

Background: Histidine 69 in the propeptide is a pH sensor that mediates compartment-specific furin activation.
Results: Histidine 69 protonation exposes the activation loop for proteolysis only within an optimal window for pH-dependent activation.
Conclusion: A small structural change functions as the trigger that regulates precise spatiotemporal activation of furin.
Significance: Our work provides insights into how individual proprotein convertases encode their unique compartment-specific activation.
Keywords: Convertases, Protein Folding, Protein Secretion, Serine Protease, Subcellular Organelles, Furin, Intramolecular Chaperone, pH Sensor, Propeptide, Protease Activation Precision
Abstract
The proprotein convertase furin requires the pH gradient of the secretory pathway to regulate its multistep, compartment-specific autocatalytic activation. Although His-69 within the furin prodomain serves as the pH sensor that detects transport of the propeptide-enzyme complex to the trans-Golgi network, where it promotes cleavage and release of the inhibitory propeptide, a mechanistic understanding of how His-69 protonation mediates furin activation remains unclear. Here we employ biophysical, biochemical, and computational approaches to elucidate the mechanism underlying the pH-dependent activation of furin. Structural analyses and binding experiments comparing the wild-type furin propeptide with a nonprotonatable His-69 → Leu mutant that blocks furin activation in vivo revealed protonation of His-69 reduces both the thermodynamic stability of the propeptide as well as its affinity for furin at pH 6.0. Structural modeling combined with mathematical modeling and molecular dynamic simulations suggested that His-69 does not directly contribute to the propeptide-enzyme interface but, rather, triggers movement of a loop region in the propeptide that modulates access to the cleavage site and, thus, allows for the tight pH regulation of furin activation. Our work establishes a mechanism by which His-69 functions as a pH sensor that regulates compartment-specific furin activation and provides insights into how other convertases and proteases may regulate their precise spatiotemporal activation.
Introduction
The requirement for single or multiple endoproteolytic cleavages of precursor proproteins as they transit the secretory pathway is an evolutionally conserved theme in the biochemistry of biologically active proteins and peptides (1). Proprotein convertases (PCs)4 are a family of calcium-dependent serine proteases that process inactive proproteins in eukaryotes (2, 3). PCs include nine endoproteases (furin, PC1/PC3, PC2, PC4, PACE4, PC5/PC6, PC7/LPC/PC8, SKI/S1P, and NARC-1/PCSK9) and constitute a subfamily within the subtilase superfamily (2, 4). PCs are synthesized on the endoplasmic reticulum (ER) and are translocated into the ER lumen where they undergo folding and intramolecular excision of their prodomains to form a propeptide-PC complex that is competent for export to late secretory compartments but is catalytically inactive because the propeptide masks the catalytic domain from substrate binding (5). Upon reaching their correct cellular destinations for processing substrates in trans, the propeptide-PC complex undergoes activation, usually through autoproteolytic cleavage of the bound propeptide (for a review, see Refs. 2, 4). Although the synthesis of proteases as inactive proenzymes enables cells to regulate their catalytic activity spatially and temporally, the molecular and cellular determinants that modulate activation of PCs remain poorly understood (4).
Furin, the most extensively studied PC, catalyzes proteolytic maturation of a diverse repertoire of growth factors, receptors, and enzyme precursors within multiple secretory pathway compartments (2, 4, 5). Consistent with its essential role in homeostasis, mice lacking furin die before embryonic day 11 because of cardiac defects (6, 7). Additionally, misregulation of furin activation has been associated with cancer invasiveness and metastasis and susceptibility to viral and parasitic infections (for a review, see Ref. 8). The furin precursor contains an 83-residue N-terminal propeptide (PROFUR) that is essential for folding the catalytic domain (MATFUR) in the ER (9, 10), putatively by stabilizing the late stages of the folding transition state (11). Because of the nature of its role in facilitating folding, the furin propeptide is considered an intramolecular chaperone (12, 13). When folded, the catalytic domain rapidly cleaves propeptide at the consensus furin site RTKR107 (t½ < 10 min (14)), which permits export of the inactive PROFUR-MATFUR inhibition complex from the ER to late secretory pathway compartments (10, 15–17). Upon reaching the mildly acidic trans-Golgi network (TGN) (pH ∼6.0), PROFUR undergoes a slow second internal proteolytic cleavage at the noncanonical furin site 69HRGVTKR75 (t½ < 100 min (14)), which disrupts the inhibition complex to allow MATFUR to exert its catalytic activity (14, 17). The necessity for exposure of PROFUR-MATFUR to the acidic pH of the TGN, coupled with the importance of tight regulation of protease activation, argues for the presence of a sensor that recognizes when the complex reaches the correct pH environment.
We reported that a conserved histidine residue (His-69) in PROFUR acts as a pH sensor that regulates the compartment-specific activation of pro-furin (18). Although a His-69-Leu substitution in the furin propeptide (H69L-PROFUR) permitted correct folding, prodomain excision, and export of the propeptide-furin complex from the ER to the TGN/endosomal system, the substitution blocked the pH-dependent second cleavage of MATFUR at Arg-75, thereby preventing furin activation (18). Moreover, we reported recently that PROFUR and the propeptide of PC1/3 (PROPC1) are sufficient to regulate the pH-dependent activation of their cognate catalytic domains, suggesting a broad role for pH sensing in activation of PCs (19) and other secreted proteases (20). Indeed, swapping propeptide domains transfers sensitivity to pH-dependent activation of the protease domain in a propeptide-dependent manner (19).
Although His-69 protonation is required for furin activation, the precise mechanism by which this pH sensor mediates activation has remained unclear. Here, we use WT-PROFUR and the deprotonated state mimic H69L-PROFUR to explore structure, stability, and pH-dependent binding, coupled with mathematical modeling and molecular dynamics, to understand how His-69 functions as a pH sensor. Taken together, our work explains the structural and mechanistic basis by which His-69 regulates compartment-specific furin activation and provides insight into how other PCs may regulate their own activation.
EXPERIMENTAL PROCEDURES
Protein Production and Purification
Sequences coding for WT-PROFUR and H69L-PROFUR variants were cloned into pET11a and expressed in BL21(DE3) Escherichia coli as described (19). Inclusion bodies were isolated and solubilized after cell lysis performed using a French press apparatus. Protein was purified using a size exclusion column and dialyzed into 6 m guanidinium HCl for long-term storage. Mature furin was overexpressed in BSC40 cells infected with recombinant Vaccinia virus encoding human furin as described (18, 19).
CD Spectroscopy
CD spectroscopy was performed on an AVIV model 215 CD spectrometer at 4 °C as described (19, 21, 22). Before taking CD measurements, purified protein was refolded in a stepwise fashion by dialysis against refolding buffer (50 mm Tris, 150 mm KCl, 1 mm CaCl2 (pH 7.4) with incrementally smaller amounts of urea, with a final dialysis step into cacodylate buffer (50 mm cacodylic acid, 150 mm KCl, 1 mm CaCl2 (pH 7.4). An additional dialysis step was carried out to adjust the pH as necessary. Refolded protein was then subjected to ultracentrifugation for 15 min to remove any particulate, and CD spectra between 190–260 nm were taken to ensure complete folding. For the protease domain, 0.1 mg/ml of mature furin was dialyzed against the cacodylate buffer at pH 5.0–7.0 and used for analyzing pH stability.
