

Background: The role of translational control mechanisms in gene expression during inflammation is incompletely understood.
Results: The proinflammatory cytokines IL-1 and IL-17 activate translation of certain mRNAs, including that of MCPIP1, a negative regulator of inflammation.
Conclusion: Translational activation of MCPIP1 contributes to changes in gene expression induced by IL-1 and IL-17.
Significance: Translational control may determine physiological and pathological consequences of inflammation.
Keywords: Cytokine, Inflammation, mRNA, MRNA Decay, Translation, IL-1, IL-17, MCPIP1
Abstract
Changes in gene expression during inflammation are in part caused by post-transcriptional mechanisms. A transcriptome-wide screen for changes in ribosome occupancy indicated that the inflammatory cytokine IL-17 activates translation of a group of mRNAs that overlaps partially with those affected similarly by IL-1. Included are mRNAs of IκBζ and of MCPIP1, important regulators of the quality and course of immune and inflammatory responses. Evidence for increased ribosome association of these mRNAs was also obtained in LPS-activated RAW264.7 macrophages and human peripheral blood mononuclear cells. Like IL-1, IL-17 activated translation of IκBζ mRNA by counteracting the function of a translational silencing element in its 3′-UTR defined previously. Translational silencing of MCPIP1 mRNA in unstimulated cells resulted from the combined suppressive activities of its 5′-UTR, which contains upstream open reading frames, and of its 3′-UTR, which silences independently of the 5′-UTR. Only the silencing function of the 3′-UTR was counteracted by IL-17 as well as by IL-1. Translational silencing by the 3′-UTR was dependent on a putative stem-loop-forming region previously associated with rapid degradation of the mRNA. The results suggest that translational control exerted by IL-1 and IL-17 plays an important role in the coordination of an inflammatory reaction.
Introduction
Post-transcriptional mechanisms play an important part in the changes of gene expression during an inflammatory response (1). A number of studies including our own have demonstrated that IL-1, a central mediator of inflammation, can induce stabilization of certain mRNAs, which encode proteins involved in inflammatory and immune reactions. More recently, we observed that IL-1 also activates translation of distinct mRNAs (2, 3). We now present evidence that translation of several of its target mRNAs is also activated by the proinflammatory cytokine IL-17.
IL-17 is a product of the Th17 subset of T-lymphocytes, but is also produced by other cell types. Through induction of various proteins, including cytokines, chemokines, and transcription factors, IL-17 plays an important part in host defense and autoimmunity (4–6) and also affects angiogenesis and tumor formation (7, 8). Marked synergy in induction of gene expression has been observed with other inflammatory stimuli, e.g. TNF (9, 10) or poly(I:C) (11). The IL-17 family contains several isoforms that homo- or heterodimerize and bind to members of a distinct family of IL-17 receptors. Signal transduction of IL-17 is incompletely understood (4). Although several pathways, including NF-κB and MAP kinase pathways, appear to contribute to transcriptional activation, it is well established that part of the effects of IL-17 on gene expression is caused by stabilization of mRNAs (12, 13). Stabilization of cyclooxygenase-2 mRNA was found to depend on p38 MAP kinase (14), whereas CXCL1 (KC) mRNA was stabilized independently of p38 MAP kinase and of AU-rich elements (AREs)3 (15, 16), through a mechanism that, according to most recent evidence, involves inducible IκB kinase, TRAF5, and SF2 (17, 18). In this study, we provide evidence that IL-17 can induce proteins by activating translation of distinct target mRNAs, including those of IκBζ and MCPIP1.
IκBζ, an atypical member of the IκB family of proteins, can modify the NF-κB response by activating expression of certain NF-κB-dependent genes while suppressing expression of others, probably due to differential interaction with NF-κB dimers, but may also regulate transcription independently. Deletion of its gene can cause proinflammatory and atopic dermatitis-like symptoms and results in impaired Th17 cell development (Ref. 19 and references therein). Induction of IκBζ by IL-1 involves post-transcriptional mechanisms (20–22), including translational activation of its mRNA (2).
MCPIP1, also named ZC3H12A or regnase-1, was identified as a protein induced by the chemokine MCP-1 (23). It can induce cell death and contributes to the pathophysiological role of MCP-1 in the development of ischemic heart disease (24). MCPIP1 has transcription factor-like activity and is involved in MCP-1-induced angiogenesis by increasing cadherin expression (25). A hallmark of MCPIP1, documented in several recent studies, is its suppression of inflammatory processes. MCPIP1 negatively affects inflammatory gene expression and NF-κB activation in response to LPS (26). Mice deficient in MCPIP1 are anemic and die within 12 weeks (27). Their macrophages show a highly increased production of IL-6 and IL-12 p40, an effect ascribed to impaired mRNA degradation. In that study, evidence was presented for RNase activity of MCPIP1, which contributed to rapid degradation of IL-6 mRNA (27), but also appears to functionally counteract DICER by cleaving the terminal loops of pre-microRNAs (28). In an independent study, MCPIP1-deficient mice developed a fatal inflammatory syndrome, which also supports an important role for MCPIP1 in limiting inflammatory and autoimmune processes (29). An inhibitory function of MCPIP1 on JNK and NF-κB activity through deubiquitination of TNF receptor-associated factor (TRAF) proteins was observed in that study.
MCPIP1 is induced upon macrophage activation with LPS (26) and in different types of cells in response to stress (30) and to IL-1 (10, 31, 32). In primary hepatocytes, MCPIP1 mRNA is induced synergistically by TNF and IL-17 (10). Transcriptional induction by IL-1β has been demonstrated to involve the NF-κB and ERK pathways and Elk-1 transcription factor (33, 34). Most recently, post-transcriptional mechanisms controlling MCPIP1 have been reported that act through the IκB kinase complex, which controls ubiquitination and degradation of MCPIP1 protein and through MCPIP1 itself, which controls degradation of its own mRNA (35).
The results presented here demonstrate that MCPIP1 expression is also limited by inhibitory effects of its 5′- and 3′-UTRs on translation. Furthermore, the inhibitory effect of the 3′-UTR is overcome by stimulation of the cells with IL-17, as well as with IL-1. Thus translational mechanisms activated by these cytokines increase the expression of factors such as MCPIP1 and IκBζ, which determine the extent and specificity of protein expression in inflammatory reactions.
