thereby inhibits PFAR.
Significance: We have clarified the mechanism by which 6AP inhibits PFAR.
Keywords: Molecular Chaperone, Prions, Protein Folding, Ribosomal RNA (rRNA), Ribosomes, 6AP
Abstract
Domain V of the 23S/25S/28S rRNA of the large ribosomal subunit constitutes the active center for the protein folding activity of the ribosome (PFAR). Using in vitro transcribed domain V rRNAs from Escherichia coli and Saccharomyces cerevisiae as the folding modulators and human carbonic anhydrase as a model protein, we demonstrate that PFAR is conserved from prokaryotes to eukaryotes. It was shown previously that 6-aminophenanthridine (6AP), an antiprion compound, inhibits PFAR. Here, using UV cross-linking followed by primer extension, we show that the protein substrates and 6AP interact with a common set of nucleotides on domain V of 23S rRNA. Mutations at the interaction sites decreased PFAR and resulted in loss or change of the binding pattern for both the protein substrates and 6AP. Moreover, kinetic analysis of human carbonic anhydrase refolding showed that 6AP decreased the yield of the refolded protein but did not affect the rate of refolding. Thus, we conclude that 6AP competitively occludes the protein substrates from binding to rRNA and thereby inhibits PFAR. Finally, we propose a scheme clarifying the mechanism by which 6AP inhibits PFAR.
Introduction
It has been shown over the past 2 decades that the ribosome is able to refold ∼20 different proteins in vitro (1–5). The protein folding activity of the ribosome (PFAR)4 is not restricted to any particular species or groups of organisms because ribosomes from various sources have been shown to possess this activity (1, 2, 6). Also, the protein substrates of PFAR are not limited to a specific protein family; proteins from diverse sources with various properties can be folded by ribosomes (4). The active site for PFAR lies in the large subunit of the ribosome (50S in bacteria and 60S in eukaryotes) and, similar to peptidyl transferase activity, involves rRNA (2, 7). In fact, both of these crucial functions of the ribosome share the same active center, i.e. domain V of the 23S rRNA in bacteria and the 25S/28S rRNA in eukaryotes (7–10). The same domain from the mitochondrial ribosome also displays activity in refolding proteins (6, 11). This RNA domain (referred hereafter as domain V rRNA) is usually free from any ribosomal protein and lies in the subunit interface of the 70S/80S ribosome. However, upon splitting of the ribosomal subunits, it is exposed on the surface of the large subunit. Thus, in vitro, 50S/60S ribosomal subunits show a more pronounced protein folding activity than the fully assembled 70S/80S ribosomes (12, 13).
Despite a series of in vitro demonstrations of PFAR, a question still remains open in the field. Is PFAR functional in the modern cells, or is it an evolutionary relic representing function of an ancient protein production machine? Although there are few reports of PFAR in living bacterial cells (14, 15), the in vivo context of PFAR has not been fully established. However, one recent finding has linked PFAR to living cells and also associated it with diseases of higher eukaryotes. It has been shown that the two unrelated compounds 6-aminophenanthridine (6AP) and guanabenz acetate, with demonstrated activity against yeast ([PSI+] and [URE3]) and mammalian prions, bind to rRNA and inhibit PFAR (15–17). The correlation between the antiprion activity of these two drugs and their ability to specifically inhibit PFAR suggests that PFAR could be involved in the establishment or maintenance of the prion processes in cells. This notion was further reinforced by the discovery that a 6AP derivative called 6APi, in which the 6-amino group of 6AP is substituted with 2-butan-1-ol, was inactive in both the reversion of the prion phenotype in vivo and inhibition of PFAR in vitro (15). In a different context, PFAR was suggested to be involved in another amyloid-based disease, oculopharyngeal muscular dystrophy, which is an inherited myodegenerative disease caused by the aggregation of PABPN1 protein into amyloid fibers within the nucleus of muscle cells (18). Thus, even though the involvement of PFAR in prion processes has yet to be directly demonstrated, 6AP and guanabenz acetate constitute valuable tools for studying PFAR (19).
In this work, we elucidated how 6AP inhibits PFAR. Using UV cross-linking followed by primer extension, we determined that the protein substrates of PFAR and 6AP (but not the inactive analog 6APi) interacted with largely overlapping sites of domain V of 23S rRNA. Mutations in the interaction sites not only abolished or changed the interaction map of both the protein substrates and 6AP but also decreased the protein folding activity of domain V of rRNA from both Escherichia coli and Saccharomyces cerevisiae. Moreover, we determined that 6AP did not affect the kinetics of PFAR but reduced the yield of the refolded protein. Our results led to a simple model for PFAR inhibition by 6AP.
EXPERIMENTAL PROCEDURES
Proteins
Human carbonic anhydrase (HCA) was expressed in E. coli and purified by column chromatography as described (20). Bovine carbonic anhydrase and bacterial dihydrofolate reductase were purchased from Sigma. His-tagged T7 RNA polymerase was purified using immobilized metal affinity chromatography after overexpression from the pET21a-T7 pol plasmid (laboratory strain).