Urea Denaturation
Thermodynamic stabilities of the wild-type propeptide (WT-PROFUR) and H69L-PROFUR were measured by perturbing the secondary structure using urea and fitting the data to either two-state or three-state unfolding models as described (21). WT-PROFUR or H69L-PROFUR (∼3 mg/ml) were refolded as above into a buffer of the desired pH, and urea was added in 0.01 m steps to a final concentration of 6 m urea using a titrator. Changes in absorbance at 222 nm were monitored as a function of denaturant concentration.
Glycerol Stability
WT-PROFUR or H69L-PROFUR (∼0.4 mg/ml) was refolded as above and then glycerol was added to a final concentration ranging from 0–30%. CD spectra between 190–260 nm were taken to assess the effect of glycerol on the stability of the isolated propeptide as described (21). Approximately 0.1 mg/ml of mature furin was dialyzed against 50 mm cacodylic acid, 150 mm KCl, and 1 mm CaCl2 (pH 7.0) containing 30% glycerol and used for analyzing the effect of glycerol on the protease conformation.
Activity Assay
Activity assays were performed to determine the inhibitory capabilities of the various propeptides as described (18). 129 μm of the fluorogenic furin substrate (Abz-RVKRGLA-Tyr[3-NO2] was incubated with serially diluted amounts of either WT-PROFUR or H69L-PROFUR (concentrations ranging from 0.15–3000 nm) in cacodylate buffer (50 mm cacodylic acid, 150 mm KCl, 1 mm CaCl2). 0.5 μl of secreted furin was added to initiate the reaction. To assess the pH dependence of IC50, the pH of the cacodylate buffer was varied between pH 5.0–7.4. For assays to measure the effect of glycerol on stability, 30% was added or not added to cacodylate buffer at pH 7.0. All activity assays were conducted in triplicate on a SpectraMax-M2 spectrofluorometer equipped with a 96-well plate reader, with the excitation wavelength set to 320 nm and emission wavelength set to 425 nm (17). Data were fitted and analyzed using GraphPad Prism to determine Ki and IC50 values.
Molecular Dynamics Simulations
A homology model of WT-PROFUR derived from the solution structure of the paralogous prohormone convertase 1 propeptide (1KN6) (23) was used as a model of PROFUR, as described earlier (19). The model for WT-PROFUR was obtained by mutating residue histidine at position 69 to a leucine using PyMOL (PyMOL molecular graphics system, version 1.5.0.4, Schrödinger, LLC) and minimized using the CHARMM22 force field (24). All hydrogen and non-protein atoms were removed, and hydrogens were added back using the autoPSF module in NAMD version 2.5 (25). Structures were solvated explicitly in water cubes in VMD (Visual Molecular Dynamics, v. 1.9.1) with a minimum distance of 12 Å to the edge of the cube. All simulations were carried out with periodic boundary conditions, particle mesh Ewald for long-range electrostatics, and a 12-Å cutoff for non-bonded interactions within the CHARMM22 force field using NAMD. Simulations were run with a step size of 1 fs with snapshots taken every 10 ps as described earlier (19). To first equilibrate the system, the protein was constrained, and solvent was minimized for 1000 steps using a conjugate gradient algorithm. The minimized system was then used for subsequent simulations. To simulate the pH-dependent protonation reactions, we approximated the pH environment by predetermining the protonation state of the histidines in the starting structure as described earlier (19). For a pH environment of 7.0, we used the HSD parameter, which represents the uncharged state, with a proton bound to the nitrogen in the Δ position. For a pH of 6.0, we used the HSP parameter to simulate the positively charged, protonated histidine with protons bound to both nitrogen atoms.
Mathematical Modeling
Modeling of the kinetics of the furin activation step was done using CellWare version 2.0 (Systems Biology Group, Bioinformatics Institute, Singapore) (26). CellWare is an integrated modeling and simulation tool for biochemical pathways and cellular processes and has been used to simulate the activation of pro-subtilisin, the bacterial prototype of pro-furin, as described earlier (22). The model used in these simulations is represented in Fig. 4A. Each intermediate is represented as a discrete molecular species, and reaction pathways were defined as reversible or irreversible, with the rate constants noted on the basis of experimental results either with furin or its homolog, subtilisin (22). Additionally, the software allows for variation of initial concentrations and simulation duration. The models were simulated using the Gillespie stochastic algorithm with 1000 experiments and 1 × 109 iterations for a total duration of 1000 time units. The appearance and disappearance of the various molecular species were monitored, and data were analyzed in GraphPad Prism.
FIGURE 4.

Mathematical modeling of furin activation. A, model of the furin activation pathway used in mathematical simulations. Fur = furin (protease), p = propeptide. Rates for each step were modeled in accordance with published values in the literature. The gray and black arrows depict reversible and irreversible reactions, respectively. B, stochastic activation of furin over 15 sample simulations. C, distribution of stochastic activation times (min) of furin over 1000 sample iterations (first panel, no free furin) and distribution of activation times (min) when active furin is present (second, third, and fourth panels). D, change in time to activation (min) as Ka, the affinity of furin for free PROFUR, is altered. The gray box indicates the physiologic range of Ka values for IMCs (intramolecular chaperones). E, the change in time to activation (min) as kcat, the catalytic efficiency, or the rate of dissociation of deg-PROFUR from degradation complexes I and II to release active furin, is altered.
RESULTS
The Constitutively Deprotonated Mimic of the pH Sensor, H69L-PROFUR, Is More Stable Than WT-PROFUR
To understand the mechanism by which His-69 functions as a pH sensor, we undertook detailed structural analyses of WT-PROFUR and the H69L-PROFUR variant reported previously, which mimics the nonprotonated state of the pH sensor, using CD and intrinsic fluorescence spectroscopy. Prior studies indicate that the H69L-PROFUR chaperones efficient folding of the catalytic domain of furin (MATFUR), as measured by autoprocessing of H69L-PROFUR to form a stable H69L-PROFUR-MATFUR complex (18). However, unlike the WT-PROFUR-MATFUR complex, H69L-PROFUR-MATFUR remains trapped in a stable state, unable to become active at acidic pH. On the other hand, the H69K-PROFUR variant, which represents a constitutively protonated state of the pH sensor, fails to fold correctly and is rapidly degraded inside the cell (18). Hence, it was not used in our analyses.
The far UV CD spectrum of the isolated WT-PROFUR revealed the existence of a significant secondary structure (Fig. 1A). Substituting the pH sensor, His-69, with Leu caused a slight increase in the secondary structure, as seen by the shift in the peak from 206 to 208 nm with a concomitant increase in negative ellipticity at 222 nm in H69L-PROFUR. Because α-helices absorb strongly at 222 nm and 208 nm (27), our results suggest that substituting residues that mimic the deprotonated state of the pH sensor marginally increase the α-helicity within the isolated H69L-PROFUR.
FIGURE 1.