EXPERIMENTAL PROCEDURES
Cells and Materials
HeLa cells constitutively expressing the tetracycline-controlled transactivator protein (36) were cultured and transfected with plasmids (see below) by the calcium phosphate method as described (37). The murine macrophage line RAW 264.7 was cultured in DMEM supplemented with 5% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Human synovial tissue samples were obtained from synovial biopsy specimens from rheumatoid arthritis and osteoarthritis patients who were undergoing joint surgery. All specimens were obtained with the approval of the Ethics Committee of the Justus-Liebig-University of Giessen. All patients gave informed consent and fulfilled the criteria of the American College of Rheumatology (38, 39). Following enzymatic digestion, primary synovial fibroblasts were isolated and cultured in supplemented DMEM as described previously (40). Human mononuclear cells were isolated from peripheral blood using Vacutainer Cell Preparation Tubes (BD Biosciences), washed, and resuspended in DMEM + 5% fetal calf serum. Recombinant human IL-1α was obtained from Promocell, recombinant human IL-17A was from R&D Systems, recombinant human TNFα was a kind gift of Guenther Adolf (Boehringer Institute Vienna), and Escherichia coli-derived LPS was from Sigma-Aldrich.
Plasmids
To express GFP-MCPIP1, the human MCPIP1 coding sequence and 3′-UTR of imaGene clone IRAUp969C1019D were inserted into the EcoRI and XbaI sites of plasmid pUHD10-3 (kindly provided by Hermann Bujard) and the PCR-amplified GFP coding sequence introduced in-frame upstream of the MCPIP1 sequence. The 3′-UTR was deleted by creation of an XbaI site downstream to the stop codon and subsequent XbaI digestion and religation. The 5′-UTR (nucleotides 2–150) and 3′-UTR of MCPIP1 (nucleotides 1952–2709, numbering according to accession number NM_025079) were amplified by RT-PCR from total RNA of HeLa cells. To express firefly luciferase mRNA with either or both UTRs, the 5′-UTR was cloned into the BamHI site upstream of the luciferase coding sequence, and the 3′-UTR was cloned into a newly created BglII site downstream of the coding sequence in pUHC13-3 (36). The 3′-UTR was also inserted into the BglII site 3′ of the β-globin coding region in ptetBBB (41). Plasmids expressing firefly luciferase mRNA with the IκBζ 3′-UTR or the IκBζ translational silencing element as well as an expression plasmid for Renilla luciferase have been described (2). Mutations of upstream start codons (ATG to ATA) in the MCPIP1 5′-UTR and deletion of a putative stem-loop-forming sequence (nucleotides 2176–2198) in the MCPIP1 3′-UTR of luciferase reporter plasmids were carried out following the QuikChangeTM site-directed mutagenesis procedure (Stratagene).
Sucrose Gradient Fractionation and RNA Isolation
Cytoplasmic lysates were prepared and centrifuged through linear sucrose gradients (10–50% sucrose), fractions were collected, and RNA was isolated as described (2). Results shown were confirmed in at least two independent experiments.
mRNA Detection
Reverse transcription and quantitative PCR (RT-qPCR) were carried out as described (2), using TaqMan assays (Applied Biosystems, assay IDs Hs00962356_m1 and Mm01182027_m1 for human and murine MCPIP1 mRNA, respectively, Hs00230071_m1 and Mm00600522_m1 for human and murine IκBζ mRNA, respectively, Hs00174131_m1 for IL-6 mRNA, and Hs99999905_m1 for GAPDH mRNA, Hs00178297_m1 for MAP3K8 mRNA, and custom-made assays for rabbit β-globin and firefly luciferase mRNAs). mRNA of the GFP-MCPIP1 fusion protein was quantified by SYBR Green-based detection (GFP-specific primers: sense, 5′-tgcagtgcttcagccgctac; antisense, 5′-tcgccctcgaacttcacctc). Values with standard deviations > 0.3 cycles were excluded from the analysis. For determination of degradation kinetics, transcription was stopped with actinomycin D or with doxycycline for Tet-Off plasmid-derived chimeric β-globin mRNAs followed by sequential RNA isolation and quantification of specific mRNAs by RT-qPCR.
Luciferase Reporter Assays
HeLa cells were co-transfected with firefly luciferase reporter plasmids and an expression plasmid for Renilla luciferase. For determining the effects of IL-1 and IL-17, the cells were stimulated on the following day with these cytokines. After lysis of the cells, firefly and Renilla luciferase activities were determined as described (2). For calculation of luciferase protein/mRNA ratios, luciferase activity (in relative light units) was divided by RNA units, calculated as 2−ΔCT × 103, based on levels of GAPDH mRNA determined as housekeeping mRNA.
Microarray Analysis
RNA of pooled sucrose gradient fractions was subjected to quality control and analyzed using whole human genome oligonucleotide microarrays (G4112F, ID 014850, Agilent Technologies) in essence as described (37). Data were filtered according to a stringent multistep approach that accounted for 1) the quality of the measurements (hybridization performance), 2) the consistency among replicate assays, 3) the intensity range, and 4) -fold change values. A detailed protocol is available upon request.
SDS-PAGE and Western Blot
Total cell lysates were separated by SDS-PAGE, and proteins were blotted onto PVDF membranes. After blocking with 5% dried milk in Tris-buffered saline-0.05% Tween 20, the blots were incubated with a mixture of two murine monoclonal antibodies against GFP (Roche Applied Science) or with monoclonal antibodies against α-tubulin (Sigma-Aldrich) followed by peroxidase-coupled secondary antibodies. Blots were developed with an ECL detection kit (ImmobilonTM, Millipore), and chemiluminescence was detected by the LAS 3000 imaging system (Fujifilm).
RESULTS
Increased Ribosome Association of Distinct mRNAs in Response to IL-17
We previously reported that translational activation contributes to induction of inflammatory proteins by IL-1 (2, 3). In view of the importance of IL-17 in various inflammatory conditions, we extended our investigation and screened for activation of mRNA translation in response to IL-17. Lysates from unstimulated and IL-17-stimulated HeLa cells were centrifuged on sucrose gradients to separate mRNAs according to their ribosome occupancy. Fractions containing ribosome-free (untranslated) mRNAs and fractions containing polysome-associated (translated) mRNAs were pooled (Fig. 1A), and the amounts of individual mRNAs in each pool were determined by high density microarrays. Several mRNAs showed a marked redistribution to polysomes in response to IL-17 (Table 1). Comparison with previous results revealed that the mRNAs listed overlap partially with those shifted to polysomes in response to IL-1 (2). In particular, the mRNAs of IκBζ (gene symbol: NFKBIZ) and MCPIP1 (gene symbol: ZC3H12A), targets of IL-1-mediated control, are those affected most strongly by IL-17 as well. On the other hand, IL-6 mRNA was not identified as a target of IL-17 under these conditions, whereas its translation is regulated by IL-1α (3). These results suggest that IL-17 induces expression of proteins in part by activating translation of the respective mRNAs and shares this activity with IL-1.
