

Background: The origin of specificity of plant α-glucosidases for long malto-oligosaccharides remains uncertain.
Results: The crystal structure and mutational analyses of sugar beet α-glucosidase revealed its substrate binding properties.
Conclusion: The long-substrate specificity was described as two structural elements, the N-loop and subdomain b2.
Significance: A slight structural difference leads to significant differences in specificity for varying chain lengths of substrate.
Keywords: Crystal Structure, Enzyme Catalysis, Enzyme Kinetics, Enzyme Structure, Plant, α-Glucosidase, Acarbose Recognition, Glycoside Hydrolase Family 31, Long-chain Specificity
Abstract
Sugar beet α-glucosidase (SBG), a member of glycoside hydrolase family 31, shows exceptional long-chain specificity, exhibiting higher kcat/Km values for longer malto-oligosaccharides. However, its amino acid sequence is similar to those of other short chain-specific α-glucosidases. To gain structural insights into the long-chain substrate recognition of SBG, a crystal structure complex with the pseudotetrasaccharide acarbose was determined at 1.7 Å resolution. The active site pocket of SBG is formed by a (β/α)8 barrel domain and a long loop (N-loop) bulging from the N-terminal domain similar to other related enzymes. Two residues (Phe-236 and Asn-237) in the N-loop are important for the long-chain specificity. Kinetic analysis of an Asn-237 mutant enzyme and a previous study of a Phe-236 mutant enzyme demonstrated that these residues create subsites +2 and +3. The structure also indicates that Phe-236 and Asn-237 guide the reducing end of long substrates to subdomain b2, which is an additional element inserted into the (β/α)8 barrel domain. Subdomain b2 of SBG includes Ser-497, which was identified as the residue at subsite +4 by site-directed mutagenesis.
Introduction
Glycoside hydrolase family 31 (GH31)3 is one of the most interesting glycoside hydrolase families and includes not only the glycoside hydrolases α-glucosidase (EC 3.2.1.20), α-1,3-glucosidase (EC 3.2.1.84), α-xylosidase (EC 3.2.1.177), and sucrase-isomaltase (EC 3.2.1.48 and EC 3.2.1.10), but also α-glucan lyase (EC 4.2.2.13) (1). The GH31 α-glucosidases (GH31AGs) are involved mainly in the metabolism of starch and its derivatives. For example, maltase-glucoamylase (MGAM) and sucrase-isomaltase in the mammalian small intestine are associated with the hydrolysis of malto-oligosaccharides first degraded by α-amylases (2). In contrast, there are GH31AGs with different roles, such as α-glucosidase II, which is localized in the endoplasmic reticulum and is involved in the quality control of nascent glycoproteins.
Most fungal α-glucosidases (e.g. those of Aspergillus niger, Schizosaccharomyces pombe, and Schwanniomyces occidentalis) (3–5) also participate in the use of malto-oligosaccharides and exhibit the highest kcat/Km for a malto-oligosaccharide with a degree of polymerization (DP) of 3 (G3), with lower values for substrates longer than G3. These are known as short chain-specific GH31AGs.
During the germination of plant seeds, starch degradation to produce glucose is one of the most important events for obtaining energy. Four types of enzymes are believed to be involved in the conversion of starch to glucose: α-amylase, β-amylase, debranching enzymes, and α-glucosidase. α-Glucosidases are thought to act on maltose and other short malto-oligosaccharides produced by amylases. This is indeed the case for barley α-glucosidase, which shows short-chain specificity (6). However, most plant α-glucosidases tend to prefer long malto-oligosaccharides. For example, buckwheat α-glucosidase (7) and sugar beet α-glucosidase (SBG) (8) show 8- and 50-fold higher kcat/Km values for maltoheptaose than for maltose, respectively. In particular, SBG has exceptional specificity for long substrates, exhibiting a 90-fold higher kcat/Km for soluble starch than for maltose. It is of interest that GH31AGs show such different chain length specificities despite the fact that the enzymes share significantly similar amino acid sequences. Understanding the basis of the substrate specificity diversity in GH31AGs is a challenging task, and few studies have focused on the molecular mechanism of the diverse chain length specificities.
The crystal structures of several GH31AGs have been determined (9–13). The major domain of GH31AGs displays a (β/α)8 barrel fold. The active site pocket is formed by the (β/α)8 barrel domain and the N-loop, which is a long loop bulging from the N-terminal β-sandwich domain. Among the GH31AGs with known structures, the C-terminal subunit of human MGAM (CtMGAM) is the only long chain-specific enzyme and has a 10 times lower Km for G5 than for G2 (13). The long-chain specificity of the C-terminal unit of the glucoamylase CtMGAM was a result of an insertion of 21 amino acids, which form subsites +2 and +3. However, SBG and other plant GH31AGs have no such insertion, and another element must be responsible for their long-chain specificity.
We previously identified Phe-236 in the N-loop of SBG as one of the important elements involved in the recognition of long-chain substrates based on a comparison of amino acid sequences and the results of site-directed mutagenesis. Substitution of Phe-236 with Ala or Ser decreased the kcat/Km values for the hydrolysis of soluble starch and malto-oligosaccharides (Gn, where n represents DP) except G2. In contrast, replacement of Thr-228, which is equivalent to Phe-236 in short chain-specific A. niger α-glucosidase, with Phe led to a shift from G3 to G4, which has the highest kcat/Km (14).