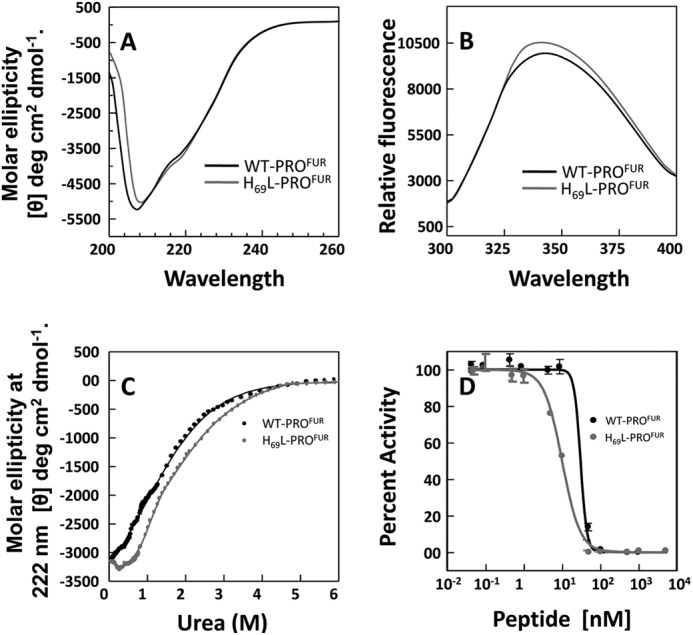
H69L-PROFUR is more structured than WT-PROFUR. A, the secondary structure of WT-PROFUR and H69L-PROFUR determined via CD spectroscopy at far UV, performed at pH 7. 0, and plotted as molar ellipticity (θ) deg/cm2/dmol. B, tertiary structure of wild-type or mutant propeptide determined by measuring intrinsic tryptophan fluorescence after excitement with λ = 295 nm. C, thermodynamic stability of the propeptides monitored by changes in ellipticity (θ) at λ = 222 nm as a function of urea concentration. Data were fit to a standard two-state equation using a Marquardt algorithm. D, normalized activity, used to estimate IC50 values, determined by monitoring cleavage of the fluorogenic peptide substrate Abz-RVKRGLA-Tyr[2-NO2] with increasing amounts of WT-PROFUR or H69L-PROFUR present. All data are averaged over three independent experiments.
Likewise, we examined the tertiary structure of the protein by exciting the protein using a wavelength of 295 nm where the tryptophan emission spectrum is dominant over the weaker tyrosine and phenylalanine fluorescence (28–30). As seen in Fig. 1B, WT-PROFUR displays a maximum peak at 342 nm. Under identical conditions, the intrinsic tryptophan fluorescence is enhanced slightly with a blue shift in its emission spectrum (maximum at 339 nm) when His-69 is substituted by leucine. This indicates that the tryptophan residues are less exposed to solvent when His-69 is replaced by a Leu, suggesting that the structure may be more packed.
To better understand the extent of stabilization, we next measured the thermodynamic stability of WT-PROFUR and its variant relative to their unfolded states. Thermodynamic stability occurs when a system is in its lowest energy state when compared with all other accessible states within the same reaction environment. It can be measured by monitoring changes in the secondary structure with progressive addition of chaotropes such as urea or guanidine hydrochloride (31). Fig. 1C compares chaotrope-induced conformational changes in WT-PROFUR or H69L-PROFUR using circular dichroism spectroscopy. The transitions were fitted using a standard Marquardt algorithm with constraints for the base line set from using the circular dichroism ellipticity of the folded and unfolded proteins (21, 22). The data demonstrate that H69L-PROFUR (ΔGNU =1.424 ± 0.12 kcal) is more stable than WT-PROFUR (ΔGNU = 0.921 ± 0.09 kcal/mol). This indicates that the constitutively deprotonated variant H69L-PROFUR is stabilized by ∼0.5 kcal/mol when compared with WT-PROFUR.
Because propeptides are bona fide temporary inhibitors of proteases (11), we next asked how an increase in thermodynamic stability affects the inhibitory function of isolated WT-PROFUR and H69L-PROFUR by comparing IC50 values as described under “Experimental Procedures” (Fig. 1D). Analysis of the data gives an estimated IC50 concentration for WT-PROFUR, at pH 6.5, of ∼33 nm, 3-fold higher than that estimated for H69L-PROFUR (IC50 ∼11 nm). This establishes a link between the increased thermodynamic stability of the H69L substitution and its ability to act as an inhibitor of MATFUR, as indicated by the decrease in IC50. Taken together, the circular dichroism and fluorescence spectra, along with the analyses of thermodynamic stabilities, suggest that the nonprotonated mimic of the pH sensor subtly increases both secondary and tertiary structure, and enhances the overall thermodynamic stability and inhibitory function of H69L-PROFUR.
The Cosolvent Glycerol Enhances the Structure of WT-PROFUR and H69L-PROFUR and Their Apparent Affinity for MATFUR
We next examined whether increasing the secondary structure and thermodynamic stability of isolated WT-PROFUR and H69L-PROFUR enhances their binding affinity for MATFUR. We had demonstrated previously that propeptides of aqualysin (PROAQU) and subtilisin E (PROSUB), which are orthologs of furin, also exist in partially folded, molten, globule-like states (19, 32). However, the progressive addition of glycerol induced and stabilized the secondary and tertiary structures within PROAQU and PROSUB and simultaneously increased their binding affinities for their cognate catalytic domains (22). Thus, we investigated whether glycerol could stabilize the secondary structures of WT-PROFUR and H69L-PROFUR. Our results demonstrate two noteworthy features that are evident in Fig. 2, A and B. First is the presence of an isosbestic point at a wavelength of 208 nm in WT-PROFUR and H69L-PROFUR. The presence of an isosbestic point (33) in the circular dichroism spectra suggests that glycerol induces the partially structured propeptide (34, 35) to fold into a more stable state, and, thus, there are two distinct states in which the propeptide can exist, depending on its local environment (28). The second feature to note is the progressive stabilization of the secondary structure with increasing amounts of glycerol within isolated WT-PROFUR and H69L-PROFUR, measured using changes in ellipticity at 222 nm (Fig. 2C). As noted previously, the secondary structure of H69L-PROFUR is marginally more stable than that of WT-PROFUR.
FIGURE 2.
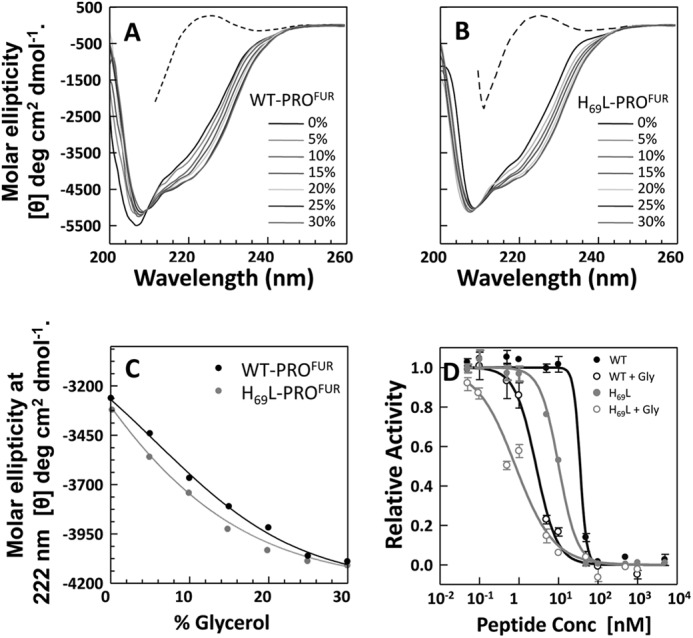
A glycerol-induced structure of the isolated propeptides enhances affinity for MATFUR. Shown is the secondary structure of WT-PROFUR (A) and H69L-PROFUR (B) measured using circular dichroism spectroscopy as a function of increasing concentration of the cosolvent glycerol. Data are plotted as molar ellipticity. C, changes in secondary structure of the isolated propeptides, monitored by changes in ellipticity at λ = 222 nm, with increasing concentration of glycerol. D, activity, normalized to maximal activity with no PRO, used to estimate IC50 values of the propeptides in the presence (○) or absence (●) of 30% glycerol. Conc, concentration.