FIGURE 1.
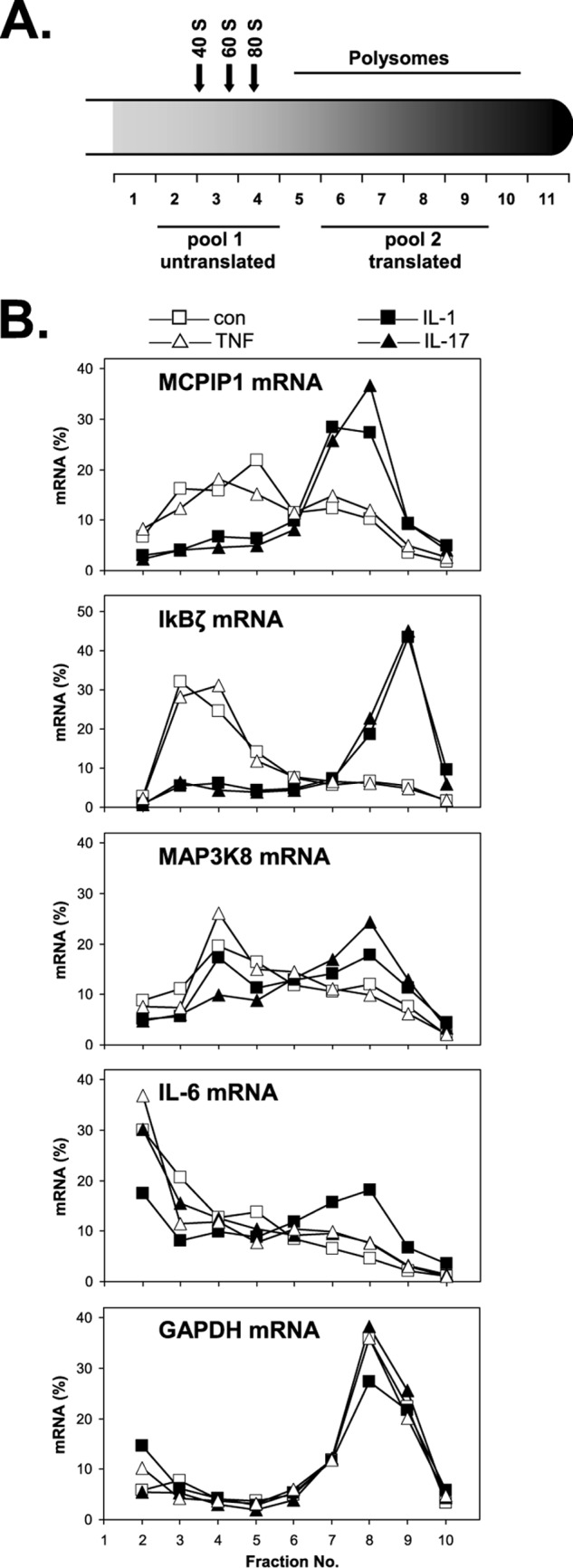
Redistribution of mRNAs to polysome fractions in response to different cytokines. After exposure of HeLa cells to IL-1α (2 ng/ml), IL-17 (25 ng/ml), or TNFα (50 ng/ml) or incubation in medium alone for 1 h, cytoplasmic lysates were prepared and fractionated on sucrose gradients (for details, see “Experimental Procedures”). A, scheme of gradient indicating the positions of ribosomal subunits, ribosomes, and polysomes according to the absorbance profile at 260 nm. Pools of untranslated and translated mRNAs were prepared from the indicated fractions of unstimulated and IL-17-stimulated cells and subjected to microarray analysis for the data presented in Table 1. B, polysome profiles of the indicated mRNAs were obtained by RT-qPCR quantification of RNA from individual fractions. con, left untreated.
TABLE 1.
mRNAs with increased ribosome association in response to IL-17
Listed are genes of mRNAs with at least 3-fold increase in polysome association (mean of two experiments) in HeLa cells stimulated with IL-17 (25 ng/ml) for 1 h as compared with unstimulated cells. Polysome association of mRNAs was calculated as the ratio of signals obtained in microarray analysis of total RNA of pooled fractions 6–9 (“translated”) over fractions 2–4 (“untranslated”) after gradient centrifugation of cytoplasmic extracts (Fig. 1A).
| Gene symbol | Description | -Fold increase |
|---|---|---|
| NFKBIZa | Homo sapiens nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, ζ, transcript variant 1, mRNA (NM_031419) | 14.06 |
| ZC3H12Aa | H. sapiens zinc finger CCCH-type containing 12A, mRNA (NM_025079) | 9.36 |
| NFKBIDa | H. sapiens T-cell activation NFκB-like protein, mRNA (NM_139239) | 4.15 |
| MAP3K8 | H. sapiens mitogen-activated protein kinase kinase kinase 8, mRNA (NM_005204) | 3.35 |
| VAT1 | H. sapiens vesicle amine transport protein 1 homolog (T. californica), mRNA (NM_006373) | 3.34 |
| FAM78A | H. sapiens family with sequence similarity 78, member A, mRNA (NM_033387) | 3.22 |
| SIPA1L3 | H. sapiens signal-induced proliferation-associated 1 like 3, mRNA (NM_015073) | 3.19 |
| APLP2 | H. sapiens amyloidβ (A4) precursor-like protein 2, mRNA (NM_001642) | 3.08 |
| ANKRD34 | H. sapiens ankyrin repeat domain 34, mRNA (NM_001039888) | 3.04 |
a Polysome association was also increased by IL-1 (2).
Comparison of Target mRNAs Redistributed to Polysomes in Response to IL-17 and IL-1
To confirm the results obtained by microarray analysis of pooled polysomal and subpolysomal fractions and to further compare target selectivity of different cytokines, mRNAs were quantified by RT-qPCR in individual fractions of the gradients (Fig. 1B). TNFα was included in this comparison and turned out not to affect distribution of the mRNAs investigated here (Fig. 1B and data not shown), whereas it induced a much stronger increase in total IL-6 mRNA as compared with IL-17, thus confirming general responsiveness of the cells to TNFα (not shown). IL-1α and IL-17 did not alter distribution of GAPDH mRNA assayed as a housekeeping control. Both cytokines induced marked redistribution of MCPIP1 and IκBζ mRNAs to polysomes. In the case of IκBζ mRNA, a previously identified translational silencing element (2) was sufficient to confer regulation by both cytokines to a luciferase reporter mRNA (supplemental Fig. S1). IL-1α only slightly affected MAP3K8 mRNA, whereas redistribution to polysomes was stronger in response to IL-17, confirming its detection as a target of IL-17 in the screening assay (Table 1). More importantly, IL-6 mRNA was redistributed to polysomes only in response to IL-1α (Fig. 1B), confirming that the target mRNAs for translational control exerted by the two cytokines overlap only partially.