In Vitro Transcription of Domain V rRNA
Plasmids pGEM4Z and pAV164, containing DNA sequences for domain V of 23S rRNA from E. coli and 25S rRNA from S. cerevisiae, respectively, were used as templates for transcription. Linear templates were prepared either by restriction digestion (e.g. pGEM4Z with EcoRI) or by PCR amplification of the target sequence. 1.5 μg of the linearized DNA template was mixed with transcription buffer (800 mm Hepes-NaOH (pH 7.5), 120 mm MgCl2, 120 mm dithiothreitol, and 8 mm spermidine). Next, 7 mm rNTP mixture, 60 units of RNase inhibitor (RiboLock, Fermentas), and 1.68 μm T7 RNA polymerase were added to start the transcription, followed by incubation for 4 h at 37 °C. DNA templates were digested with RNase-free DNase I. RNA was precipitated with 3 m sodium acetate (pH 5.2) and ethanol after extraction with phenol and chloroform (1:1). Next, RNA was made free of nucleotides using the NucleoSpin RNA cleanup kit (Macherey-Nagel). RNA concentrations were measured using a NanoDropTM 1000 spectrophotometer (Thermo Scientific V3.6), and the quality was checked by running on a 4% denaturing urea-polyacrylamide gel.
Mutagenesis of Domain V rRNA
Plasmids pGEM4Z and pAV164 were used for introducing base mutations on domain V of 23S rRNA from E. coli and 25S rRNA from S. cerevisiae, respectively. 19 variants of domain V of 23S rRNA and nine variants of domain V of 25S rRNA were created by QuikChange mutagenesis (Stratagene). The mutations were confirmed by DNA sequencing, and the corresponding RNAs were transcribed and purified as described above.
HCA Refolding Assay
HCA (30 μm) was denatured with 6 m guanidine hydrochloride by overnight incubation at 37 °C. Refolding of HCA was done typically for 30 min at room temperature by 100× dilution of the denatured mixture in refolding buffer (20 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 100 mm NaCl, and 0.05 mm ZnSO4) as described (20) without (self-folding) or with domain V rRNA. For time course experiments, samples were withdrawn from the refolding mixture at different time points and assayed for HCA activity. The typical concentration of domain V rRNA in these experiments was 350 nm. The enzymatic activity of HCA was used as a measure of refolding, taking the native enzymatic activity (stored undiluted in ice) as 100%.
UV Cross-linking
6 m guanidine hydrochloride-denatured proteins (HCA, bovine carbonic anhydrase, and dihydrofolate reductase, all at 30 μm) were diluted 100 times in refolding buffer containing domain V of 23S rRNA from E. coli (300 nm), and UV cross-linking was performed immediately in a Bio-Rad GS Gene LinkerTM instrument, with 254 nm UV irradiation (600 mJ) (21). For cross-linking with 6AP, 300 nm domain V of 23S rRNA and 0.5 mm 6AP (or 6APi) were mixed and subjected to the same procedure as described above. In both cases, the samples were kept on ice during irradiation to prevent heat damage of the RNA. The irradiated samples were precipitated by salt/ethanol and washed with 70% ethanol for primer extension.
Primer Extension Assay
Primer 5′-ACCCCGGATCCGCGCCCACGGCAGATAGG-3′ was labeled with [γ-32P]dATP at 37 °C using T4 polynucleotide kinase (Fermentas) for 1 h by the 5′-end labeling method (22). The labeled primer was incubated with the cross-linked RNA-protein or RNA-6AP/6APi complex (∼10 μg) at 65 °C for 5 min. Primer extension was initiated by the addition of ThermoScript reverse transcriptase (Invitrogen) at 55 °C for ∼1 h, followed by incubation for 15 min at 72 °C for completion of the reaction. The products were precipitated, washed with 70% ethanol, and run on a 6.5% polyacrylamide gel with 8 m urea next to a sequencing ladder of domain V rDNA obtained using the same primer by Thermo Sequenase DNA polymerase (Thermo SequenaseTM sequencing kit, USB Corp.).
RESULTS
Refolding of HCA with Domain V rRNAs
HCA denatured in 6 m guanidine hydrochloride was subjected to refolding in the presence of in vitro transcribed domain V of 23S rRNA from E. coli and 25S rRNA from S. cerevisiae. In comparison to self-folding, which resulted in ∼25% native HCA activity, both domain V rRNAs increased the refolding to 38–40% (Fig. 1A). Titration with domain V rRNAs showed an optimal RNA concentration at ∼1:1 ratio with the HCA concentration during refolding. Furthermore, HCA refolding with both domain V rRNAs could be fully inhibited by 6AP to the level of self-folding (Fig. 1A, inset). Consistent with an earlier report (20), 6AP had no effect on self-folding (Fig. 1B).
FIGURE 1.