We next examined whether the increase in the secondary structure induced by glycerol translates into a tighter binding affinity between the propeptide and the catalytic domain. As seen in Fig. 2D, the addition of 30% glycerol enhanced the binding affinity, as measured using IC50 values. The IC50 of WT-PROFUR decreased from 33 nm to ∼2 nm and that of H69L-PROFUR from 11 nm to ∼0.8 nm. Taken together, our results support the hypothesis that both the increased structural (Fig. 2, A–C) and thermodynamic stability (Fig. 1C) of the propeptides enhances their affinity for their cognate catalytic domains.
H69L-PROFUR Is More Stable toward pH-dependent Unfolding
Because pro-furin undergoes its primary cleavage in the neutral environment of the ER to form a cleaved, non-covalently associated PROFUR-MATFUR complex that transits in to the mildly acidic TGN to become active, we next examined how changes in pH affect the structure, stability, and binding affinity of WT-PROFUR and H69L-PROFUR. Propeptides were purified, refolded, and analyzed for their secondary structure content using CD spectroscopy as described (“Experimental Procedures”). The results show that WT-PROFUR undergoes pH-dependent unfolding with an isosbestic point at ∼208 nm (Fig. 3A). A plot of the changes in CD signal at 222 nm as a function of pH suggests that WT-PROFUR undergoes a cooperative sigmoidal transition to a more unstructured state. Interestingly, the midpoint of this transition occurs at pH ∼6.0, close to the optimal pH for activation of furin. Under identical conditions, H69L-PROFUR is more stable. Although it does undergo some pH-dependent unfolding, with a midpoint of transition, likewise, at pH ∼6.0, it is critical to note that not only it is more stable at neutral pH than the WT, but also that it is not unfolded to the same extent, suggesting that the unfolding response to pH is blunted. In comparison, the change in structure of isolated MATFUR across this pH range is not significant (Fig. 3C, colored lines), nor does the addition of glycerol markedly change the structure of MATFUR (gray line). Taken together, this suggests that the protonation status of His-69 may drive pH-dependent conformational changes in the isolated furin propeptide. Our results indicate that lowering the pH triggers a transition between a folded state at pH 7.4 and a less folded, but not completely unstructured, state at pH 5.0. Mutations of titratable group His-69 to leucine marginally increase secondary structure at pH 7.4 and, to a larger extent, at pH 5.0 (Fig. 3B), suggesting that protonation of His-69 is essential for the pH-dependent transition between the two states.
FIGURE 3.

H69L-PROFUR is more stable than WT-PROFUR to pH-induced unfolding. A, pH-dependent secondary structure of WT-PROFUR performed at pH 7.0–5.0 and plotted as molar ellipticity. B, changes in secondary structure of the isolated propeptides monitored by changes in ellipticity at λ = 222 nm and plotted as a function of increasing pH. The midpoint of the unfolding transition for both peptides occurs at pH ∼6.0. C, CD structure of MATFUR at varying pH (colored lines) and with the addition of 30% glycerol (gray line). The dotted line represents the spectra of denatured MATFUR. D, thermodynamic stability of WT-PROFUR and H69L-PROFUR as a function of pH, given in calories (Cal). E, IC50 values for WT-PROFUR (top panel) and H69L-PROFUR (bottom panel) as a function of pH. F, activity of furin in the absence of the propeptide at varying pH. All values are given as a percentage of maximum activity and are the average of three independent experiments.
Next we measured the changes in thermodynamic stability of the WT-PROFUR and H69L-PROFUR, as described earlier (Fig. 1C), under conditions of varied pH. Our data suggest that the overall thermodynamic stability of the proteins decreases when the pH becomes more acidic (Fig. 3D). The greater change in thermodynamic stability of H69L-PROFUR as a function of pH suggested that the H69L substitution enhanced the thermodynamic stability in the isolated propeptide when compared with WT-PROFUR.
Because the concentration of protons affects the conformation of WT-PROFUR and H69L-PROFUR, we next measured how this conformational change affects the IC50 values as a function of pH (Fig. 3D). The data demonstrate that the IC50 values for WT-PROFUR and H69L-PROFUR change as a function of pH, with the maximum inhibitory concentration required for both proteins at pH 6.0 (Fig. 3D) and the midpoint of the conformational transition being ascertained using CD spectroscopy (B). Moreover, three important features in Fig. 3D are noteworthy. 1) The IC50 value for WT-PROFUR at pH 7.4 (∼12 nm) is about 4-fold higher than at pH 6.0 (IC50 ∼ 50 nm); 2) the IC50 value for H69L-PROFUR at pH 6.0 (∼17 nm) is about 3-fold lower than that for WT-PROFUR (IC50 ∼50 nm); and 3) when the pH is lower than the optimum for activation (pH ∼6.0), the IC50 value drops to lower concentrations of propeptides for both WT-PROFUR and H69L-PROFUR, suggesting an apparent increase in binding affinity. To examine whether pH denatures or inactivates MATFUR, we also monitored changes in the secondary structure (Fig. 3C) and the activity of furin across this pH range with no propeptide present (F). It is worth noting that although the activity of furin does decrease as pH drops, it remains active, with an activity at pH 5.0 roughly 50% of that observed at pH 6.0, where IC50 is highest. This suggests that furin remains structurally stable, indicating that increased affinity at pH 5.0 is likely a chemical phenomenon. Hence, the change in IC50, which is roughly 40-fold lower at pH 5.0 than at pH 6.0, cannot be explained by changes in activity alone. We are currently unable to examine directly how pH affects the propeptide-furin complex because of the high concentrations of mature furin required to create stoichiometric complexes. We used mathematical simulations to test whether small changes in binding affinities could account for in the inability of the variant to undergo activation, as described in the next section.