We recently reported that IL-1 can activate translation of IL-6 and IL-1α mRNAs, which both contain AREs, by counteracting a translational silencing effect of the ARE-binding protein KSRP (3). However, silencing of MCPIP1 and IκBζ mRNAs in unstimulated cells appeared independent of KSRP because its knockdown did not affect the ribosome association of these mRNAs, which are devoid of AREs (supplemental Fig. S2, A and B). These results argue for the existence of an additional, ARE- and KSRP-independent mechanism of silencing, which can be relieved in response to both IL-1 and IL-17 (supplemental Fig. S2C).
Increased Ribosome Association of mRNAs in Cells of Fibroblast and Macrophage Origin
Evidence for this type of translational regulation was also obtained for other cells and inducers. Synovial fibroblasts from patients with osteoarthritis or rheumatoid arthritis show an increase in ribosome occupancy of MCPIP1 mRNA in response to IL-1α (Fig. 2A), demonstrating that this effect of IL-1 occurs in pathophysiologically relevant target cells. Furthermore, cells of the murine macrophage line RAW.264.7 respond to LPS stimulation with redistribution of MCPIP1 and IκBζ mRNAs to polysome fractions (Fig. 2B). Redistribution of MCPIP1 mRNA in response to LPS is also observed in mononuclear cells isolated from human peripheral blood (Fig. 2B). This also suggests enhanced translation of MCPIP1 and IκBζ mRNAs in response to TLR4 activation.
FIGURE 2.
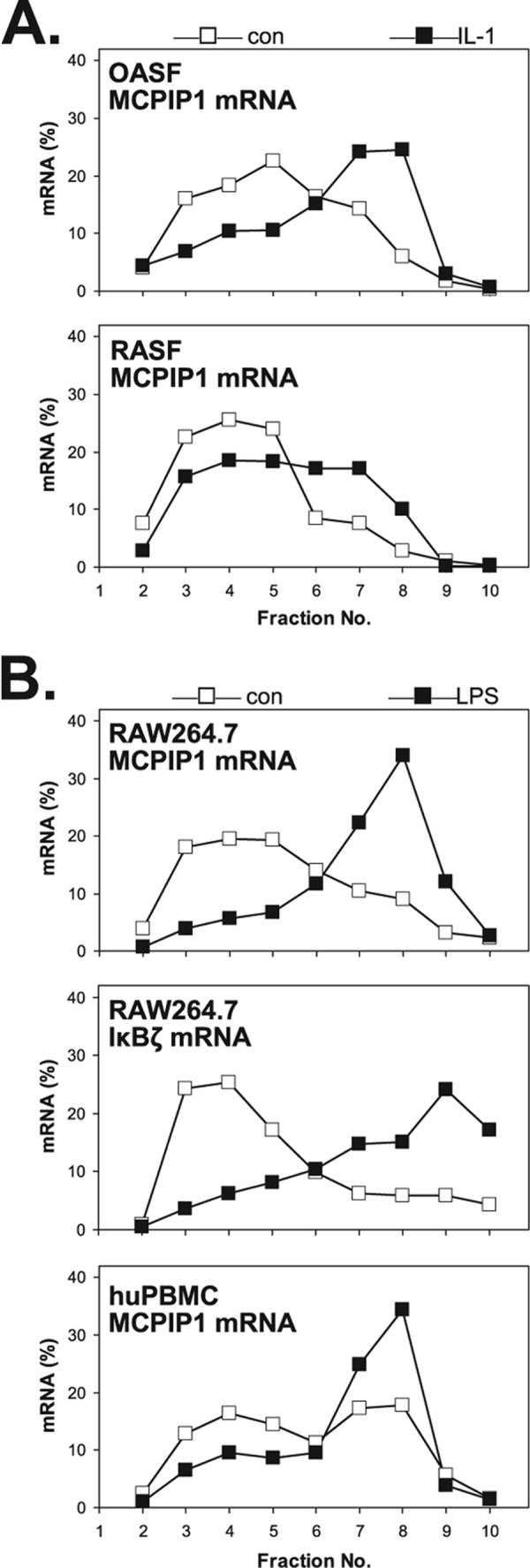
Redistribution of MCPIP1 mRNA to polysomes in synovial fibroblasts and macrophages. A, synovial fibroblasts from a patient with osteoarthritis (OASF) and from a patient with rheumatoid arthritis (RASF) were incubated with IL-1α (2 ng/ml) for 1 h or left untreated (con). B, cells of the murine macrophage line RAW264.7 and human peripheral blood mononuclear cells (huPBMC) were incubated with LPS (1 μg/ml) or left untreated (con). Polysome profiles of MCPIP1 and IκBζ mRNAs were obtained as in Fig. 1B.
3′-UTR-dependent Control of MCPIP1 mRNA Degradation by IL-17
Because MCPIP1 has important roles in shaping and limiting the response to inflammatory stimuli, we sought to elucidate the mechanism involved in post-transcriptional control of its expression in further detail. MCPIP1 expression has been reported to be controlled at the level of mRNA stability (35). As shown in Fig. 3A, IL-17 induced marked stabilization of MCPIP1 mRNA and shared this activity with IL-1α. As for translational activation (Fig. 1B), TNFα was also inactive in this respect (Fig. 3A). IL-6 mRNA was stabilized by IL-1α as expected (42), but marginally affected by IL-17 and TNF. Thus the selectivity for MCPIP1 mRNA of IL-17-induced stabilization corresponds to that observed for IL-17-induced redistribution to polysomes (Fig. 1B) and supports a distinct pathway of post-transcriptional control shared by IL-1 and IL-17 (supplemental Fig. S2C). A reporter mRNA that contains the MCPIP1 3′-UTR inserted after the stop codon of β-globin mRNA (Fig. 3B, scheme) was degraded in untreated cells with similar kinetics as endogenous MCPIP1 mRNA (Fig. 3B). Treatment with IL-1α or IL-17 induced transient stabilization of the chimeric mRNA. Thus both cytokines control MCPIP1 expression on the level of mRNA stability by a mechanism depending on 3′-UTR sequences.
FIGURE 3.