In this study, we determined the crystal structure of SBG in an effort to obtain structural insights into the long-chain specificity. This is the first crystal structure of a plant GH31AG reported to date. A complex structure bound with acarbose, a pseudotetrasaccharide inhibitor, reveals that the N-loop forms subsites +2 and +3. Furthermore, we identified the residue forming subsite +4 is Ser-497 by site-directed mutagenesis.
EXPERIMENTAL PROCEDURES
Purification of α-Glucosidase from Sugar Beet Seeds
Sugar beet (Beta vulgaris L. cv. Abend) seeds with pericarp (1 kg) were milled, suspended in 4 liters of 0.1 m sodium acetate buffer (pH 5.4) (buffer A), and stirred for 10 h at 4 °C. The crude extract was obtained from the suspension by filtration using a nylon net, centrifugation at 11,300 × g for 10 min, and Celite 535 (Wako Pure Chemical Industries, Osaka, Japan). Proteins were precipitated by treatment with 90% saturated ammonium sulfate for 30 h at 4 °C, collected by centrifugation at 11,300 × g for 20 min, and dissolved in 20 mm buffer A containing 12% ammonium sulfate. The samples were loaded onto a Toyopearl butyl-650M column (3 cm (inner diameter) × 38 cm; Tosoh, Tokyo, Japan) equilibrated with 20 mm buffer A containing 12% ammonium sulfate. After washing the column with equilibration solution, the bound proteins were eluted with a linear gradient of 12 to 0% ammonium sulfate in 20 mm buffer A. The active fractions were collected, dialyzed against 20 mm buffer A, and loaded onto a CM-Sepharose Fast Flow column (3 cm (inner diameter) × 38 cm; GE Healthcare) equilibrated with 20 mm buffer A. The column was washed, and the bound proteins were eluted with a linear gradient of 0–1 m sodium chloride in 20 mm buffer A. The active fractions were concentrated using a Centriprep YM-50 unit (Millipore, Billerica, MA) and loaded onto a Toyopearl HW-55F column (2.7 cm (inner diameter) × 80 cm; Tosoh) equilibrated with 20 mm buffer A containing 100 mm sodium chloride. The purified native SBG fractions were collected and dialyzed against 20 mm buffer A.
Cloning of the Gene Encoding SBG
Sugar beet (cv. Abend) seeds were germinated for 9 days at 30 °C. Their shoots were collected, immediately frozen, and crushed in liquid nitrogen. The whole DNA was extracted from the crushed shoots (100 mg) using ISOPLANT II (Nippon Gene, Toyama, Japan). The extracted DNA was used for PCR with a pair of synthesized primers: 5′-TCCTAAAAGCTCAACATTTATCGAGGGTTT-3′ and 5′-CACACACAAAATCAGAAAAACTCCAAGG-3′. These primers were designed according to a reported α-glucosidase cDNA cloned from sugar beet seeds (strain NK185-BR2; GenBankTM accession number AB698976). PCR was performed using PrimeSTAR Max DNA polymerase (Takara Bio, Otsu, Japan). The PCR product was ligated using a Ver. 2 ligation kit (Takara Bio) into the EcoRV site of pBluescript II SK(+) (Stratagene, La Jolla, CA) and propagated in Escherichia coli strain DH5α. The amplified DNA strand was sequenced using an automated DNA sequencer (Applied Biosystems 310 Genetic Analyzer and BigDye Terminator v3.1). The nucleotide sequence was deposited in the GenBankTM with the accession number AB699590. The exons of the amplified DNA were predicted by Spidey (15) using the reported SBG cDNA (GenBankTM accession number AB698976) as a template.
Crystallization, Data Collection, and Refinement
Purified native SBG (3.53 mg) was incubated in 20 mm buffer A (7 ml) containing 70 milliunits of endoglycosidase F3 (Endo-F3) (Calbiochem). After incubation for 65 h at 4 °C, Endo-F3 was removed by CM-Sepharose Fast Flow column chromatography as described for purification of native SBG. Endo-F3-treated native SBG was dialyzed against 10 mm CHES (pH 9.0) and concentrated using an Amicon Ultra-15 unit to 30,000 nominal molecular weight limits (Millipore).
In all cases, crystallization was performed by the hanging-drop vapor-diffusion method at 25 °C for ∼1 month. Several crystals of the ligand-free form were obtained in a drop consisting of 6 μl of Endo-F3-treated native SBG (2.16 mg/ml) and 3 μl of reservoir solution (50 mm sodium acetate buffer (pH 4.5), 100 mm ammonium sulfate, and 18% polyethylene glycol monomethyl ether 2000). Several co-crystals with acarbose were obtained in a drop consisting of 3 μl of Endo-F3-treated native SBG (2.16 mg/ml), 3 μl of reservoir solution (50 mm sodium acetate buffer (pH 4.0), 50 mm ammonium sulfate, and 16% polyethylene glycol monomethyl ether 2000), and 1 μl of 100 mm acarbose.
Data sets were collected under a stream of nitrogen at 100 K from a single crystal at beamline BL41XU of SPring-8 (Hyogo, Japan) at a wavelength of 1.000 Å. Each crystal was flash-cooled after soaking in each reservoir solution containing 20% glycerol (and 14 mm acarbose for the co-crystal) for several minutes. Diffraction data sets were collected using an MX225HE CCD detector (Rayonix, Norderstedt, Germany). The diffraction data were indexed, integrated, scaled, and merged with XDS (16).