Mathematical Modeling of Furin Activation
To better understand the mechanism of furin activation, we modeled the activation the of pro-furin complex using CellWare in a manner similar to our previous work on the activation of pro-subtilisin (22). The CellWare package offers a multialgorithmic environment for modeling and simulation of kinetic networks using both deterministic and stochastic algorithms. The software allows modeling of elementary molecular interactions in terms of rate equations, and the temporal changes in molecular species or their stationary state values can be studied using stochastic and deterministic algorithms. Such models enable the analysis of evolution of molecular species and how they can affect the overall behavior of the system. The main advantage offered by such models is that it allows us to evaluate how modulating individual interactions affects the overall process outcome and was used to substantiate the experimentally observed stochastic activation of pro-subtilisin (22). Using CellWare version 2.0, we modeled the activation pathway furin. The autoprocessed PROFUR-MATFUR complex, free folded PROFUR, the PROFUR-MATFUR degradation complex, degraded PROFUR, and free active MATFUR were represented as individual molecular species. Interactions between them were defined using the law of mass action, and rate constants were defined from experimentally determined rates that have been published previously to recapitulate the established timing of furin activation (14, 22). Fig. 4A depicts the complete pathway. Starting with 10,000 autoprocessed PROFUR-MATFUR complexes, the evolution of individual molecular species along the maturation pathway was simulated using the Gillespie algorithm (“Experimental Procedures”). Although mature furin already exists in the cellular environment, our first simulation assumed that furin initially exists only as part of the inhibition complex inside the TGN. In this reaction, the complex has to dissociate into isolated furin and PROFUR molecules, which can then reassociate in a different configuration to form the degradation complex I (Fig. 4A, reactions 1 and 2, respectively). Numerical values for rates of dissociation of the complex and the formation of degradation complex I were approximated using affinity constants of PROFUR (14 nm, Refs. 18, 36) and Km for furin substrate reactions (1.99 μm, Refs. 14, 18). Using this model, the time-dependent activation of mature furin follows a stochastic model and concurs with experimental data for subtilisin, a bacterial homolog of furin (22). The rapid decrease in accumulated autoprocessed particles coincided with a none-to-all increase in mature furin and is indicative of rapid, autocatalytic activation (Fig. 4B). Although the release of free protease is extremely random, a graph of the number of active furin molecules versus time of activation for 1000 sample iterations is distributed broadly (Fig. 4C, first panel), with the maximum number of molecules being activated at ∼120 min. Thus, by fitting a minimalist mathematical model to the protease activation pathway of furin using a stochastic algorithm, our results appear consistent with our earlier experimental data that suggest that the time of activation of furin is ∼120 min (14, 17) However, if active furin already exists when the inhibition complex enters the TGN, which is the likely scenario inside a cell, the distribution of furin molecules versus time is altered, occurring almost instantaneously when even one free molecule is present (Fig. 4C, second, third, and fourth panels). As in this scenario, should the cleavage loop be accessible for cleavage in trans, the breakdown of the inhibition complex is rapid and establishes an activation paradigm that is dependent on initial precursor concentration, a case that does not match experimental observations, indicating that there must be some conformational change that modulates access to the cleavage site before processing and activation can occur. We next simulated the activation pathway for different values of Ka (the affinity between the propeptide and protease domain, Fig. 4A, reaction 1) and analyzed the distribution in time of activation (D). For each value of Ka, the mean time of activation for 1000 iterations was estimated. At low Ka, activation is fairly uniform and rapid, whereas a higher affinity results in the simultaneous increase in both time of activation and the associated stochastics (Fig. 4D), similar to that observed in the activation of isolated pro-subtilisin (22). Furthermore, the range of experimentally determined affinities depicted in Fig. 4D suggests that an ∼3-fold change in apparent affinity from ∼50 nm (WT-PROFUR) to ∼17 nm (H69L-PROFUR) appears to be insufficient to account for the experimentally observed inability of H69L-PROFUR-MATFUR to undergo activation at an acidic pH. Taken together, our simulations suggest that changes in affinity of the magnitudes seen in our results are not sufficient to account for the inability of H69L-PROFUR to undergo activation in the secretory pathway.
Another noteworthy observation of our experimental results was the fact that the apparent affinity of the propeptide for the protease domain increases below the optimal pH for activation, despite the decrease in thermodynamic stability of the isolated propeptide. An earlier report suggested that at a pH of 5.0, the catalytic efficiency (kcat) of furin is reduced to 35% of its maximum at optimal pH (37, 38), which may explain the apparently higher affinity seen in our results. To explore this, we again used our model to monitor the time of activation for a complex when the rate of degradation of the intramolecular chaperone (degradation complexes I and II, Fig. 4A, reactions 4 and 5) is altered. We simulated up to a 10-fold increase and decrease in rate of processing of the intramolecular chaperone as a surrogate for protease activity. Our results show that changes in activity have little effect on the overall time of activation (data not shown), indicating that a change in activity is unlikely to account for the differences in affinity observed in our studies.
Molecular Dynamics of the Furin Propeptide:
Molecular dynamics (MD) simulations can provide information that complements biophysical and biochemical studies of propeptide-mediated protease activation in eukaryotes. MD simulations have been used successfully to model time-dependent changes on residue resolved scale, and the results obtained are consistent with experimental data (19, 24, 39–44). To investigate how the H69L substitution may affect pH-mediated structural changes, we employed MD simulations.
We have shown previously that protonation of histidine side chains in the PRO of furin leads to drastic loss of structure during a 10-ns MD simulation (19). We hypothesized that introduction of the H69L into our model should stabilize the structure. To test this hypothesis, we first compared the root mean square fluctuations (RMSF) values of WT-PROFUR at pH 7.0 and pH 6.0 (Fig. 5A). Although histidine protonation increased fluctuations at almost all residues, the highest increase was observed in the loop flanked by β2 and β3 in proximity of residue 61 and within the C-terminal half of the cleavage loop. The largest increase was observed for His-80, whereas His-69 did not show any change upon protonation and remained stable. The H69L substitution reduced conformational fluctuations during the simulation at pH 7, with an even greater stabilization observed at pH 6. Interestingly, in H69L-PROFUR, His-80 appears to be the most stabilized compared with residues at pH 6.
FIGURE 5.

Molecular dynamics indicate that protonation of His-69 destabilizes the core of PROFUR. MD simulations were performed on WT-PROFUR and H69L-PROFUR using NAMD as described under “Experimental Procedures.” A, top panel, RMSF as a function of residue number for WT-PROFUR. Bottom panel, differences between the RMSF of simulation with H69L-PROFUR and the WT (ΔRMSF). Negative values indicate reduced fluctuations, and positive values indicate increased fluctuations because of the H69L point mutation. Values obtained under a simulated pH of 7 are shown in black, whereas values obtained under simulated pH of 6 are shown in red. B, RMSF values for the core region (all except for the loop) are shown by dashed lines, and loop regions (residues 70–79) are depicted by solid lines and plotted as a function of simulation time. Black lines represent WT-PROFUR, and red lines represent H69L-PROFUR. C, ribbon representation of the starting (red) and final (blue) structures of the simulations. The secondary cleavage site, Arg-75, is indicated in the cleavage loop (blue), and the pH sensor, His-69, is indicated in green. All simulations were done over 10 ns.
Analysis of the root mean square deviation values compared with the starting structure (Fig. 5B) as well as ribbon representation of the starting and end structures (C) revealed that during the simulation, the core region remains largely stable at pH 7.0 in the WT and H69L-PROFUR. It is important to note that the loop regions, which had a very high root mean square deviation during the simulation of WT-PROFUR, were stabilized substantially by the H69L variant. At pH 6, the core domain of the WT lost its native structure, indicated by rising root mean square deviation values. Compared with WT-PROFUR, the H69L variant stabilized the core and loop region significantly, although a slight increase in root mean square deviation was still observed.
Hence, our MD simulations suggest that although the loop region shows a high degree of flexibility during simulations using both protonated and unprotonated histidines in WT-PROFUR, the core remains stable in simulations using unprotonated histidines but loses structure in simulations using protonated histidines. Introduction of the H69L substitution into our model greatly increased stability of the core region during our simulation using protonated histidines, confirming that protonation of His-69 alone plays an important role in the pH-mediated structural changes. Interestingly, H69L mutations also lead to an increase in stability in the loop regions that stayed buried during the simulation. Taken together, the MD simulations are consistent with our hypothesis that changes in physical properties of the side chain at the pH sensor position has a strong influence on the structure of the activation loop and also suggest that leucine, because of its greater hydrophobicity, may not be a faithful representation of unprotonated histidine.