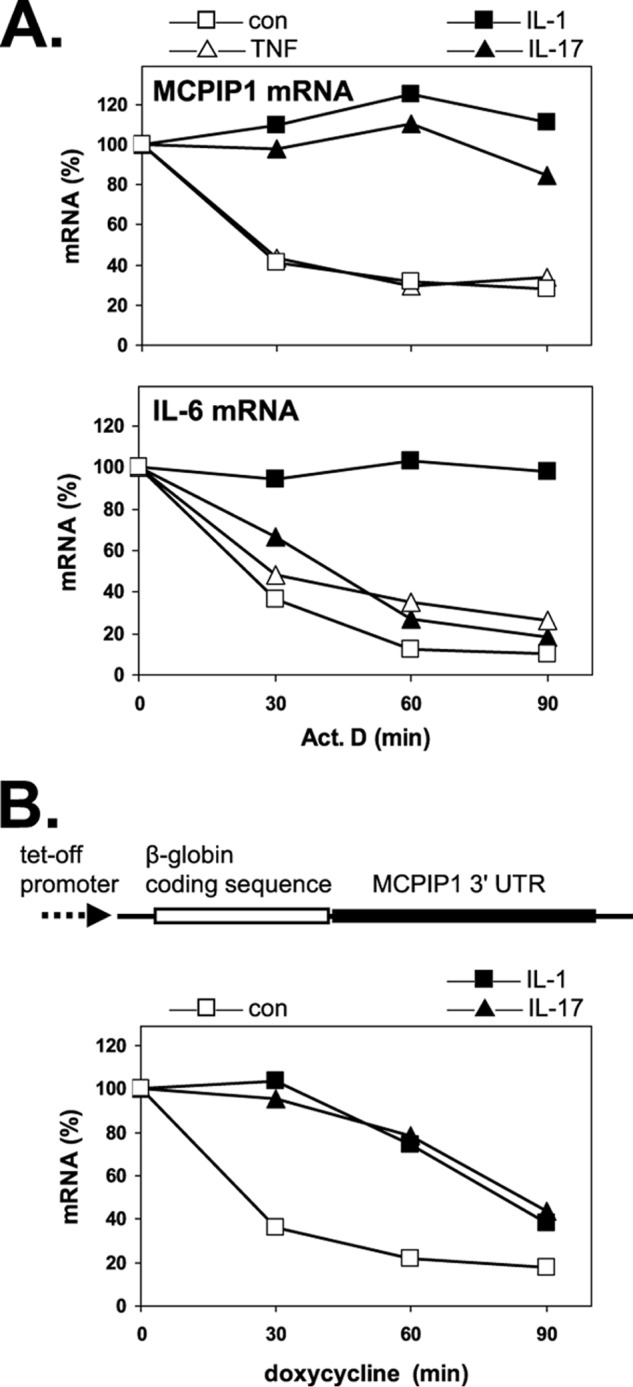
Effect of different cytokines on the degradation of MCPIP1 and IL-6 mRNAs. A, HeLa cells were incubated without (con) or with the indicated cytokines for 15 min followed by the addition of actinomycin D (5 μg/ml). Total RNA was isolated at the indicated times thereafter, and mRNAs were quantified by RT-qPCR. B, HeLa cells were transfected with a Tet-Off plasmid expressing chimeric β-globin mRNA containing the 3′-UTR of MCPIP1 (see scheme). After incubation without (con) or with cytokines, transcription from the plasmid was stopped by adding doxycycline (3 μg/ml), and degradation of the mRNA was determined as in A, using a primer set specific for β-globin cDNA.
Suppression of Translation by 5′- and 3′-UTR Sequences of MCPIP1 mRNA
Unlike the control of MCPIP1 mRNA stability, translational control of MCPIP1 mRNA to our knowledge has not been investigated so far. Initial experiments to localize the mRNA region responsible for translational control were performed with reporter constructs that express firefly luciferase mRNA under the control of a constitutive promoter. They contain the 5′-UTR or the 3′-UTR or both UTRs placed into the respective positions (Fig. 4A). Firefly luciferase activity, normalized to luciferase mRNA, was markedly suppressed in cells transfected with the construct containing both UTRs, as compared with the construct without MCPIP1 mRNA sequences (Fig. 4B). Normalization to Renilla luciferase activity, which does not consider changes in mRNA amounts, showed about 16-fold suppression (not shown). Thus suppression reflects decreased mRNA amounts, resulting from increased degradation, as well as translational silencing. Suppression was counteracted by stimulating the cells with IL-1α as well as with IL-17 (Fig. 4C). Increased luciferase activity, normalized to its mRNA, supports translational activation by these cytokines and indicates that it is dependent on 5′- and/or 3′-UTR sequences.
FIGURE 4.

Suppression of luciferase expression by the 5′-UTR of MCPIP1 mRNA. A, scheme of luciferase (Luc)-MCPIP1 reporter mRNAs investigated in this study. They contain insertions of the 5′-UTR and 3′-UTR of MCPIP1, the 5′-UTR alone, or the 3′-UTR alone. The positions of a putative stem-loop-forming sequence in the 3′-UTR (35) and its deletion are indicated. B, cells were transfected with the firefly luciferase expression plasmid containing the 5′-UTR and 3′-UTR of MCPIP1 (M5′-Luc-M3′). Firefly luciferase activity normalized to its own mRNA as quantified by RT-qPCR is expressed as -fold suppression as compared with values for the firefly luciferase construct without insertions (set as 1) (mean and S.D. of four independent experiments). C, cells transfected with the M5′-Luc-M3′ plasmid were left untreated (con) or stimulated with IL-1α (2 ng/ml) or IL-17 (25 ng/ml) for the indicated times. Shown is the -fold change in luciferase activity (normalized to its mRNA) of stimulated over untreated cultures (mean and S.D. for one of two experiments with similar results). D, luciferase activity per mRNA of cells expressing firefly luciferase mRNA with wild-type or mutated MCPIP1 5′-UTR (see scheme) is expressed as -fold change as compared with luciferase mRNA without insertions (set as 1) (mean and S.D. of four independent experiments).
Upstream Open Reading Frame-dependent Translational Silencing by the MCPIP1 5′-UTR
The 5′-UTR of MCPIP1 mRNA contains two AUG codons upstream of the start codon of its main open reading frame (Fig. 4D, scheme), marking two upstream ORFs, one ending 5′ of the main ORF, the second extending into it with a different frame. Upstream ORFs are known as elements limiting translation (43). Indeed, luciferase activity in cells transfected with the construct containing only the 5′-UTR of MCPIP1 mRNA was strongly reduced as compared with the control plasmid without MCPIP1 mRNA sequences (Fig. 4D). Mutation of both upstream AUGs or of the first AUG fully restored luciferase activity (Fig. 4D). This indicates that MCPIP1 mRNA translation is limited by the presence of upstream ORFs.