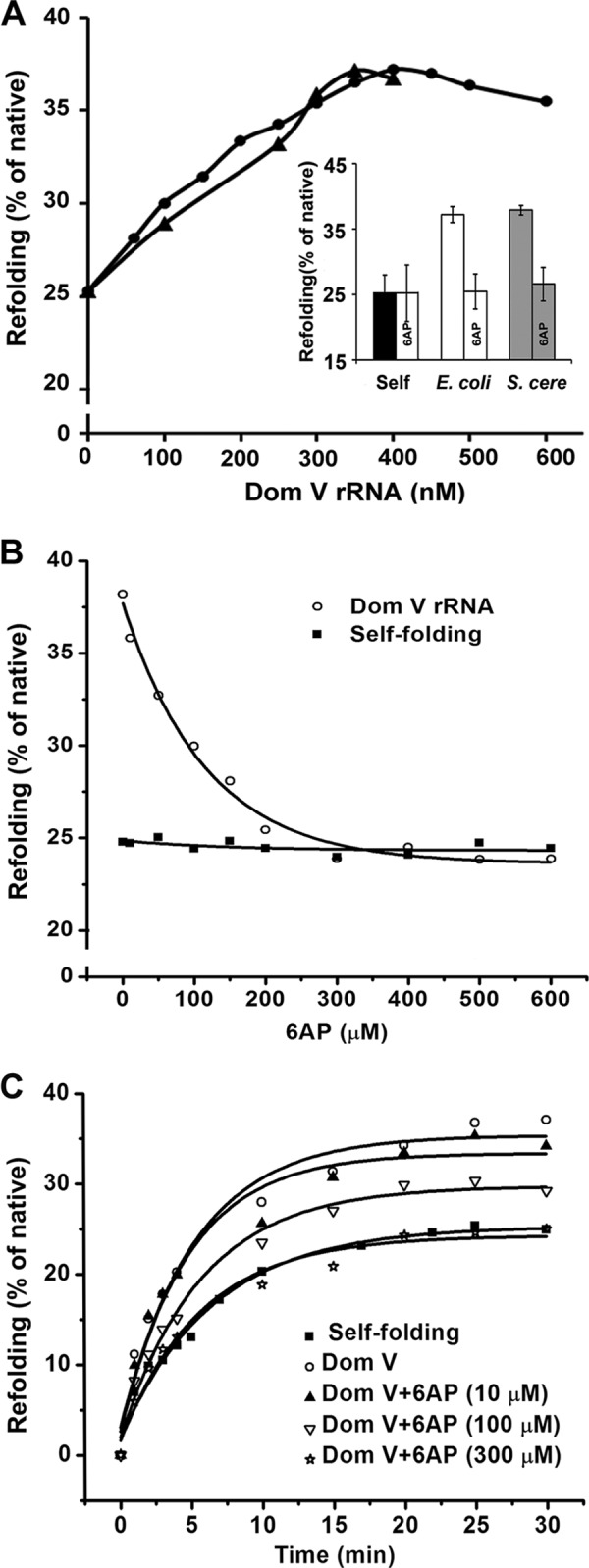
Refolding of HCA with domain V rRNA and effect of 6AP. A, refolding of denatured HCA (final concentration of 300 nm) with various concentrations of domain V (Dom V) of 23S rRNA (E. coli; ●) and 25S rRNA (S. cerevisiae; ▴). The extent of refolding (30 min at room temperature) was estimated as the percentage of native HCA activity (stored undiluted in ice). The inset shows the effect of 6AP (300 μm) on HCA refolding without and with domain V rRNAs from E. coli and S. cerevisiae (S. cere). The results are the average of a minimum of three individual measurements, and error bars represent S.D. B, refolding of HCA (300 nm, 30 min at room temperature) in the presence of increasing concentrations of 6AP without (self-refolding; ■) or with (○) domain V of E. coli 23S rRNA (300–400 nm). The IC50 (half-maximal inhibitory concentration) was determined from the x-intercept drawn at half-maximal refolding, taking the difference between the self-refolding and 23S rRNA domain V-assisted refolding as 100%. C, time course of HCA refolding without (self-refolding; ■) or with (○) domain V of 23S rRNA at different concentrations of 6AP (▴, ▿, and ☆). The curves were fitted with a single exponential equation using Origin 8.0, and the rates (mean of a minimum of three experiments) were estimated from the respective fits.
6AP-mediated Inhibition of the Protein Folding Activity of Domain V of 23S rRNA
Because domain V of rRNA constitutes the active center for PFAR (7), we studied the mode of action of 6AP as an inhibitor using domain V of 23S rRNA as a folding modulator. With increasing concentrations of 6AP, domain V-assisted refolding of HCA decreased gradually from 38 to 25% (Fig. 1, B and C). The data were fitted with a hyperbolic function using Origin 8.0 software. The half-maximal inhibitory concentration (IC50) was estimated from the fitted curve as 90 μm, similar to earlier measurements with other ribosome-borne folding modulators, e.g. 70S ribosome (20).
Next, refolding of HCA was monitored as a function of time without (self-folding) or with domain V of 23S rRNA, and the rates were determined by fitting the resulting curves with a single exponential function. The estimated rates in both reactions were similar (Fig. 1C and Table 1), which is consistent with a previous study in which 23S rRNA was used as the folding modulator (20). The addition of 6AP in gradually increasing concentrations did not alter the rate (Fig. 1C and Table 1), although the extent of refolding was reduced as seen in the single time point assays (Fig. 1B).
TABLE 1.
Rate and percent refolding (native = 100%) of HCA without (self-folding) or with domain V of 23S rRNA in the absence and presence of increasing concentrations of 6AP
| Kobs | Refolding | |
|---|---|---|
| min−1 | % | |
| Domain V RNA-assisted folding | 0.18 ± 0.03 | 38.5 ± 1.5 |
| Domain V + 6AP (10 μm) | 0.18 ± 0.03 | 36 ± 1 |
| Domain V + 6AP (100 μm) | 0.17 ± 0.02 | 30 ± 1.5 |
| Domain V + 6AP (300 μm) | 0.17 ± 0.04 | 22 ± 2 |
| Self-folding | 0.15 ± 0.02 | 23 ± 2 |
Binding Sites of Three Protein Substrates on Domain V of 23S rRNA
The protein-binding sites on the central loop and lower part of domain V of 23S rRNA have been reported previously for bovine carbonic anhydrase, lysozyme, malate dehydrogenase, and lactate dehydrogenase protein substrates using UV cross-linking followed by primer extension (21, 23). We extended these observations using HCA and dihydrofolate reductase as protein substrates. In good agreement with previously published results (21, 23), our results showed a similar interaction map for all three proteins. Five major interaction sites spanning residues U2474–A2476, U2492–G2494, G2553–C2556, A2560–A2564, and U2585–G2588 were identified (Fig. 2A). When zoomed into the tertiary structure of the ribosome, these sites lie in close proximity to each other, creating a binding pocket (Fig. 2B). Because the same sets of nucleotides were identified as the interaction sites for various proteins, these sites must be of general importance for PFAR.