DISCUSSION
In this work, we have employed biophysical, biochemical, and computational approaches to investigate pH-dependent activation of furin, the canonical proprotein convertase (5). Our results provide insight into the way in which mature furin recognizes and responds to the changing pH of the secretory pathway and allow us to propose a mechanism for regulated activation. We also demonstrated that the propeptide provides furin a defined “activation window” wherein the steps of regulated proteolysis occur within a specific pH range, outside of which the chemical and structural properties of the propeptide appear to block activation of the protease, thereby preventing activation within downstream compartments.
Mechanism of pH-dependent Activation of Pro-furin
There are at least three possible mechanisms (Fig. 6) through which the stoichiometric inhibition complex (PROFUR-MATFUR) can become active upon reaching the TGN. The first mechanism posits that protonation of the pH sensor, along with other histidine residues, can induce dissociation of PROFUR from MATFUR, which then triggers the second proteolytic cleavage (23, 45). The second mechanism postulates that pH causes partial unfolding of the propeptide, which enables the second cleavage site to access the active site, promoting proteolysis in a cis reaction. The third mechanism hypothesizes that protonation of the pH sensor induces conformational changes that allow a mature furin molecule to access the loop that harbors the second internal cleavage site in trans (4, 18). In these cases, subsequent cleavage facilitates propeptide dissociation (Fig. 6A).
FIGURE 6.
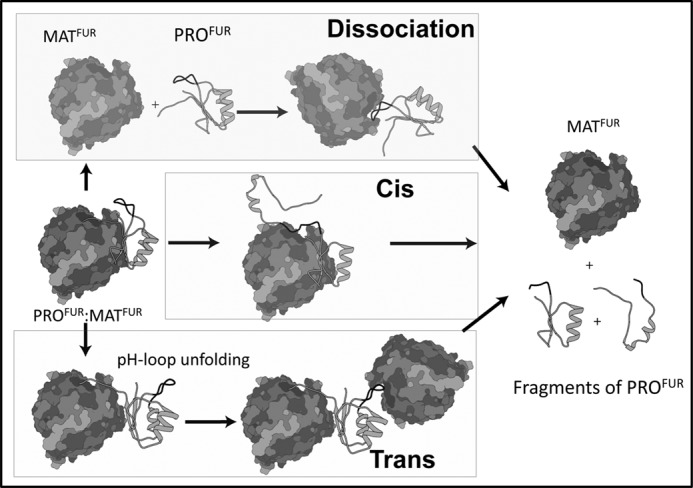
Models of furin activation. Three potential models of furin activation are considered. The dissociation model (top panel) posits that PROFUR dissociates from MATFUR after the pH sensor is protonated and then reassociates in a different orientation so that the propeptide can be cleaved. The cis and trans models of processing (center and bottom panels, respectively), in contrast, both suggest that protonation of the pH sensor drives an unfolding event that allows the cleavage site loop to become accessible to the active site, either for its own MATFUR to be cleaved in cis (center panel), or to a second molecule of MATFUR to be cleaved in trans. Only after the propeptide is cleaved does it dissociate from the protease.
To test these possibilities, we undertook various in vitro and in vivo experiments. Our results are most consistent with the hypothesis that the cleavage loop is critical to the overall structure and stability of the propeptide for the following reasons.
The data demonstrate that a 25-fold increase in proton concentration observed between pH 7.5–6.0 causes an ∼3.5-fold change in apparent binding affinity for WT-PROFUR. Moreover, the H69L pH sensor variant affects the apparent binding affinity ∼3.5-fold. To further test this, we built a mathematical model that assumed that protonation of the pH sensor promoted dissociation, allowing it to be processed. By varying Ka, we determined the effect of varying the affinity between the propeptide and protease on the rate of activation, which is a stochastic process in bacterial subtilisin (22). Our simulation results demonstrate that ∼10-fold changes in the affinity between PROFUR and MATFUR have only a minimal effect on the rate of activation within physiologically relevant range of values. It is noteworthy that our experimental data establish that the change in affinity because of the substitution is ∼3-fold, which argues that dissociation alone cannot account for lack of activation of the H69L-PROFUR-MATFUR complex observed in cell-based studies.
MD simulations suggest that His-69 protonation affects furin activation by increasing the conformational dynamics of the cleavage loop. At acidic pH, histidine residues within WT-PROFUR are protonated, including the pH sensor, His-69. However, when His-69 is replaced by Leu, the conformational dynamics of the loop are reduced dramatically, despite all of the remaining histidine residues being protonated (Fig. 5). Hence, the protonation status of His-69 alone affects the dynamics of the activation loop of the wild-type and mutant propeptides at two different pHs. Our simulations suggest that at pH 6.0, where the imidazole side chain of histidine is protonated, a dramatic movement in the loop region of PROFUR precedes the overall unfolding of the propeptide domain. This movement is reduced substantially in the case of the H69L variant, resulting in diminished unfolding of the propeptide as seen in Fig. 5.
The addition of the cosolvent glycerol induces a greater structure in WT-PROFUR as compared with H69l-PROFUR, indicating that the mutant is more “native-like,” perhaps because of the hydrophobic packing of the loop into the core of the propeptide. This increased structural stability correlates with an increase in affinity of the propeptide for the protease domain, as evidenced by the lower IC50 values calculated from experiments both with the constitutively deprotonated mutant propeptide and with the wild-type as pH is lowered. It is important to note that His-69 is solvent-accessible yet abuts a pocket formed partly by the hydrophobic core residues. Above the pH optimum of activation (pH > 7.0), the packing of the deprotonated His-69 into the core maintains a well packed structure that favors the bound state. However, upon protonation, the imidazole ring becomes charged, disrupting the packing and resulting in destabilization and local unfolding that exposes the cleavage site.
Hence, the results of our experimental and simulated data indicate that structural changes alter the accessibility of the cleavage site, thus raising the question of how the cleavage site becomes available to the active site. Although we cannot definitely distinguish between the possibility that the loop movement simply moves the cleavage site into a position more accessible to the active site in cis, or that there is a larger destabilization of the packing of the hydrophobic core that allows processing by a second molecule of furin in trans (Fig. 6), findings reported previously may lend some insight. In earlier work we observed that when the pH sensor, His-69, was mutated to leucine, no activation of furin takes place under basal conditions (18). Experiments where excess active furin was added to the inhibition complex indicated that at a non-permissive pH, exogenous furin was unable to affect activation of the inhibited furin. This suggests that the cleavage loop is inaccessible to free furin molecules at a pH outside of its optimum (18). Therefore, we argue that activation is mediated by proteolysis permitted by movement in the cleavage loop that only occurs upon protonation of the pH sensor and that dissociation occurs subsequently to processing.
Given this model, it is interesting to consider the possibility that activation is not concomitant with processing but, rather, that the C-terminal part of the propeptide sits in the substrate binding pocket and likely remains bound there for a period of time before it too dissociates to release inhibition. This is consistent with studies that demonstrate that the C-terminal propeptides fragments are potent inhibitors of furin (36, 46). We do not know whether, upon cleavage, there is a change in affinity or structure or if another protease plays a role in the dissociation (47). We can speculate that after the propeptide is cleaved at Arg-75, the shorter peptide fragment that lies in the substrate binding pocket is simply too short to make efficient contacts with residues of the protease and dissociates or, alternatively, that the cleavage allows a structural change to take place that promotes dissociation. A final alternative possibility is that the peptide fragment then becomes a substrate for cleavage in trans by another protease, such as carboxypeptidase (48). Although we cannot yet distinguish between these possibilities, future work will undoubtedly shed further light on this step of activation. The mathematical modeling MD simulations, along with experimentally measured changes in secondary structure and binding affinities, are consistent with our model that proteolytic processing precedes propeptide dissociation (Fig. 6).