Translational Silencing Mediated by the MCPIP1 3′-UTR
Further experiments showed that in addition to the 5′-UTR, other regions in MCPIP1 mRNA silence its expression. In polysome profiling of HeLa cells transfected with a construct that lacks the 5′-UTR but contains the GFP coding sequence followed by MCPIP1 coding sequences and 3′-UTR (GFP-Mcds-M3′), the mRNA localized preferentially to subpolysomal fractions (Fig. 5A), thus mimicking the behavior of endogenous MCPIP1 mRNA. However, after also deleting the 3′-UTR, the resulting GFP-MCPIP1 mRNA (GFP-Mcds) was shifted to polysome fractions. Similar redistribution was also observed with a construct expressing MCPIP1 coding sequence and 3′-UTR without GFP sequences (not shown). Thus the 3′-UTR appears to inhibit translation independently of the 5′-UTR, whereas the coding region alone allows normal ribosome loading. Corresponding to these results, the amount of protein expressed from the GFP-Mcds-M3′ plasmid was strongly suppressed as compared with the GFP-Mcds plasmid lacking the 3′-UTR (Fig. 5B). This indicates that the 3′-UTR can silence translation of MCPIP1 mRNA independently of the 5′-UTR.
FIGURE 5.

Suppression of GFP-MCPIP1 expression by the 3′-UTR of MCPIP1 mRNA. A, HeLa cells were transfected with plasmids containing sequences for a GFP-MCPIP1 fusion protein followed by the MCPIP1 3′-UTR (GFP-Mcds-M3′) or not (GFP-Mcds). Polysome profiles were obtained as described in Fig. 1B using RT-qPCR specific for GFP and GAPDH mRNAs. Similar results were obtained in three independent assays. B, amounts of GFP-MCPIP1 fusion protein were compared by Western blot with antibodies to GFP. Detection of α-tubulin is shown as a control.
Suppression Exerted by the MCPIP1 3′-UTR Is Counteracted by IL-1 and IL-17 and Dependent on a Putative Stem-Loop-forming Sequence
Luciferase reporter constructs containing the 5′-UTR or the 3′-UTR or both UTRs of MCPIP1 mRNA were employed to find out which of the sequences exerted suppression that was overcome by stimulation of the cells (Fig. 6A). Luciferase activity was suppressed by the 5′-UTR, as shown above in Fig. 4D, and independently by the 3′-UTR, confirming the results obtained with GFP-MCPIP1 mRNAs (Fig. 5). Maximal suppression was observed by the combined presence of both UTRs (M5′-Luc-M3′). Most importantly, treatment of the cells with IL-1α partially relieved suppression exerted by the 3′-UTR or by both UTRs in combination, whereas suppression by the 5′-UTR was not affected by IL-1α (Fig. 6A). Expression of the MCPIP1 3′-UTR-containing luciferase reporter mRNA (Luc-M3′) was also activated in response to IL-17 (Fig. 6B). These results suggest that IL-1 and IL-17 induce expression of MCPIP1 in part by activating translation of its mRNA through mechanisms antagonizing a silencing function of the 3′-UTR.
FIGURE 6.

MCPIP1 3′-UTR-mediated suppression is counteracted by IL-1 and IL-17 and dependent on a putative stem-loop-forming sequence. A, cells transfected with plasmids for firefly luciferase (Luc) without or with the MCPIP1 UTRs as indicated (see scheme in Fig. 4A) were left untreated (con) or stimulated with IL-1α for 4 h (IL-1). Firefly luciferase activity was normalized to co-expressed Renilla activity and is expressed as -fold suppression as compared with activity derived from a plasmid without MCPIP1 sequences in unstimulated cells (set as 1) (mean and S.D. of three independent experiments; n.s., not significant (p > 0.1, Student's t test). rel. luciferase activity, relative luciferase activity. B, cells were transfected with plasmid for firefly luciferase containing the MCPIP1 3′-UTR and left untreated or stimulated with IL-1α or IL-17 for 4 h as indicated. Firefly luciferase activity was normalized to Renilla activity and is presented as -fold increase in the cytokine-stimulated cultures as compared with unstimulated cultures (set as 1) (mean and S.D. of triplicate determinations). C, luciferase activity and mRNA were quantified in cells transfected with luciferase vectors without (Luc) or with the intact 3′-UTR (Luc-M3′) or a deletion mutant lacking the stem-loop-forming sequence (Luc-M3′dsl) as well as with the corresponding vectors containing in addition the MCPIP1 5′-UTR (M5′-Luc-M3′ and M5′-Luc-M3′dsl). Results are expressed as -fold suppression of the luciferase activity/mRNA ratio as compared with the vector without MCPIP1 sequences (set as 1) (mean and S.D. of triplicate determinations).
Recently, a putative stem-loop-forming sequence in the murine MCPIP1 3′-UTR has been shown to limit expression of luciferase reporters in an MCPIP1-dependent manner, presenting an autoregulatory feedback mechanism (35), which was ascribed to accelerated degradation of the mRNA. Deletion of the corresponding sequence in the luciferase reporter constructs containing the MCPIP1 3′-UTR alone or combined with the 5′-UTR caused a marked decrease in translational silencing (Fig. 6C), indicating that this sequence is also involved in the regulation of MCPIP1 mRNA translation.
DISCUSSION
An inherent property of inflammatory stimuli is to activate mechanisms that counteract their activity, serving to limit and finally resolve the inflammatory reaction. The recently identified negative feedback regulator MCPIP1 is expressed in response to various inducers of inflammation. Here we provide evidence that induction of MCPIP1 by IL-1 and IL-17 involves activated translation of its mRNA. MCPIP1 expression is limited by suppressive effects of the 5′- and 3′-UTRs on translation. Unlike the EGR2 mRNA, which contains an internal ribosomal entry site element in its 5′-UTR that is activated by IL-1 (44), the MCPIP1 5′-UTR suppressed translation in a manner not affected by treatment with IL-1 (Fig. 6A). This function may serve to prevent formation of excess MCPIP1, which has been shown to cause cell death (23), and thus to balance MCPIP1 levels during inflammatory responses. Such conditions can diminish suppression mediated by the 3′-UTR of MCPIP1, as shown here for stimulation with IL-1 or IL-17.