FIGURE 2.

Interaction sites of the protein substrates on domain V of 23S rRNA. A, primer extension analysis of domain V of 23S rRNA after UV cross-linking without any protein (lane 1) or with HCA (lane 2), dihydrofolate reductase (lane 3), and bovine carbonic anhydrase (lane 4). The first four lanes show sequencing ladders as indicated on top of the lanes. B, illustration of the common interaction sites of the protein substrates on the three-dimensional structure of the 50S subunit from E. coli in bronze (Protein Data Bank code 3UOS). The stem-loop part of domain V of 23S rRNA (A2009–A2434) is shown in green, and the circular central loop (A2435–G2668) is shown in blue, with the nucleotides corresponding to the protein interaction sites (identified in A) shown in yellow. The interaction sites are labeled in the zoomed panel above.
Binding Sites of 6AP on Domain V of 23S rRNA
We mapped the interaction sites of 6AP on domain V of 23S rRNA in the same way as for the protein substrates. As shown in Fig. 3A, 10 6AP-binding sites were identified by UV cross-linking followed by primer extension assay. These are U2473–C2475, U2491–G2494, C2499–C2501, A2513–C2515, G2553–C2556, U2561–U2562, A2564–A2565, U2586–G2588, A2598–G2599, and C2601–A2602 (Fig. 3, A and B). Interestingly, six of these sites (U2473–C2475, U2491–G2494, G2553–C2556, U2561–U2562, A2564–A2565, and U2586–G2588) showed partial or complete overlap with the protein-binding sites mentioned above (Fig. 2A). This overlap of binding sites on the rRNA suggests that 6AP competes with the protein substrates for the binding sites on domain V of 23S rRNA.
FIGURE 3.

Interaction sites of 6AP on domain V of 23S rRNA. A, primer extension analysis of domain V of 23S rRNA after UV cross-linking in the absence (lane 1) or presence of 6AP (lane 2) and 6APi (lane 3). Lanes 4–6 are control experiments for lanes 1–3 without UV treatment. The first four lanes show sequencing ladders as indicated on top of the lanes. B, mapping of the 6AP interaction sites (green) on the secondary structure of domain V of 23S rRNA. The nucleotides interacting with protein substrates (shown in Fig. 2) are indicated by black boxes. C, alignment of domain V rRNA sequences from E. coli (NC000913) and S. cerevisiae (S. cere; U53879) showing strong overlap and high conservation of the sequences involved in the interaction with protein substrates (black boxes) and antiprion drugs (green boxes). The yellow highlighting indicates the sequences subjected to mutagenesis; those that showed loss of protein folding activity are shown in red (see Fig. 4).
When 6APi was reacted in the same way as 6AP, only a full-length product was obtained in the primer extension experiment, similar to the control reaction with just domain V of 23S rRNA (Fig. 3A). This result shows that, in contrast to 6AP, 6APi does not interact with domain V rRNA, which also explains its inactivity in inhibition of PFAR. In another control experiment, primer extension was done without UV treatment. In this case also, only full-length products were seen with both 6AP and 6APi (Fig. 3A). This confirms that the bands seen after UV cross-linking with 6AP were indeed due to 6AP binding to domain V rRNA and not degradation of the labeled transcript. The possibility of RNase contamination was further eliminated by running the reactions on a standard urea-acrylamide gel, which showed only intact RNAs (data not shown).
Effect of Mutations in Domain V of 23S/25S rRNA on HCA Refolding
The domain V rRNA, also the center for ribosomal peptidyl transferase activity, is a highly conserved region of the ribosome. Compared in distant species such as E. coli and S. cerevisiae, most of the protein- and 6AP-binding sites showed a high degree of conservation (Fig. 3C). To understand the importance of specific nucleotides in PFAR, we mutated several bases of domain V rRNA from both E. coli (23S) and S. cerevisiae (25S), especially those forming 6AP- and protein-binding sites, and tested the resulting mutants in the HCA refolding assay.
As shown in Fig. 4A, most of the mutants of domain V of 23S rRNA showed highly reduced refolding activity: <50% compared with the WT (Table 2). The highest defect (> 85%) was seen with individual or group mutations at UUG2492–2494, UU2561–2562, and UAG2586–2588 on 23S rRNA (Fig. 4A and Table 2). In comparison, mutations at G2472, UC2474–2475, UA2563–2564, UU2554–2555, and UC2500–2501 showed a lesser degree of defect (50–75%). When 25S rRNA mutants were tested in the same assay, mutations at UGUC2873–2876 were most defective (Fig. 4B and Table 2). UUG2861–2863 mutants also showed a significant defect (∼75%); these nucleotides correspond to UUG2492–2494 on 23S rRNA (Fig. 3C), suggesting that these residues play crucial role in PFAR. The control mutations CGG2486–2488AUU on 23S rRNA and A2820C and G2841A on 25S rRNA, which did not correspond to the protein or 6AP interaction sites, showed no reduction in HCA refolding (Fig. 4, A and B, and Table 2). Thus, these results not only pinpointed the nucleotides important for PFAR but also confirmed that the cross-linking sites represent actual interaction sites for the protein substrate on domain V rRNA. It is interesting to note that no preference for base alteration was seen in the HCA refolding assay. In the case of functionally important residues (such as U2492–G2494 in E. coli), changes from purine to purine or from purine to pyrimidine and vice versa caused similar deleterious effects on rRNA assistance for HCA refolding (Fig. 4A and Table 2).