The structure of the acarbose complex was determined by the molecular replacement method with AutoMR in PHENIX (17) using the N-terminal subunit of MGAM (NtMGAM; Protein Data Bank code 2QLY) as a search model. The ligand-free structure was determined using the acarbose complex structure as a search model. After several cycles of manual model corrections with Coot (18) and refinement with REFMAC5 (19) in CCP4 and phenix.refine (17), the refinement converged. Ramachandran plot analysis was performed using RAMPAGE (20) in CCP4. Coordinates and structure factors have been deposited in the Protein Data Bank with codes 3W37 and 3W38. Graphical representations were prepared using PyMOL (21).
Production of Recombinant Enzymes
Site-directed mutagenesis was performed using a PrimeSTAR mutagenesis basal kit (Takara Bio). The SBG-carrying pGAPZαA vector was used as the PCR template with primers 5′-AGCTTCGCTAGGGACCTTAACTTGTAT-3′ and 5′-GTCCCTAGCGAAGCTAGCAATGTCAGC-3′ for N237A and primers 5′-AATAATGCTGGAGGCCGTGTACCAATA-3′ and 5′-GCCTCCAGCATTATTGATCTTATATGG-3′ for S497A, where the underlined nucleotides indicate the mutated codons. The expression and purification of the mutant enzymes were performed according to a previous report (14).
Biochemical Assays
α-Glucosidase activity, protein concentration, and the effects of pH were measured as described previously (14). Substrates for measuring kinetic parameters were G2–G7 (a series of malto-oligosaccharides with DP = 2–7; Nihon Shokuhin Kako Co., Ltd., Tokyo, Japan), G18 (Amylose EX-I, average DP of 18; Hayashibara, Okayama, Japan), and soluble starch (Nacalai Tesque, Kyoto, Japan), whose concentration of nonreducing termini (0.136 μmol/mg) was estimated by Smith degradation (22). The initial rates for eight substrate concentrations (1/3·Km − 5·Km) were measured. The kinetic parameters kcat and Km were determined from s-v plots fitted to the Michaelis-Menten equation using KaleidaGraph 3.6J (Synergy Software, Reading, PA). The enzyme concentrations used were 0.790–1.90 nm N237A and 0.746–1.49 nm S497A.
Size-exclusion Chromatography
Size-exclusion chromatography was performed by HPLC using a TSKgel G3000SWXL column (7.8 mm (inner diameter) × 30 cm; Tosoh) equilibrated with 50 mm sodium acetate buffer (pH 4.5) containing 150 mm sodium chloride. Native SBG (0.517 μg, 10 μl) was applied to the column and eluted at a flow rate of 0.7 ml/min while the absorbance was monitored at 280 nm. The molecular mass of native SBG was estimated from its elution coefficient relative to those of the molecular mass marker proteins (gel filtration standard, Bio-Rad): thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1.35 kDa).
RESULTS AND DISCUSSION
Crystal Structure Analysis of SBG
SBG was purified from sugar beet seeds. Three amino acid differences (N423D, V871I, and R876L) were found in SBG as deduced by comparing the genomic DNA sequence with the reported sequence (14). Purified SBG was deglycosylated by treatment with Endo-F3, and deglycosylated SBG was crystallized and co-crystallized with acarbose. The crystals of SBG belong to the space group P212121 (unit cell parameters a = 83.5, b = 95.5, and c = 107.7 Å for the apo enzyme crystal and a = 86.4, b = 98.2, and c = 108.8 Å for the acarbose complex), with one protein molecule present in each asymmetric unit. This observation was in agreement with the results of size-exclusion chromatography of SBG, indicating that it exists as a monomer in solution. Crystal structures were determined at 2.8 Å (ligand-free structure) and 1.7 Å (acarbose complex) resolution, respectively, with the molecular replacement method using the structure of NtMGAM (10) as a search model (Fig. 1 and Table 1). All 913 residues were built based on the electron density with the exception of residues 1–38, 119–137, 859–884, and 910–913 in both structures. Both crystal structures were almost identical (root mean square deviation with Cα = 0.4 Å calculated by the Dali pairwise server (23)).
FIGURE 1.
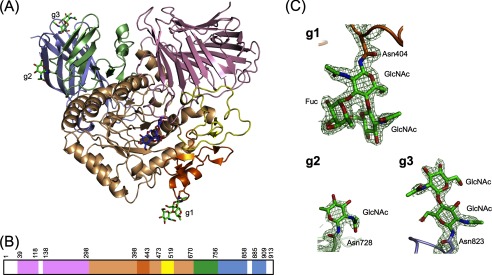
Overall structure of SBG bound to acarbose. A, ribbon diagram of SBG with stick representation of acarbose (purple) and N-glycans (green). B, linear schematic representation of the domain composition with amino acid numbers. In A and B, the N-terminal domain is pink, the catalytic domain is light orange, subdomain b1 is orange, subdomain b2 is yellow, the proximal C-terminal domain is light green, and the distal C-terminal domain is light blue. The white spaces in B indicate a residue was not built because of poor electron density (Met-1–Gly-38, Ser-119–Gln-137, Glu-859–Gly-884, and Gly-910–Arg-913). C, electron density maps of N-glycans at Asn-404 (g1), Asn-728 (g2), and Asn-823 (g3).
TABLE 1.