The signaling induced by IL-1 and IL-17 originates from different types of receptors and associated molecules. The similarities between the response to IL-17 and IL-1, which both stabilize and activate translation of MCPIP1 and IκBζ mRNAs, suggest that they share some common downstream mechanism. It remains to be found out at which point the pathways converge to effect post-transcriptional control. The mechanisms controlling MCPIP1 and IκBζ mRNAs appear distinct from a second type of control, which is activated only by IL-1 and targets IL-6 mRNA. This is suggested by the following observations. 1) Neither IκBζ nor MCPIP1 mRNA contain typical AREs in their 3′-UTRs, which distinguishes them from IL-6 mRNA, whose translation and stability are regulated by IL-1 but not by IL-17 (Figs. 1B and 3A) (3). 2) IL-6 mRNA but not MCPIP1 mRNA is among the top 10 mRNAs identified as targets for destabilization by the ARE-binding protein KSRP (37). Translation of IL-6 mRNA is controlled by KSRP as well; its siRNA-mediated depletion increased translation of IL-6 mRNA (3), but had no significant effect on the ribosome association of MCPIP1 mRNA (supplemental Fig. S2B). 3) Correspondingly, the group of mRNAs redistributed to polysomes upon depletion of KSRP (3) did not overlap with those redistributed to polysomes in response to IL-17 (Table 1).
We speculate that KSRP function is targeted by IL-1 but not by IL-17. Our observations complement earlier studies showing that instability of CXCL1 mRNA, which responds to IL-17-induced stabilization, was independent of the presence of an ARE and of KSRP (15, 16). In the latter study, evidence was also presented against a role for the ARE-binding protein tristetraprolin, which is known for its destabilizing function but also involved in translational silencing of TNF mRNA (45). To what extent stabilization and redistribution to polysomes are mechanistically linked remains unclear at present.
It is also not clear whether MCPIP1 and IκBζ mRNAs are regulated by the same mechanism. Both mRNAs harbor regulatory elements in their 3′-UTRs. The previously defined translational silencing element of IκBζ mRNA contains putative stem-loop structures essential for its function (2). A stem-loop-forming sequence in the MCPIP1 3′-UTR has been reported to mediate control of its stability (35). As shown in Fig. 6C, deletion of that sequence also impaired translational silencing. Because MCPIP1 itself has been implied in the degradation of its own mRNA (35), it will be important to find out whether MCPIP1 also controls translation of its own mRNA and whether it targets IκBζ mRNA as well. Of note, we detected no obvious sequence homology between the MCPIP1 3′-UTR and the IκBζ translational silencing element or a region in CXCL1 mRNA essential for IL-17 regulation (16).
Our results so far suggest that at least two different mechanisms of post-transcriptional control are activated by IL-1, ARE- and KSRP-dependent and -independent, and that the latter is activated by IL-17 as well (supplemental Fig. S2C). This latter mechanism is likely to contribute to negative feedback regulation of inflammatory stimuli by increasing expression of MCPIP1. By inducing MCPIP1, IL-17 may limit the extent and/or duration of its own effects, but may also limit the response to other agents. MCPIP1 has a protective role in ischemic stroke, and its induction contributes to the tolerance to ischemic stroke induced by preconditioning with LPS (46). In view of our results, it appears likely that translational activation of MCPIP1 plays an important role in such conditions.
Supplementary Material
Acknowledgments
We thank Monika Barsch and Heike Schneider for skillful technical assistance.
This work was supported by Grants SFB 566/A10, SFB 566/Z2, and Ho 1116/5 from the Deutsche Forschungsgemeinschaft and by a grant from the Deutsche Krebshilfe.

This article contains supplemental Figs. S1 and S2.
- ARE
- AU-rich element
- RT-qPCR
- reverse transcription and quantitative PCR
- KSRP
- K homology-type splicing regulatory protein.
REFERENCES
- 1. Anderson P. (2010) Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 10, 24–35 [DOI] [PubMed] [Google Scholar]
- 2. Dhamija S., Doerrie A., Winzen R., Dittrich-Breiholz O., Taghipour A., Kuehne N., Kracht M., Holtmann H. (2010) IL-1-induced post-transcriptional mechanisms target overlapping translational silencing and destabilizing elements in IκBζ mRNA. J. Biol. Chem. 285, 29165–29178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dhamija S., Kuehne N., Winzen R., Doerrie A., Dittrich-Breiholz O., Thakur B. K., Kracht M., Holtmann H. (2011) Interleukin-1 activates synthesis of interleukin-6 by interfering with a KSRP-dependent translational silencing mechanism. J. Biol. Chem. 286, 33279–33288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaffen S. L. (2011) Recent advances in the IL-17 cytokine family. Curr. Opin. Immunol. 23, 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milner J. D. (2011) IL-17 producing cells in host defense and atopy. Curr. Opin. Immunol. 23, 784–788 [DOI] [PubMed] [Google Scholar]
- 6. Zhu S., Qian Y. (2012) IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin. Sci. (Lond.) 122, 487–511 [DOI] [PubMed] [Google Scholar]
- 7. Maniati E., Soper R., Hagemann T. (2010) Up for mischief? IL-17/Th17 in the tumour microenvironment. Oncogene 29, 5653–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murugaiyan G., Saha B. (2009) Protumor vs antitumor functions of IL-17. J. Immunol. 183, 4169–4175 [DOI] [PubMed] [Google Scholar]
- 9. Katz Y., Nadiv O., Beer Y. (2001) Interleukin-17 enhances tumor necrosis factor α-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheum. 44, 2176–2184 [DOI] [PubMed] [Google Scholar]
- 10. Sparna T., Rétey J., Schmich K., Albrecht U., Naumann K., Gretz N., Fischer H. P., Bode J. G., Merfort I. (2010) Genome-wide comparison between IL-17 and combined TNF-α/IL-17 induced genes in primary murine hepatocytes. BMC Genomics 11, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryzhakov G., Lai C. C., Blazek K., To K. W., Hussell T., Udalova I. (2011) IL-17 boosts proinflammatory outcome of antiviral response in human cells. J. Immunol. 187, 5357–5362 [DOI] [PubMed] [Google Scholar]
- 12. Cai X. Y., Gommoll C. P., Jr., Justice L., Narula S. K., Fine J. S. (1998) Regulation of granulocyte colony-stimulating factor gene expression by interleukin-17. Immunol. Lett. 62, 51–58 [DOI] [PubMed] [Google Scholar]
- 13. Hartupee J., Liu C., Novotny M., Li X., Hamilton T. (2007) IL-17 enhances chemokine gene expression through mRNA stabilization. J. Immunol. 179, 4135–4141 [DOI] [PubMed] [Google Scholar]
- 14. Faour W. H., Mancini A., He Q. W., Di Battista J. A. (2003) T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 3′-untranslated region of COX-2 mRNA. J. Biol. Chem. 278, 26897–26907 [DOI] [PubMed] [Google Scholar]