FIGURE 4.

Effect of mutations in domain V rRNA on the domain V rRNA-assisted refolding of HCA. A and B, refolding of HCA without (self-folding) or with wild-type domain V (Dom V) of E. coli 23S rRNA (A) and S. cerevisiae 25S rRNA (B) or with mutations in various base positions as indicated. The grid lines indicate the level of self-folding (lower grid), wild-type domain V rRNA-assisted folding (upper grid), and the midpoint between the upper and lower grids (middle grid) for easy inspection of the degree of defect with the individual mutants.
TABLE 2.
Domain V rRNA mutants
Domain V rRNA mutants were analyzed and sorted according to their refolding efficiency estimated as (refoldingdomain V mutant − self-folding)/(refoldingWT domain V − self-folding) from the results presented in Fig. 4 (A and B) and expressed as the percentage relative to the wild-type domain V rRNA (100%). The third column represents degree of defect (100% − refolding efficiency).
| Variants of E. coli 23S domain V rRNA | Refolding efficiency | Degree of defect |
|---|---|---|
| % of WT | % | |
| WT | 100 | 0 ± 10 |
| CGG2486–2488AUU | 104.4 | |
| UC2474–2475AU | 56.2 | 10–50 |
| UC2500–2501AU | 44.3 | 50–75 |
| UA2563–2564AU | 41.1 | |
| UU2554–2555AA | 31.6 | |
| G2472U | 30.7 | |
| UAG2586–2588CCA | 22.2 | 75–85 |
| A2587C | 19.7 | |
| U2493C | 14.1 | 85–90 |
| G2494C | 13.4 | |
| G2588A | 13.1 | |
| U2586C | 11.2 | |
| G2494A | 7.8 | 90–95 |
| UUG2492–2494CCA | 7.2 | |
| U2492G | 3.5 | >95 |
| U2492C | 1.6 | |
| UU2561–2562AA | 1.5 | |
| UUG2492–2494GGC | 1.1 | |
| U2493G | −5.5 |
| Variants of S. cerevisiae 25S domain V rRNA | Refolding efficiency | Degree of defect |
|---|---|---|
| % of WT | % | |
| WT | 100 | 0 ± 10 |
| G2841A | 100.6 | |
| A2820C | 97.6 | |
| AG2956–2957GU | 31.2 | 50–75 |
| UU2861–2862CC | 27.1 | |
| G2863A | 22.4 | 75–85 |
| UU2954–2955CC | 21.6 | |
| UC2875–2876GA | 16.2 | |
| UG2873–2874CA | 15.4 | |
| U2932C | 11.3 | >85 |
To ensure that the mutations did not introduce any global change in the structure of domain V rRNA, the wild-type domain V rRNA and three mutant variants (CGG2486–2488AUU, UU2561–2562AA, and UAG2586–2588CAA) were subjected to CD analysis. Furthermore, using CD at 270 nm, the temperature melting profiles of these RNAs were recorded. All the of tested RNAs produced essentially identical CD spectra and temperature melting profiles (Fig. 5), confirming that the base mutations did not alter the secondary structure of the RNA. The difference in CD spectra between 20 and 85 °C indicates loss of folding in all tested RNAs with an increase in temperature. It also confirms that the in vitro transcribed domain V rRNAs are structured under our experimental conditions (20 °C).
FIGURE 5.

CD analysis of wild-type and mutant domain V rRNAs. Wild-type domain V of 23S rRNA and three variants carrying mutations CCG2486–2488AUU, UU2561–2562AA, and UAG2586–2588CCA were subjected to CD analysis to judge the secondary structure. The measurements were done in water using a quartz cuvette with a path length of 1 mm in a Jasco J-815 CD spectrometer with an RNA concentration of 0.5 μm. A and B, CD spectra at 20 and 85 °C, respectively. The dashed line in B indicates the spectrum of wild-type domain V at 20 °C as a reference. C, CD profiles of temperature-dependent melting of wild-type and mutant RNAs where the change in CD signal was monitored at a fixed wavelength (270 nm) with a temperature gradient of 1°/min. mdeg, millidegrees.
Loss of Both Protein and Drug Interaction Sites Due to Mutations in Domain V of 23S rRNA
The loss of protein folding activity due to mutations on domain V rRNA called for a cross-check of the interaction sites using mutant rRNAs. Among the mutations tested in the HCA refolding assay, UUG2492–2494CCA and UAG2586–2588CCA showed major loss of PFAR (Fig. 4A). When these mutant RNAs were subjected to UV cross-linking and primer extension, distinct changes in the interaction patterns were seen for both HCA and 6AP. For UUG2492–2494CCA rRNA, the bands corresponding to these nucleotides were completely missing for both HCA and 6AP (Fig. 6, A and B, upper panels), whereas other bands remained intact. Similarly, in the case of UAG2586–2588CCA, a major change in the banding pattern was observed at this site (Fig. 6, A and B, lower panels). For HCA, the bands corresponding to the U2586–G2588 nucleotides became weaker compared with the WT (Fig. 6A, lower panel), whereas for 6AP, these bands were completely missing (Fig. 6B, lower panel). These results confirmed that UUG2492–2494 and UAG2586–2588 indeed interact with both the protein substrate and 6AP. Thus, a correlation can be envisaged between PFAR and the mode of action of 6AP.
FIGURE 6.