Data collection and refinement statistics for SBG data sets
| Crystal |
||
|---|---|---|
| Ligand-free | Acarbose complex | |
| Data collection | ||
| Space group | P212121 | P212121 |
| Unit cell parameters (a, b, c; Å) | 83.5, 95.5, 107.7 | 86.4, 98.2, 108.8 |
| Resolution range (Å) | 43.6–2.79 (2.96–2.79) | 43.2–1.70 (1.79–1.70) |
| No. of unique reflections | 21,842 (3360)a | 102,030 (14446) |
| Rmerge | 0.133 (0.581) | 0.103 (0.880) |
| Completeness (%) | 99.1 (96.1) | 99.9 (100) |
| 〈I/σ(I)〉 | 12.13 (3.64) | 11.84 (2.97) |
| Multiplicity | 5.72 (5.71) | 5.56 (5.57) |
| Refinement | ||
| Rwork | 0.2283 | 0.1481 |
| Rfree | 0.2623 | 0.1775 |
| No. of protein atoms | 6583 | 6784 |
| No. of water molecules | 35 | 739 |
| No. of acarbose molecules | 0 | 1 |
| No. of sugar residues of N-glycans | 4 | 6 |
| r.m.s.d.b values from ideal | ||
| Bond lengths (Å) | 0.0116 | 0.014 |
| Bond angles | 1.44° | 1.57° |
| Ramachandran plot analysis | ||
| Favored region (%) | 96.71 | 97.28 |
| Allowed region (%) | 3.29 | 2.48 |
| Outlier region (%) | 0 | 0.24 |
a Values in parentheses are for the highest resolution shell.
b r.m.s.d., root mean square deviation.
The crystal structure of the acarbose complex revealed that SBG is partially N-glycosylated, with electron density visible for β-N-acetylglucosaminyl-(1→4)-(α-fucosyl-(1→3))-β-N-acetylglucosaminyl-Asn-404, β-N-acetylglucosaminyl-Asn-728, and β-N-acetylglucosaminyl-(1→4)-β-N-acetylglucosaminyl-Asn-823 (Fig. 1C). These electron densities are unclear in the ligand-free structure with low resolution. SBG has six potential N-glycosylation sites (Asn-Xaa-(Ser/Thr), where Xaa is not Pro). Among them, Asn-404 and Asn-728 form the Asx turn. The Asx turn is preferentially recognized by an oligosaccharyltransferase, which is located in the endoplasmic reticulum and catalyzes N-glycan transfer (24). Asn-823 is not involved in the Asx turn; however, the carbonyl side chain of Asn-823 forms a water-mediated hydrogen bond with the hydroxy group of Thr-825. This hydrogen bond may induce the nitrogen to form an imidate tautomer, which is a competent nucleophile (25). Other conserved amino acids, Asn-54, Asn-495, and Asn-517, have no such secondary structure element and hydrogen bond. The crystal structure of SBG indicates that the N-glycan was retained even though the enzyme was treated with Endo-F3 before crystallization. This apparent contradiction may be explained by the substrate specificity of Endo-F3, which has high activity for α-1,6-fucosylated N-glycans but not for α-1,3-fucosylated N-glycans (26).
Overall Structure
The overall structure of SBG was divided into four major domains and two subdomains similar to other GH31AGs (Fig. 1, A and B): the N-terminal β-sandwich domain (residues 39–298), the (β/α)8 barrel domain (residues 299–670), insertion subdomain b1 (residues 399–443) and subdomain b2 (residues 474–519), the proximal C-terminal domain (residues 671–756), and the distal C-terminal domain (residues 757–909). The N-terminal β-sandwich domain consists of four antiparallel β-sheets. Several strands are connected with long loops, one of which, from Trp-229 to Ser-245, forms part of the active site pocket. This is the so-called “N-loop” and plays a crucial role in substrate binding, as discussed below.
The (β/α)8 barrel domain is the major domain of SBG. The active site pocket of SBG is formed mainly by the (β/α)8 barrel domain and is extended by the N-loop. The (β/α)8 barrel fold has two insertions as subdomains b1 and b2. These two subdomains form part of the active site pocket, as in other GH31AGs. Subdomain b1, inserted into β→α loop 3, is well conserved among the GH31AGs for which structures are known, except CtMGAM, which contains four small strands and one small helix (13). Subdomain b2, inserted into β→α loop 4, has no typical secondary structure element. The overall structure of subdomain b2 is similar to other GH31AGs for which structures are known, but those of Sulfolobus solfataricus α-glucosidase (MalA) (9) and Ruminococcus obeum α-glucosidase (12) are quite distinct from that of SBG, which has an α-helix element.
The proximal C-terminal domain consists of three antiparallel β-sheets and two small α-helices. The distal C-terminal domain forms a nine-stranded antiparallel β-sandwich structure. Neither C-terminal domain has any interaction with the active site pocket. These domains appear to contribute to stabilization of the (β/α)8 barrel catalytic domain rather than substrate binding.
The overall structure of SBG is similar to those of other α-glucosidases in GH31, with root mean square deviations calculated by the Dali server (27) of 1.5 Å for 786 of 871 residues (the N-terminal subunit of human sucrase-isomaltase, Protein Data Bank code 3LPP, chain A), 1.5 Å for 789 of 863 residues (NtMGAM, code 2QMJ), 1.7 Å for 781 of 890 residues (CtMGAM, code 3TOP, chain B), 2.2 Å for 645 of 691 residues (MalA, code 2G3M, chain A), and 2.3 Å for 615 of 665 residues (R. obeum α-glucosidase, code 3NXM, chain B).
A glycoside hydrolase that is able to attack polysaccharides generally bears an extra carbohydrate-binding domain and/or surface binding site for polysaccharide. In GH31 enzymes, Cellvibrio japonicas α-xylosidase has an extra PA14 domain in the N-terminal part of the enzyme to accommodate long xylo-oligosaccharides (28). Gracilariopsis lemaneiformis α-1,4-glucan lyase (Protein Data Bank code 2X2I) possesses a second substrate-binding site in the N-terminal domain (29). However, neither the extra domain nor the surface binding site is found in the crystal structure of SBG. The electron density of acarbose is visible only at the active site (Fig. 2).