The physical and Chemical Properties of the Propeptide Provide an Optimal Window for pH-dependent Furin Activation
Another interesting facet of furin activation is the apparent optimal pH window. Just as furin is not activated until it reaches the optimal pH environment of the TGN, it is not active at a lower pH unless it has transitioned appropriately through its activation window. We demonstrate that the propeptide defines this activation window. As indicated by the seeming discordance between pH-dependant changes in thermodynamic stability and affinity for the protease domain, there are multiple factors at play that determine the inhibitory behavior of the propeptide.
Although the thermodynamic stability analyses were conducted on isolated propeptides, the inhibition experiments require an association between the propeptide and protease domains, an interaction that we cannot, at this time, study directly. Although the isolated propeptide may lose stability at an acidic pH, its association with the protease domain may be enhanced because of changes in protonation states of charges side chains in the complex. As depicted in Fig. 7, there are several potential ionic interactions that may affect the interface between the propeptide and the protease domain. Also, given the local hydrophobicity at the interface, the pKa values of the charged groups are likely to be perturbed.
FIGURE 7.

The interface between MATFUR and PROFUR. Shown is a ribbon diagram showing the potential interaction of residues at the interface of MATFUR (green) and PROFUR (pink). Acidic residues are indicated in red, and basic residues are indicated in blue.
An alternative possibility for the apparently higher affinity at low pH could be a result of the apparent reduction in enzyme activity because of lower catalytic efficiency, as seen in Fig. 3F and as reported by others (37, 38). It is noteworthy that although furin activity drops ∼50% at pH 5.0 when compared with pH 6.0, the corresponding changes in IC50 are ∼40-fold over the same pH range. Hence, we posit that furin remains structurally stable (as seen by activity and CD studies), indicating that this is likely a chemical phenomenon. We tested this hypothesis further using mathematical simulations, simulating changes in the catalytic efficiency of the enzyme and examining its effects on the time of activation. Our results suggest that in physiologic ranges of the binding affinity, 10-fold changes in the catalytic efficiencies do not significantly alter the time of activation (data not shown). Hence, we argue that the observed optimal window for activation is a result of the apparent increase in affinity between the propeptide and protease domain. Although this may be tested experimentally using isothermal titration calorimetry, the high concentrations of active furin required make these experiments difficult at this time.
Hence, on the basis of our biophysical and biochemical experiments and computational simulations, we argue that the right balance of protonation and destabilization of PROFUR must be struck for efficient proteolysis to occur within the secondary cleavage site within the propeptide. We propose that this balance is only found within the strict confines of the activation window, thus preventing inappropriate activation of the protease within downstream compartments.
Implications of the pH Sensor in the Activation of Proprotein Convertases
The data presented here suggest an overarching model for activation of the PCs using furin as an example. Upon entering its window of activation, His-69, the pH-sensor in furin, is protonated to destabilize the hydrophobic pocket in which the pH sensor sits. Destabilization pushes the cleavage loop outward, thus allowing the catalytic site access to the secondary cleavage site. At the same time, a certain amount of flexibility in the association of the propeptide with the protease domain is preserved, which allows this cleavage to act as a finely tuned trigger. Above the pH optimum for activation, the stability of the propeptide, because of the hydrophobic packing of the pocket, keeps the propeptide tightly associated with the protease, and it is likely that below this window, secondary associations form as acidic residues, such as glutamates and aspartates, and are protonated, thus forming salt bridges that keep the propeptide tightly associated. Hence, it is only within a small window of pH that furin is able to be activated.
We have demonstrated that the propeptides of furin and PC1 alone contain information necessary for their compartment-specific activation (19). However, the residue that corresponds to His-69 in furin is conserved within all PCs, suggesting that additional factors must augment the subtle differences between the pH optima of individual PCs. This may be in part mediated by the distribution of additional histidines and other charged residues within the propeptides of PCs. Whether such evolutionary enrichment in His content is evident in other protease families is a subject of ongoing research in our laboratory.
Acknowledgments
We thank J. Dikeakos for reading the manuscript and Dr. David Farrens for insightful discussions.
This work was supported, in whole or in part, by a National Institutes of Health training grant (to D. M. W.) and National Institutes of Health Grant CA151564 (to G. T.). This work was also supported by National Science Foundation CAREER Award MCB0746589 and a grant in aid from the American Heart Foundation (to U. S.) and by American Heart Association Predoctoral Training Grant 12PRE11470005 (to J. E.).
- PC
- proprotein convertase
- ER
- endoplasmic reticulum
- TGN
- trans-Golgi network
- MD
- molecular dynamics
- RMSF
- root mean square fluctuation
- PRO
- propeptide.
REFERENCES
- 1. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008) The activation and physiological functions of the proprotein convertases. Int. J Biochem. Cell Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G. (2011) The proprotein convertases, 20 years later. Methods Mol. Biol. 768, 23–57 [DOI] [PubMed] [Google Scholar]
- 3. Artenstein A. W., Opal S. M. (2011) Proprotein convertases in health and disease. N. Engl. J. Med. 365, 2507–2518 [DOI] [PubMed] [Google Scholar]
- 4. Shinde U., Thomas G. (2011) Insights from bacterial subtilases into the mechanisms of intramolecular chaperone-mediated activation of furin. Methods Mol. Biol. 768, 59–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas G. (2002) Furin at the cutting edge. From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scamuffa N., Calvo F., Chrétien M., Seidah N. G., Khatib A. M. (2006) Proprotein convertases. Lessons from knockouts. FASEB J. 20, 1954–1963 [DOI] [PubMed] [Google Scholar]
- 7. Kim W., Essalmani R., Szumska D., Creemers J. W., Roebroek A. J., D'Orleans-Juste P., Bhattacharya S., Seidah N. G., Prat A. (2012) Loss of endothelial furin leads to cardiac malformation and early postnatal death. Mol. Cell. Biol. 32, 3382–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seidah N. G. (2011) What lies ahead for the proprotein convertases? Ann. N.Y. Acad. Sci. 1220, 149–161 [DOI] [PubMed] [Google Scholar]
- 9. Rehemtulla A., Dorner A. J., Kaufman R. J. (1992) Regulation of PACE propeptide-processing activity. Requirement for a post-endoplasmic reticulum compartment and autoproteolytic activation. Proc. Natl. Acad. Sci. U.S.A. 89, 8235–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leduc R., Molloy S. S., Thorne B. A., Thomas G. (1992) Activation of human furin precursor processing endoprotease occurs by an intramolecular autoproteolytic cleavage. J. Biol. Chem. 267, 14304–14308 [PubMed] [Google Scholar]
- 11. Shinde U., Inouye M. (2000) Intramolecular chaperones. Polypeptide extensions that modulate protein folding. Semin. Cell Dev. Biol. 11, 35–44 [DOI] [PubMed] [Google Scholar]