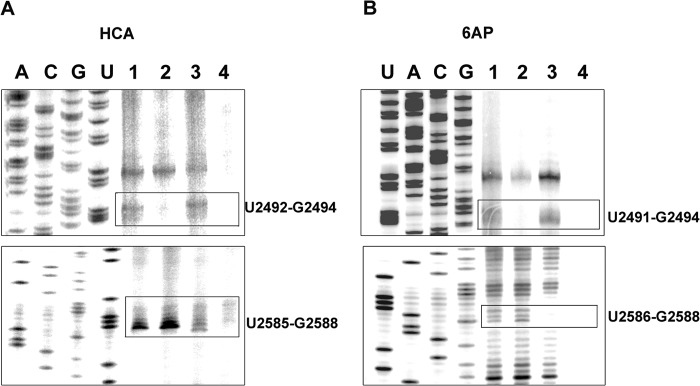
Effect of mutations in domain V rRNA on the interaction map of HCA and 6AP. Shown are the interaction sites of HCA (A) and 6AP (B) on wild-type domain V of 23S rRNA (E. coli; lane 1) and mutants UUG2492–2494CCA (lane 2) and UAG2586–2588CCA (lane 3). Lane 4 is the control with only domain V rRNA without HCA (A) or 6AP (B). The bands corresponding to the mutation sites are boxed.
DISCUSSION
Domain V rRNA Is the Universal Active Center for PFAR
It was shown previously that the active site of PFAR lies in domain V of 23S rRNA in the bacterial ribosome (7, 8, 24). It was also shown that PFAR is a universal activity borne by prokaryotic, eukaryotic, and mitochondrial ribosomes (4, 7, 25). This is not surprising considering the high degree of sequence conservation of the domain V rRNA through evolution. However, it was unknown whether or not in vitro transcribed domain V of the eukaryotic rRNA retains protein folding activity. Our results put an affirmative end to this discussion, as domain V of 25S rRNA from S. cerevisiae could refold HCA as efficiently as domain V of 23S rRNA from E. coli (Fig. 1A). Furthermore, the protein folding activity of both domain V rRNAs was inhibited similarly when treated with 6AP (Fig. 1A, inset). Thus, we conclude that, similar to peptidyl transferase activity, domain V-based PFAR is conserved from bacteria to eukaryotes.
6AP Inhibits Protein Folding by Domain V rRNA by Direct Competition
The inhibition of PFAR by 6AP has been reported previously using various ribosomal components as protein folding modulators (70S, 50S, and 23S rRNAs from E. coli and 80S rRNA from S. cerevisiae) (15, 20). Based on the refolding kinetics, it was suggested that 6AP competes with the protein substrates for the binding sites on rRNA, although direct proof in support of this claim was unavailable (20). Our present results show that the binding sites of 6AP on domain V of rRNA highly overlap with those of the protein substrates (Figs. 2 and 3). Moreover, specific base mutations in the overlapping sites not only alter the interaction of both the protein substrate and 6AP with domain V rRNA (Fig. 6, A and B) but also decrease PFAR (Fig. 4, A and B). Thus, we conclude that 6AP competitively occludes the protein substrates from gaining access to the functionally important interaction sites on the domain V rRNA and thereby inhibits PFAR.
How 6AP preferentially inhibits PFAR is an open question. Previous results have shown that there is no effect of 6AP on global protein synthesis (15). When tested on di- or tripeptide formation, no decrease in the level of ML dipeptide or MLL tripeptide was seen in 60 s even with high concentrations of 6AP, which would completely inhibit PFAR (data not shown). However, a detailed understanding of the action of 6AP in protein synthesis requires fast kinetics or single molecule-based analysis, which are foreseeable steps in the future investigation of this topic. Also, it is important to characterize the structure of the ribosome in complex with 6AP, which will certainly add to the understanding of how 6AP selectively targets PFAR.
Whether 6AP binds to other domains of the rRNA is yet another open question. As 6AP has planar structure, which presumably makes it prone to layered stacking between the RNA bases, a detailed understanding of the chemistry of the 6AP-rRNA interaction requires further investigation beyond the scope of this work.5
Model of Domain V rRNA-assisted Refolding and 6AP Inhibition
We propose a simple model to explain the mechanism of protein folding with domain V rRNA and its inhibition by 6AP (Fig. 7). According to this model, the unfolded protein substrate (U) collapses to an early folding intermediate (I) immediately after dilution of the denaturant. In the case of self-folding, a fraction of the intermediate I (∼25%) rapidly primes to a productive folding intermediate (I*), which slowly folds to the active folded state (F). The remaining ∼75% is trapped in a misfolded state (mF), possibly through a different intermediate (I−). The rate-limiting step in this folding pathway is I* → F, with an average time (t½ = 1/k) of 6–7 min. It is clear from our results that the rate of refolding of HCA does not change in the presence of domain V rRNA (Fig. 1B and Table 1). Hence, the role of the domain V rRNA as a protein folding modulator is probably to increase the fraction of the productive intermediate I* (∼40%) through transient trapping of I rather than to modulate the actual folding step (I* → F). 6AP inhibits the “trapping” reaction by competing with I for the binding sites on domain V rRNA. As a result, the protein follows the self-folding pathway. That is why at a high concentration of 6AP, which presumably blocks all protein interaction sites on domain V rRNA, refolding only to the extent of self-folding could be achieved. This model can be extrapolated to elucidate PFAR in general, as the other ribosomal folding modulators (e.g. 70S ribosome, 50S subunit, and 23S rRNA), similar to domain V rRNA, modulate only the extent of refolding, but the rate of refolding remains unaffected (20).
FIGURE 7.