FIGURE 2.

Acarbose recognition by SBG. Shown are stereo diagrams with electron density of acarbose bound to the active site pocket of SBG. A, amino acid residues and water molecules (red spheres) interacting with acarbose. The catalytic nucleophile and acid/base are Asp-469 and Asp-568, respectively. B, structure around the glucose moiety at the reducing end of acarbose. The contour level of the 2Fo − Fc map is 1σ, and the structure is colored as described in the legend to Fig. 1.
Subsites −1 and +1
The structures of subsites −1 and +1 are almost identical to those of other GH31AGs. Two catalytic aspartic acid residues, Asp-469 and Asp-568, are located in β→α loops 4 and 6, respectively. The active site pocket is occupied by acarbose (Fig. 2). The valienamine unit (ring A) and the 4-amino-4,6-dideoxy-α-d-glucose unit (ring B) of acarbose, occupying subsites −1 and +1, respectively, are enclosed by a number of hydrogen bonds and van der Waals interactions (Fig. 3). Asp-357, Arg-552, Asp-568, and His-626 formed hydrogen bonds with the hydroxy groups of ring A. Asp-398, Trp-432, and Asp-597 interact with the hydroxy groups of ring A through water-bridging hydrogen bonds. Ile-396, Trp-467, and Trp-565 are located at the bottom of the active site pocket. Trp-329, Ile-358, Trp-432, Phe-476, and Phe-601 are located at the entrance of the active site pocket and seem to form a hydrophobic barrier. Asp-232 in the N-loop and Arg-552 in the (β/α)8 barrel domain interact with ring B at subsite +1 through hydrogen bonds. Met-470, which is present in two conformations, appears to make contact with ring B. All of the above residues are invariant among GH31AGs except Trp-329. The equivalence of Trp-329 is conserved as Trp or Tyr in GH31AGs. The difference of this aromatic residue was reported to be related to the substrate preference of α-1,4- and α-1,6-glucosidic linkages in several GH31AGs. For example, R. obeum α-glucosidase, possessing Trp-169 at this position, exhibits α-1,6-glucoside specificity, and the substitution of Trp-169 with Tyr switches the substrate preference of R. obeum α-glucosidase from α-1,6-glucoside to α-1,4-glucoside (12). In addition, for both NtMGAM and CtMGAM, the specificity constant kcat/Km for α-1,6-glucoside was increased by replacement of the Tyr residue with Trp at this position (13). The relatively high specificity of SBG for the α-1,6-glucosidic linkage (the kcat/Km for isomaltose is one-fifth of that for maltose) (8) is likely because of Trp-329.
FIGURE 3.

Schematic representation of acarbose recognition by SBG. Subsites −1 to +3 bind to rings A–D of acarbose, respectively. The dashed lines indicate hydrogen bonds. Asp-469 and Asp-568 with asterisks are catalytic residues. Boxed residues are invariant among GH31AGs.
Subsites +2 and +3
In contrast to the numerous interactions at subsites −1 and +1, a few interactions hold two glucose moieties (rings C and D) of acarbose at subsites +2 and +3 (Figs. 2 and 3). Subsites +2 and +3 are composed of residues provided by the N-loop. The nitrogen atom of Ala-234 and Nδ2 of Asn-237 interact with O6 and the ring oxygen (O5) of ring D through hydrogen bonds. Ile-233 and Phe-236 form a hydrophobic lining for rings C and D. We previously proposed that Phe-236 in the N-loop contributes to the formation of subsites +2 and +3 via a London dispersion force interaction based on the results of the site-directed mutagenesis study without the tertiary structure information (14). The present structural study confirmed these suggestions and also indicates a contribution from the side chain of Asn-237 to the substrate binding at subsite +3.
Site-directed Mutagenesis of Asn-237
To evaluate the contribution of Asn-237 to the substrate specificity of SBG, we produced the N237A mutant enzyme using a Pichia pastoris expression system and assessed the kinetic properties of this mutant enzyme for a series of malto-oligosaccharides (G2–G7), amylose (G18, average DP = 18), and soluble starch (Table 2). The N237A kcat values for all substrates were ∼75% of wild-type recombinant SBG (rSBG). The reduction in kcat values is likely because of the change in the optimum pH. The optimum pH of N237A was pH 5.3, but the kinetic parameters were determined under the same reaction conditions as used for rSBG at pH 4.8 to better compare the kinetic constants. The N237A Km values for G2 and G3 were almost the same as those of rSBG, whereas the Km values for G4–G7 were 1.9–2.7 times higher than those of rSBG. The reduction in kcat/Km values for malto-oligosaccharides longer than G3 was larger than those for G2 and G3. These results indicate that the substitution of Asn-237 with Ala decreased the affinity for malto-oligosaccharides longer than G3. It is noteworthy that N237A displayed a smaller kcat/Km for G4 (49.6 s−1 mm−1) than for G3 (62.6 s−1 mm−1), whereas wild-type rSBG exhibited a larger kcat/Km value for G4 than for G3. This result indicates that the N237A mutant lost the increment in binding energy at subsite +3 and that Asn-237 contributed to the formation of subsite +3. A reduction in affinity at subsite +3 should increase the Km values for G4–G7 because subsite +3 would contribute to binding G4–G7.
TABLE 2.