- 12. Shinde U., Li Y., Chatterjee S., Inouye M. (1993) Folding pathway mediated by an intramolecular chaperone. Proc. Natl. Acad. Sci. U.S.A. 90, 6924–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shinde U., Inouye M. (1993) Intramolecular chaperones and protein folding. Trends Biochem. Sci 18, 442–446 [DOI] [PubMed] [Google Scholar]
- 14. Anderson E. D., Molloy S. S., Jean F., Fei H., Shimamura S., Thomas G. (2002) The ordered and compartment-specific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J. Biol. Chem. 277, 12879–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Creemers J. W., Vey M., Schäfer W., Ayoubi T. A., Roebroek A. J., Klenk H. D., Garten W., Van de Ven W. J. (1995) Endoproteolytic cleavage of its propeptide is a prerequisite for efficient transport of furin out of the endoplasmic reticulum. J. Biol. Chem. 270, 2695–2702 [DOI] [PubMed] [Google Scholar]
- 16. Vey M., Schäfer W., Berghöfer S., Klenk H. D., Garten W. (1994) Maturation of the trans-Golgi network protease furin. Compartmentalization of propeptide removal, substrate cleavage, and COOH-terminal truncation. J. Cell Biol. 127, 1829–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anderson E. D., VanSlyke J. K., Thulin C. D., Jean F., Thomas G. (1997) Activation of the furin endoprotease is a multiple-step process. Requirements for acidification and internal propeptide cleavage. EMBO J. 16, 1508–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feliciangeli S. F., Thomas L., Scott G. K., Subbian E., Hung C. H., Molloy S. S., Jean F., Shinde U., Thomas G. (2006) Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J. Biol. Chem. 281, 16108–16116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dillon S. L., Williamson D. M., Elferich J., Radler D., Joshi R., Thomas G., Shinde U. (2012) Propeptides are sufficient to regulate organelle-specific pH-dependent activation of furin and proprotein convertase 1/3. J. Mol. Biol. 423, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elferich J., Williamson D. M., Krishnamoorthy B., Shinde U. (2013) FASEB J., in press [Google Scholar]
- 21. Subbian E., Yabuta Y., Shinde U. (2004) Positive selection dictates the choice between kinetic and thermodynamic protein folding and stability in subtilases. Biochemistry 43, 14348–14360 [DOI] [PubMed] [Google Scholar]
- 22. Subbian E., Yabuta Y., Shinde U. P. (2005) Folding pathway mediated by an intramolecular chaperone. Intrinsically unstructured propeptide modulates stochastic activation of subtilisin. J. Mol. Biol. 347, 367–383 [DOI] [PubMed] [Google Scholar]
- 23. Tangrea M. A., Bryan P. N., Sari N., Orban J. (2002) Solution structure of the pro-hormone convertase 1 pro-domain from Mus musculus. J. Mol. Biol. 320, 801–812 [DOI] [PubMed] [Google Scholar]
- 24. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM. The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dhar P., Meng T. C., Somani S., Ye L., Sairam A., Chitre M., Hao Z., Sakharkar K. (2004) CellWare. A multi-algorithmic software for computational systems biology. Bioinformatics 20, 1319–1321 [DOI] [PubMed] [Google Scholar]
- 27. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenfield N. J. (2006) Analysis of the kinetics of folding of proteins and peptides using circular dichroism. Nat. Protoc. 1, 2891–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nair S. K., Thomas T. J., Greenfield N. J., Chen A., He H., Thomas T. (2005) Conformational dynamics of estrogen receptors α and β as revealed by intrinsic tryptophan fluorescence and circular dichroism. J. Mol. Endocrinol. 35, 211–223 [DOI] [PubMed] [Google Scholar]
- 30. Lakowicz J. R., Maliwal B. P., Cherek H., Balter A. (1983) Rotational freedom of tryptophan residues in proteins and peptides. Biochemistry 22, 1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greenfield N. J. (2006) Determination of the folding of proteins as a function of denaturants, osmolytes or ligands using circular dichroism. Nat. Protoc. 1, 2733–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marie-Claire C., Yabuta Y., Suefuji K., Matsuzawa H., Shinde U. (2001) Folding pathway mediated by an intramolecular chaperone. The structural and functional characterization of the aqualysin I propeptide. J. Mol. Biol. 305, 151–165 [DOI] [PubMed] [Google Scholar]
- 33. Sawicki C. A., Gibson Q. H. (1976) Quaternary conformational changes in human hemoglobin studied by laser photolysis of carboxyhemoglobin. J. Biol. Chem. 251, 1533–1542 [PubMed] [Google Scholar]
- 34. Bhattacharjya S., Xu P., Wang P., Osborne M. J., Ni F. (2007) Conformational analyses of a partially-folded bioactive prodomain of human furin. Biopolymers 86, 329–344 [DOI] [PubMed] [Google Scholar]
- 35. Bhattacharjya S., Xu P., Xiang H., Chrétien M., Seidah N. G., Ni F. (2001) pH-induced conformational transitions of a molten-globule-like state of the inhibitory prodomain of furin. Implications for zymogen activation. Protein Sci. 10, 934–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Basak A., Lazure C. (2003) Synthetic peptides derived from the prosegments of proprotein convertase 1/3 and furin are potent inhibitors of both enzymes. Biochem. J. 373, 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jean F., Basak A., Rondeau N., Benjannet S., Hendy G. N., Seidah N. G., Chrétien M., Lazure C. (1993) Enzymic characterization of murine and human prohormone convertase-1 (mPC1 and hPC1) expressed in mammalian GH4C1 cells. Biochem. J. 292, 891–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dufour E. K., Désilets A., Longpré J. M., Leduc R. (2005) Stability of mutant serpin/furin complexes. Dependence on pH and regulation at the deacylation step. Protein Sci. 14, 303–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brooks C. L., 3rd. (1992) Characterization of “native” apomyoglobin by molecular dynamics simulation. J. Mol. Biol. 227, 375–380 [DOI] [PubMed] [Google Scholar]
- 40. Daggett V., Levitt M. (1992) A model of the molten globule state from molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 89, 5142–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daggett V., Levitt M. (1993) Protein unfolding pathways explored through molecular dynamics simulations. J. Mol. Biol. 232, 600–619 [DOI] [PubMed] [Google Scholar]
- 42. Huang W., Eichenberger A. P., van Gunsteren W. F. (2012) Molecular dynamics simulation of thionated hen egg white lysozyme. Protein Sci. 21, 1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karplus M., Kuriyan J. (2005) Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U.S.A. 102, 6679–6685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Salimi N. L., Ho B., Agard D. A. (2010) Unfolding simulations reveal the mechanism of extreme unfolding cooperativity in the kinetically stable α-lytic protease. PLoS Comput. Biol. 6, e1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tangrea M. A., Alexander P., Bryan P. N., Eisenstein E., Toedt J., Orban J. (2001) Stability and global fold of the mouse prohormone convertase 1 pro-domain. Biochemistry 40, 5488–5495 [DOI] [PubMed] [Google Scholar]
- 46. Basak A., Jean F., Seidah N. G., Lazure C. (1994) Design and synthesis of novel inhibitors of prohormone convertases. Int. J. Pept. Protein Res. 44, 253–261 [DOI] [PubMed] [Google Scholar]
- 47. Day R., Lazure C., Basak A., Boudreault A., Limperis P., Dong W., Lindberg I. (1998) Prodynorphin processing by proprotein convertase 2. Cleavage at single basic residues and enhanced processing in the presence of carboxypeptidase activity. J. Biol. Chem. 273, 829–836 [DOI] [PubMed] [Google Scholar]
- 48. Tanco S., Zhang X., Morano C., Avilés F. X., Lorenzo J., Fricker L. D. (2010) Characterization of the substrate specificity of human carboxypeptidase A4 and implications for a role in extracellular peptide processing. J. Biol. Chem. 285, 18385–18396 [DOI] [PMC free article] [PubMed] [Google Scholar]