Simple model for PFAR and 6AP action. The unfolded state (U) collapses to an intermediate (I) upon dilution of the denaturant. For self-folding, I transforms either to a folding-competent intermediate (I*), which folds slowly to the native state (F), or to another intermediate (I−), which leads to the misfolded state (mF). The domain V (Dom V) rRNA-assisted refolding proceeds through a fast trapping reaction, which facilitates conversion of I to I*, thereby driving more I molecules to the productive folding pathway (I* → F). 6AP inhibits the trapping reaction by binding to the overlapping sites on domain V rRNA. As a result, the domain V rRNA-assisted pathway gets blocked, and the proteins fold via the self-folding pathway.
Our current knowledge about the nature of the interaction between protein substrates and the domain V rRNA is quite limited. The common interaction map of various protein substrates on domain V of 23S rRNA suggests a general mechanism of PFAR for all protein substrates. The multiple interaction sites on domain V rRNA indicate that the transition I → I* might involve multiple steps of interaction between the protein substrate and the domain V rRNA, the sequence of which remains open for future investigations.
Correlation between PFAR and Prion
As discussed above, the binding of both 6AP and PFAR involves common sets of nucleotides on the domain V rRNA (Figs. 2 and 3), and both are inhibited when these nucleotides are altered by mutation (Figs. 4 and 6). Because 6AP possesses well documented antiprion activity but does not interact directly with the prion protein (16, 26), our results, in line with earlier reports (15, 20), suggest indirectly that PFAR might be involved in prion formation. However, the possibility of alternative mechanisms by which 6AP inhibits prion phenotype cannot be completely overruled.
The involvement of nucleic acids in formation of prions is not a new concept. Independent studies have shown that nucleic acid interactions facilitate prion fibril formation (27–29), although the mechanism of such processes is not known. Detailed knowledge about the in vivo substrates of PFAR and direct kinetic analysis will be required to corroborate our hypothesis about the involvement of PFAR in prion mechanisms.
Acknowledgments
We acknowledge Lars Hellman (Uppsala University) for allowing us to use the UV cross-linker. We also thank Susan Liebman (University of Illinois, Chicago, IL) for providing the construct for 25S rRNA. Xueliang Ge and Petar Kovachev are acknowledged for help in manuscript preparation.
This work was supported by the Swedish Research Council (individual grants from the M and NT sections, VR-SIDA (Swedish Research Link), and a Linnaeus grant to the Uppsala RNA Research Center); the Carl Tryggers Stiftelse; a postdoctoral stipendium from the Wenner Gren Stiftelse (to D. B.); the Knut and Wallice Wallenberg Foundation (to RiboCORE), Vinnova/DBT (India), and the SSF-Dalen (Sweden-France Bilateral Collaboration) Program (to S. S.); a scholarship from the Chinese Scholarship Council (to Y. P.); and INSERM, CRITT Santé Bretagne (Région Bretagne), and ANR “Blanche” of the French Government (to M. B. and C. V.).
D. Banerjee, manuscript in preparation.
- PFAR
- protein folding activity of the ribosome
- 6AP
- 6-aminophenanthridine
- HCA
- human carbonic anhydrase.
REFERENCES
- 1. Bera A. K., Das B., Chattopadhyay S., DasGupta C. (1994) Protein folding by ribosome and its RNA. Curr. Sci. 66, 230–232 [Google Scholar]
- 2. Das B., Chattopadhyay S., Bera A. K., DasGupta C. (1996) In vitro protein folding by ribosomes from Escherichia coli, wheat germ and rat liver–the role of the 50S particle and its 23S rRNA. Eur. J. Biochem. 235, 613–621 [DOI] [PubMed] [Google Scholar]
- 3. Argent R. H., Parrott A. M., Day P. J., Roberts L. M., Stockley P. G., Lord J. M., Radford S. E. (2000) Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 275, 9263–9269 [DOI] [PubMed] [Google Scholar]
- 4. Das D., Das A., Samanta D., Ghosh J., Dasgupta S., Bhattacharya A., Basu A., Sanyal S., Das Gupta C. (2008) Role of the ribosome in protein folding. Biotechnol. J. 3, 999–1009 [DOI] [PubMed] [Google Scholar]
- 5. Kudlicki W., Coffman A., Kramer G., Hardesty B. (1997) Ribosomes and ribosomal RNA as chaperones for folding of proteins. Fold Des. 2, 101–108 [DOI] [PubMed] [Google Scholar]
- 6. Sulijoadikusumo I., Horikoshi N., Usheva A. (2001) Another function for the mitochondrial ribosomal RNA: protein folding. Biochemistry 40, 11559–11564 [DOI] [PubMed] [Google Scholar]
- 7. Chattopadhyay S., Das B., DasGupta C. (1996) Reactivation of denatured proteins by 23S ribosomal RNA: role of domain V. Proc. Natl. Acad. Sci. U.S.A. 93, 8284–8287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanyal S. C., Pal S., Chowdhury S., DasGupta C. (2002) 23S rRNA assisted folding of cytoplasmic malate dehydrogenase is distinctly different from its self-folding. Nucleic Acids Res. 30, 2390–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pal D., Chattopadhyay S., Chandra S., Sarkar D., Chakraborty A., Das Gupta C. (1997) Reactivation of denatured proteins by domain V of bacterial 23S rRNA. Nucleic Acids Res. 25, 5047–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pal S., Chandra S., Chowdhury S., Sarkar D., Ghosh A. N., Das Gupta C. (1999) Complementary role of two fragments of domain V of 23 S ribosomal RNA in protein folding. J. Biol. Chem. 274, 32771–32777 [DOI] [PubMed] [Google Scholar]