Kinetic parameters of the SBG variants
The kcat or Km with S.D. is the average value of triplicate measurements.
| Substrate | rSBG (Ref. 13) |
N237A |
S497A |
||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| s−1 | mm | s−1 mm−1 | s−1 | mm | s−1 mm−1 | s−1 | mm | s−1 mm−1 | |
| G2 | 245 ± 5 | 17.9 ± 1.0 | 13.7 | 180 ± 1 | 19.2 ± 0.3 | 9.36 | 258 ± 2 | 19.2 ± 0.4 | 13.4 |
| G3 | 340 ± 10 | 3.32 ± 0.08 | 102 | 224 ± 1 | 3.58 ± 0.07 | 62.6 | 306 ± 2 | 3.46 ± 0.05 | 88.5 |
| G4 | 293 ± 4 | 2.16 ± 0.02 | 136 | 199 ± 3 | 4.02 ± 0.04 | 49.6 | 313 ± 5 | 2.78 ± 0.02 | 113 |
| G5 | 347 ± 4 | 0.690 ± 0.020 | 503 | 245 ± 7 | 1.83 ± 0.06 | 134 | 334 ± 4 | 1.43 ± 0.03 | 234 |
| G6 | 341 ± 4 | 0.426 ± 0.009 | 802 | 261 ± 2 | 1.04 ± 0.03 | 251 | 316 ± 2 | 0.859 ± 0.005 | 368 |
| G7 | 328 ± 5 | 0.378 ± 0.007 | 868 | 258 ± 1 | 0.927 ± 0.011 | 279 | 322 ± 2 | 0.748 ± 0.001 | 431 |
| G18 | 328 ± 3 | 0.382 ± 0.007 | 859 | 258 ± 3 | 0.969 ± 0.011 | 266 | 317 ± 3 | 0.806 ± 0.017 | 394 |
| Soluble starcha | 301 ± 1 | 0.246 ± 0.004 | 1230 | 242 ± 3 | 0.631 ± 0.013 | 384 | 302 ± 1 | 0.512 ± 0.006 | 589 |
a The Km for soluble starch is its concentration of nonreducing termini.
Additional Subsites
Previous subsite mapping analysis indicated that SBG possesses subsites from −1 to +6 (14). In addition, the kcat/Km values for G5–G7, G18, and soluble starch of N237A gradually increased. These results indicate that SBG has other subsites unrelated to Asn-237. However, the present acarbose complex structure does not provide information on additional subsites. Thus, we expected the position of other subsites to be located in the direction of the anomeric hydroxy group of ring D of the bound acarbose. The structure of the SBG-acarbose complex shows that ring D at subsite +3 is an α-glucosyl moiety (Fig. 2B). Another refinement was performed by placing a β-glucosyl moiety in this position, but no electron density of the equatorial O1 with β-configuration was observed. The axial hydroxy group of ring D is oriented toward subdomain b2, and thus, additional subsites likely exist in subdomain b2.
Site-directed Mutagenesis of Ser-497
Among the residues in subdomain b2, we anticipated that Ser-497 (with dual conformation), in which Oγ is at a distance of 6.7 Å from the anomeric hydroxy group, contributes to the formation of other subsites. To confirm this, the S497A mutant enzyme was produced and characterized. The optimum pH of the S497A mutant enzyme was the same as that of wild-type rSBG. S497A exhibited almost the same kinetic parameters for substrates G2, G3, and G4 as rSBG; however, the mutant enzyme exhibited a 2.1-fold larger Km and a 2.1-fold smaller kcat/Km for G5 compared with rSBG (Table 2). These results indicate that the substitution of Ser-497 with Ala had a negative effect on substrate binding at subsite +4. The substitution of Ser-497 decreased the specificity for substrates longer than G5, increasing Km values and decreasing kcat/Km for the substrates. The reduction of affinity at subsite +4 may affect the long-chain specificity of SBG.
The site-directed mutagenesis study provided evidence that Ser-497 contributed to the formation of subsite +4 and that the reducing end of the longer substrate moved to subdomain b2. It raised the possibility that subdomain b2 includes additional subsites beyond subsite +4. To our knowledge, this is the first example showing that subdomain b2 is involved in substrate binding in GH31AGs. The function of subdomain b2 in GH31AGs has been reported in only MalA and was related not to substrate binding but to maintenance of the quaternary assembly of the hexamer (9). Plant GH31AGs possess a similar subdomain b2 and a serine residue equivalent to Ser-497 (see Fig. 5A) except for barley GH31AG, which prefers shorter substrates (7, 8, 30–32). The Ser residues and subdomain b2 may contribute to the long-chain specificity of plant GH31AGs.
FIGURE 5.

Sequence alignment of plant GH31AGs. The multiple sequence alignment at subdomain b2 (A) and around the N-loop (B) was produced by MUSCLE (33) and was depicted with the secondary structure of SBG by ESPript 2.2 (34). The inverted black triangles indicate the positions of Asn-237 and Ser-497 of SBG. SOG, spinach α-glucosidase (O04893); BWG, buckwheat α-glucosidase (H. Mori, unpublished data); ONG1, rice α-glucosidase isozyme 1 (Q653V4); ONG2, rice α-glucosidase isozyme 2 (Q653V7); BAG, barley α-glucosidase (Q43763).