- 15. Hartupee J., Liu C., Novotny M., Sun D., Li X., Hamilton T. A. (2009) IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J. Immunol. 182, 1660–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Datta S., Novotny M., Pavicic P. G., Jr., Zhao C., Herjan T., Hartupee J., Hamilton T. (2010) IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J. Immunol. 184, 1484–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun D., Novotny M., Bulek K., Liu C., Li X., Hamilton T. (2011) Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 12, 853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bulek K., Liu C., Swaidani S., Wang L., Page R. C., Gulen M. F., Herjan T., Abbadi A., Qian W., Sun D., Lauer M., Hascall V., Misra S., Chance M. R., Aronica M., Hamilton T., Li X. (2011) The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat. Immunol. 12, 844–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayden M. S., Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamazaki S., Muta T., Matsuo S., Takeshige K. (2005) Stimulus-specific induction of a novel nuclear factor-κB regulator, IκB-ζ, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J. Biol. Chem. 280, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 21. Watanabe S., Takeshige K., Muta T. (2007) A cis-element in the 3′-untranslated region of IκB-ζ mRNA governs its stimulus-specific expression. Biochem. Biophys. Res. Commun. 356, 785–791 [DOI] [PubMed] [Google Scholar]
- 22. Ohba T., Ariga Y., Maruyama T., Truong N. K., Inoue J., Muta T. (2012) Identification of interleukin-1 receptor-associated kinase 1 as a critical component that induces post-transcriptional activation of IκB-ζ. FEBS J. 279, 211–222 [DOI] [PubMed] [Google Scholar]
- 23. Zhou L., Azfer A., Niu J., Graham S., Choudhury M., Adamski F. M., Younce C., Binkley P. F., Kolattukudy P. E. (2006) Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 98, 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu J., Kolattukudy P. E. (2009) Role of MCP-1 in cardiovascular disease: molecular mechanisms and clinical implications. Clin. Sci. (Lond.) 117, 95–109 [DOI] [PubMed] [Google Scholar]
- 25. Niu J., Azfer A., Zhelyabovska O., Fatma S., Kolattukudy P. E. (2008) Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 283, 14542–14551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang J., Wang J., Azfer A., Song W., Tromp G., Kolattukudy P. E., Fu M. (2008) A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J. Biol. Chem. 283, 6337–6346 [DOI] [PubMed] [Google Scholar]
- 27. Matsushita K., Takeuchi O., Standley D. M., Kumagai Y., Kawagoe T., Miyake T., Satoh T., Kato H., Tsujimura T., Nakamura H., Akira S. (2009) Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki H. I., Arase M., Matsuyama H., Choi Y. L., Ueno T., Mano H., Sugimoto K., Miyazono K. (2011) MCPIP1 ribonuclease antagonizes Dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell 44, 424–436 [DOI] [PubMed] [Google Scholar]
- 29. Liang J., Saad Y., Lei T., Wang J., Qi D., Yang Q., Kolattukudy P. E., Fu M. (2010) MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J. Exp. Med. 207, 2959–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qi D., Huang S., Miao R., She Z. G., Quinn T., Chang Y., Liu J., Fan D., Chen Y. E., Fu M. (2011) Monocyte chemotactic protein-induced protein 1 (MCPIP1) suppresses stress granule formation and determines apoptosis under stress. J. Biol. Chem. 286, 41692–41700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jura J., Wegrzyn P., Korostyński M., Guzik K., Oczko-Wojciechowska M., Jarzab M., Kowalska M., Piechota M., Przewłocki R., Koj A. (2008) Identification of interleukin-1 and interleukin-6-responsive genes in human monocyte-derived macrophages using microarrays. Biochim. Biophys. Acta 1779, 383–389 [DOI] [PubMed] [Google Scholar]
- 32. Qi Y., Liang J., She Z. G., Cai Y., Wang J., Lei T., Stallcup W. B., Fu M. (2010) MCP-induced protein 1 suppresses TNFα-induced VCAM-1 expression in human endothelial cells. FEBS Lett. 584, 3065–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Skalniak L., Mizgalska D., Zarebski A., Wyrzykowska P., Koj A., Jura J. (2009) Regulatory feedback loop between NF-κB and MCP-1-induced protein 1 RNase. FEBS J. 276, 5892–5905 [DOI] [PubMed] [Google Scholar]
- 34. Kasza A., Wyrzykowska P., Horwacik I., Tymoszuk P., Mizgalska D., Palmer K., Rokita H., Sharrocks A. D., Jura J. (2010) Transcription factors Elk-1 and SRF are engaged in IL1-dependent regulation of ZC3H12A expression. BMC Mol. Biol. 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iwasaki H., Takeuchi O., Teraguchi S., Matsushita K., Uehata T., Kuniyoshi K., Satoh T., Saitoh T., Matsushita M., Standley D. M., Akira S. (2011) The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat. Immunol. 12, 1167–1175 [DOI] [PubMed] [Google Scholar]
- 36. Gossen M., Bujard H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winzen R., Thakur B. K., Dittrich-Breiholz O., Shah M., Redich N., Dhamija S., Kracht M., Holtmann H. (2007) Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell Biol. 27, 8388–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altman R., Asch E., Bloch D., Bole G., Borenstein D., Brandt K., Christy W., Cooke T. D., Greenwald R., Hochberg M., et al. (1986) Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 29, 1039–1049 [DOI] [PubMed] [Google Scholar]
- 39. Arnett F. C., Edworthy S. M., Bloch D. A., McShane D. J., Fries J. F., Cooper N. S., Healey L. A., Kaplan S. R., Liang M. H., Luthra H. S., et al. (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 31, 315–324 [DOI] [PubMed] [Google Scholar]
- 40. Neumann E., Judex M., Kullmann F., Grifka J., Robbins P. D., Pap T., Gay R. E., Evans C. H., Gay S., Schölmerich J., Müller-Ladner U. (2002) Inhibition of cartilage destruction by double gene transfer of IL-1Ra and IL-10 involves the activin pathway. Gene Ther. 9, 1508–1519 [DOI] [PubMed] [Google Scholar]
- 41. Xu N., Loflin P., Chen C. Y., Shyu A. B. (1998) A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 26, 558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C. Y., Shyu A. B., Müller M., Gaestel M., Resch K., Holtmann H. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18, 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Calvo S. E., Pagliarini D. J., Mootha V. K. (2009) Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U.S.A. 106, 7507–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rübsamen D., Blees J. S., Schulz K., Döring C., Hansmann M. L., Heide H., Weigert A., Schmid T., Brüne B. (2012) IRES-dependent translation of egr2 is induced under inflammatory conditions. RNA 18, 1910–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tiedje C., Ronkina N., Tehrani M., Dhamija S., Laass K., Holtmann H., Kotlyarov A., Gaestel M. (2012) The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 8, e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liang J., Wang J., Saad Y., Warble L., Becerra E., Kolattukudy P. E. (2011) Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J. Neuroinflammation 8, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.