- 11. Das A., Ghosh J., Bhattacharya A., Samanta D., Das D., Das Gupta C. (2011) Involvement of mitochondrial ribosomal proteins in ribosomal RNA-mediated protein folding. J. Biol. Chem. 286, 43771–43781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basu A., Samanta D., Bhattacharya A., Das A., Das D., DasGupta C. (2008) Protein folding following synthesis in vitro and in vivo: association of newly synthesized protein with 50S subunit of E. coli ribosome. Biochem. Biophys. Res. Commun. 366, 592–597 [DOI] [PubMed] [Google Scholar]
- 13. Basu A., Samanta D., Das D., Chowdhury S., Bhattacharya A., Ghosh J., Das A., DasGupta C. (2008) In vitro protein folding by E. coli ribosome: unfolded protein splitting 70S to interact with 50S subunit. Biochem. Biophys. Res. Commun. 366, 598–603 [DOI] [PubMed] [Google Scholar]
- 14. Chattopadhyay S., Pal S., Pal D., Sarkar D., Chandra S., Das Gupta C. (1999) Protein folding in Escherichia coli: role of 23S ribosomal RNA. Biochim. Biophys. Acta 1429, 293–298 [DOI] [PubMed] [Google Scholar]
- 15. Tribouillard-Tanvier D., Dos Reis S., Gug F., Voisset C., Béringue V., Sabate R., Kikovska E., Talarek N., Bach S., Huang C., Desban N., Saupe S. J., Supattapone S., Thuret J. Y., Chédin S., Vilette D., Galons H., Sanyal S., Blondel M. (2008) Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs. PloS ONE 3, e2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bach S., Talarek N., Andrieu T., Vierfond J. M., Mettey Y., Galons H., Dormont D., Meijer L., Cullin C., Blondel M. (2003) Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat. Biotechnol. 21, 1075–1081 [DOI] [PubMed] [Google Scholar]
- 17. Tribouillard-Tanvier D., Béringue V., Desban N., Gug F., Bach S., Voisset C., Galons H., Laude H., Vilette D., Blondel M. (2008) Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions. PLoS ONE 3, e1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barbezier N., Chartier A., Bidet Y., Buttstedt A., Voisset C., Galons H., Blondel M., Schwarz E., Simonelig M. (2011) Antiprion drugs 6-aminophenanthridine and guanabenz reduce PABPN1 toxicity and aggregation in oculopharyngeal muscular dystrophy. EMBO Mol. Med. 3, 35–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Voisset C., Thuret J. Y., Tribouillard-Tanvier D., Saupe S. J., Blondel M. (2008) Tools for the study of ribosome-borne protein folding activity. Biotechnol. J. 3, 1033–1040 [DOI] [PubMed] [Google Scholar]
- 20. Dos Reis S., Pang Y., Vishnu N., Voisset C., Galons H., Blondel M., Sanyal S. (2011) Mode of action of the antiprion drugs 6AP and GA on ribosome assisted protein folding. Biochimie 93, 1047–1054 [DOI] [PubMed] [Google Scholar]
- 21. Samanta D., Mukhopadhyay D., Chowdhury S., Ghosh J., Pal S., Basu A., Bhattacharya A., Das A., Das D., DasGupta C. (2008) Protein folding by domain V of Escherichia coli 23S rRNA: specificity of RNA-protein interactions. J. Bacteriol. 190, 3344–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lewis M. E., Sherman T. G., Burke S., Akil H., Davis L. G., Arentzen R., Watson S. J. (1986) Detection of proopiomelanocortin mRNA by in situ hybridization with an oligonucleotide probe. Proc. Natl. Acad. Sci. U.S.A. 83, 5419–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das D., Samanta D., Hasan S., Das A., Bhattacharya A., Dasgupta S., Chakrabarti A., Ghorai P., Das Gupta C. (2012) Identical RNA-protein interactions in vivo and in vitro and a scheme of folding the newly synthesized proteins by ribosomes. J. Biol. Chem. 287, 37508–37521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chowdhury S., Pal S., Ghosh J., DasGupta C. (2002) Mutations in domain V of the 23S ribosomal RNA of Bacillus subtilis that inactivate its protein folding property in vitro. Nucleic Acids Res. 30, 1278–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voisset C., Saupe S. J., Blondel M. (2011) The various facets of the protein-folding activity of the ribosome. Biotechnol. J. 6, 668–673 [DOI] [PubMed] [Google Scholar]
- 26. Gug F., Oumata N., Tribouillard-Tanvier D., Voisset C., Desban N., Bach S., Blondel M., Galons H. (2010) Synthesis of conjugates of 6-aminophenanthridine and guanabenz, two structurally unrelated prion inhibitors, for the determination of their cellular targets by affinity chromatography. Bioconjug. Chem. 21, 279–288 [DOI] [PubMed] [Google Scholar]
- 27. Gomes M. P., Vieira T. C., Cordeiro Y., Silva J. L. (2012) The role of RNA in mammalian prion protein conversion. Wiley Interdiscip. Rev. RNA 3, 415–428 [DOI] [PubMed] [Google Scholar]
- 28. Macedo B., Millen T. A., Braga C. A., Gomes M. P., Ferreira P. S., Kraineva J., Winter R., Silva J. L., Cordeiro Y. (2012) Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 51, 5402–5413 [DOI] [PubMed] [Google Scholar]
- 29. Silva J. L., Vieira T. C., Gomes M. P., Rangel L. P., Scapin S. M., Cordeiro Y. (2011) Experimental approaches to the interaction of the prion protein with nucleic acids and glycosaminoglycans: Modulators of the pathogenic conversion. Methods 53, 306–317 [DOI] [PubMed] [Google Scholar]