Divergence of Substrate Recognition in GH31AGs
As mentioned above, the structures of subsites −1 and +1 are almost identical among GH31AGs. However, those of subsites +2 and +3 are divergent. Subsites +2 and +3 of SBG contain the N-loop; however, NtMGAM and CtMGAM have different architecture at subsites +2 and +3 compared with SBG (Fig. 4). Subsites +2 and +3 of CtMGAM contain mainly Trp-1369 in the specific 21-amino acid insertion and Phe-1560 in β→α loop 7 of the catalytic domain. Pro-1159 on the N-loop is situated near subsite +3, but its contribution to substrate binding seems to be modest. NtMGAM has few interactions with sugar molecules bound at subsites +2 and +3, and its N-loop is unrelated to substrate binding. This structural feature reflects the difference in the Ki values for acarbose, i.e. the Ki of NtMGAM (62 μm) is higher than those of SBG (6.68 μm) and CtMGAM (14 μm) (11). In addition, the N-loop of SBG is likely to possess a different role, which is related to the long-chain specificity of SBG, i.e. the side chains of Phe-236 and Asn-237 in the N-loop make the reducing end of the long-chain substrates move toward subdomain b2, where subsite +4 and possible additional subsites exist (Fig. 4A). CtMGAM has no such machinery, although it displays specificity for longer substrates. The acarbose molecule in CtMGAM twists around Phe-1560; thus, the reducing end of acarbose is oriented toward the other direction of subdomain b2 (Fig. 4B).
FIGURE 4.
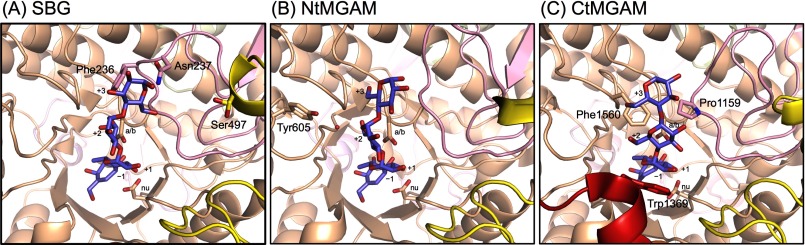
Comparison of acarbose-binding sites (subsites −1 to +3) in GH31AGs. A, SBG. B, NtMGAM (Protein Data Bank code 2QMJ). C, CtMGAM (code 3TOP). The catalytic residues and the residues related to substrate binding at subsites +2 and +3 are shown in stick representation. The catalytic nucleophile and acid/base are labeled with nu and a/b, respectively. Numbers indicate subsite numbers. The red sphere indicates the water molecule. The red helix in C represents the 21-amino acid insertion in CtMGAM, and other color coding is as described in the legend to Fig. 1.
The structural comparison indicates that the N-loop is the key structural element that governs the long-chain specificity. GH31AGs, even short chain-specific enzymes, possess the N-loop; however, its amino acid sequence is divergent. This divergence probably causes the difference in the affinity for the substrate and determines the destination of the reducing end of the long-chain substrates. Plant GH31AGs with higher substrate specificity for longer substrates possess a conserved N-loop and an Asn residue equivalent to Asn-237 (Fig. 5B). This Asn residue is likely important for determining the binding of the reducing end of the longer substrate and contributes to the long-chain specificity. It is of interest that short chain-specific barley GH31AG has a Tyr residue in place of the Asn residue. The Tyr residue may not be adequate to hold the substrate and organize the direction of the reducing end of the longer substrates.
In conclusion, we have presented a mechanism that explains the long-chain specificity of SBG. The N-loop and subdomain b2 appear to be associated with long-chain specificity. In particular, it is likely that Phe-236 and Asn-237 in the N-loop play key roles in determining the long-chain specificity by forming subsites +2 and +3 and guiding the reducing end of long substrates to subdomain b2, in which we identified subsite +4 as well as other possible more distant subsites.
Acknowledgments
We thank T. Hirose (Instrumental Analysis Division, Creative Research Institution, Hokkaido University) for amino acid analysis. We also thank the staff of beamline BL41XU at SPring 8 for help with data collection.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s)AB699590.
The atomic coordinates and structure factors (codes 3W37 and 3W38) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GH31
- glycoside hydrolase family 31
- GH31AG
- GH31 α-glucosidase
- MGAM
- maltase-glucoamylase (human)
- DP
- degree of polymerization
- G2–G7
- malto-oligosaccharides with DP = 2–7, respectively
- SBG
- sugar beet α-glucosidase
- CtMGAM
- C-terminal subunit of MGAM
- Endo-F3
- endoglycosidase F3
- CHES
- N-cyclohexyl-2-aminoethanesulfonic acid
- NtMGAM
- N-terminal subunit of MGAM
- G18
- amylose with average DP = 18
- rSBG
- recombinant SBG.
REFERENCES
- 1. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quezada-Calvillo R., Robayo-Torres C. C., Opekun A. R., Sen P., Ao Z., Hamaker B. R., Quaroni A., Brayer G. D., Wattler S., Nehls M. C., Sterchi E. E., Nichols B. L. (2007) Contribution of mucosal maltase-glucoamylase activities to mouse small intestinal starch α-glucogenesis. J. Nutr. 137, 1725–1733 [DOI] [PubMed] [Google Scholar]
- 3. Kita A., Matsui H., Somoto A., Kimura A., Takata M., Chiba S. (1991) Substrate specificity and subsite affinities of crystalline α-glucosidase from Aspergillus niger. Agric. Biol. Chem. 55, 2327–2335 [Google Scholar]
- 4. Okuyama M., Tanimoto Y., Ito T., Anzai A., Mori H., Kimura A., Matsui H., Chiba S. (2005) Purification and characterization of the hyper-glycosylated extracellular α-glucosidase from Schizosaccharomyces pombe. Enzyme Microb. Technol. 37, 472–480 [Google Scholar]
- 5. Sato F., Okuyama M., Nakai H., Mori H., Kimura A., Chiba S. (2005) Glucoamylase originating from Schwanniomyces occidentalis is a typical α-glucosidase. Biosci. Biotechnol. Biochem. 69, 1905–1913 [DOI] [PubMed] [Google Scholar]
- 6. Im H., Henson C. A. (1995) Characterization of high pI α-glucosidase from germinated barley seeds: substrate specificity, subsite affinities and active-site residues. Carbohydr. Res. 277, 145–159 [Google Scholar]
- 7. Chiba S., Kanaya K., Hiromi K., Shimomura T. (1979) Substrate specificity and subsite affinities of buckwheat α-glucosidase. Agric. Biol. Chem. 43, 237–242 [Google Scholar]
- 8. Matsui H., Chiba S., Shimomura T. (1978) Substrate specificity of an α-glucosidase in sugar beet seed. Agric. Biol. Chem. 42, 1855–1860 [Google Scholar]
- 9. Ernst H. A., Lo Leggio L., Willemoës M., Leonard G., Blum P., Larsen S. (2006) Structure of the Sulfolobus solfataricus α-glucosidase: implications for domain conservation and substrate recognition in GH31. J. Mol. Biol. 358, 1106–1124 [DOI] [PubMed] [Google Scholar]
- 10. Sim L., Quezada-Calvillo R., Sterchi E. E., Nichols B. L., Rose D. R. (2008) Human intestinal maltase-glucoamylase: crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J. Mol. Biol. 375, 782–792 [DOI] [PubMed] [Google Scholar]
- 11. Sim L., Willemsma C., Mohan S., Naim H. Y., Pinto B. M., Rose D. R. (2010) Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J. Biol. Chem. 285, 17763–17770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan K., Tesar C., Wilton R., Keigher L., Babnigg G., Joachimiak A. (2010) Novel α-glucosidase from human gut microbiome: substrate specificities and their switch. FASEB J. 24, 3939–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ren L., Qin X., Cao X., Wang L., Bai F., Bai G., Shen Y. (2011) Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2, 827–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tagami T., Okuyama M., Nakai H., Kim Y.M., Mori H., Taguchi K., Svensson B., Kimura A. (2013) Key aromatic residues at subsites +2 and +3 of glycoside hydrolase family 31 α-glucosidases contribute to recognition of long-chain substrates. Biochim. Biophys. Acta 1834, 329–335 [DOI] [PubMed] [Google Scholar]
- 15. Wheelan S. J., Church D. M., Ostell J. M. (2001) Spidey: A tool for mRNA-to genomic alignments. Genome Res. 11, 1952–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I. W., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Genet. 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 21. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC, San Carlos, CA [Google Scholar]
- 22. Hizukuri S., Osaki S. (1978) A rapid Smith-degradation for the determination of nonreducing, terminal residues of (1→4)α-d-glucans. Carbohydr. Res. 63, 261–264 [Google Scholar]
- 23. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 24. Imperiali B., Hendrickson T. L. (1995) Asparagine-linked glycosylation: specificity and function of oligosaccharyl transferase. Bioorg. Med. Chem. 3, 1565–1578 [DOI] [PubMed] [Google Scholar]
- 25. Imperiali B., Shannon K. L., Unno M., Rickert K. W. (1992) A mechanistic proposal for asparagine-linked glycosylation. J. Am. Chem. Soc. 114, 7944–7945 [Google Scholar]
- 26. Tarentino A. L., Plummer T. H., Jr. (1994) Enzymatic deglycosylation of asparagine-linked glycans: purification, properties, and specificity of oligosaccharide-cleaving enzymes from Flavobacterium meningosepticum. Methods Enzymol. 230, 44–57 [DOI] [PubMed] [Google Scholar]
- 27. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Larsbrink J., Izumi A., Ibatullin F. M., Nakhai A., Gilbert H. J., Davies G. J., Brumer H. (2011) Structural and enzymatic characterization of a glycoside hydrolase family 31 α-xylosidase from Cellvibrio japonicus involved in xyloglucan saccharification. Biochem. J. 436, 567–580 [DOI] [PubMed] [Google Scholar]
- 29. Yu S. (2008) The anhydrofructose pathway of glycogen catabolism. IUBMB Life 60, 798–809 [DOI] [PubMed] [Google Scholar]
- 30. Sugimoto M., Furui S., Suzuki Y. (1995) Multiple molecular forms of α-glucosidase from spinach seeds, Spinacia oleracea L. Biosci. Biotechnol. Biochem. 59, 673–677 [Google Scholar]
- 31. Nakai H., Ito T., Hayashi M., Kamiya K., Yamamoto T., Matsubara K., Kim Y. M., Jintanart W., Okuyama M., Mori H., Chiba S., Sano Y., Kimura A. (2007) Multiple forms of α-glucosidase in rice seeds (Oryza sativa L., var Nipponbare). Biochimie 89, 49–62 [DOI] [PubMed] [Google Scholar]
- 32. Nakai H., Tanizawa S., Ito T., Kamiya K., Kim Y. M., Yamamoto T., Matsubara K., Sakai M., Sato H., Imbe T., Okuyama M., Mori H., Sano Y., Chiba S., Kimura A. (2007) Function-unknown glycoside hydrolase family 31 proteins, mRNAs of which were expressed in rice ripening and germinating stages, are α-glucosidase and α-xylosidase. J. Biochem. 142, 491–500 [DOI] [PubMed] [Google Scholar]
- 33. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]