

Abstract
Animal cells strictly control the distribution of cholesterol in their organelle membranes. This regulation requires an efficient machinery to transport insoluble cholesterol between organelles. In the present study, we generate an 125I-labeled mutant version of Perfringolysin O (PFO), a cholesterol-binding protein, and use it to measure cholesterol in the plasma membrane of intact cells. We also purify plasma membranes from the same cells, which allows us to directly relate cholesterol concentration to 125I-PFO binding. We show that cholesterol is organized in the plasma membrane in a manner that makes it inaccessible to PFO until its concentration exceeds a threshold of 35 mol% of total lipids. This 35% threshold is in striking contrast to the 5% threshold previously found for PFO binding to endoplasmic reticulum membranes. The 125I-PFO probe also proved useful in monitoring the movement of LDL-derived cholesterol from lysosomes to plasma membranes. Using three different mutant cell lines, we show that this transport requires receptor-mediated uptake of LDL, hydrolysis of LDL-cholesteryl esters in lysosomes, and transfer of the liberated cholesterol to the plasma membrane.
Cholesterol is an essential and tightly regulated lipid of mammalian cells that is distributed unevenly among cell membranes. The majority (65–90%) of cholesterol resides in the plasma membrane (1, 2). A major source of cellular cholesterol derives from lipoprotein particles, such as LDL, that have a core rich in cholesteryl esters (3). LDL particles are internalized to endosomes and lysosomes by receptor-mediated endocytosis where acid lipase hydrolyses the cholesteryl esters of LDL. The liberated cholesterol is then transported from lysosomes to plasma membrane and endoplasmic reticulum (ER) (4, 5). Although much has been learned about endocytosis and lysosomal digestion, our understanding of how cholesterol is trafficked from lysosomes to cell membranes and of how cholesterol is organized within membranes remains limited.
Any approach to understanding cholesterol trafficking requires tools to accurately track cholesterol in living cells. Currently, several methods are used for probing cellular cholesterol movement, but each method has drawbacks. These methods are based on: (i) modification of plasma membrane cholesterol by cholesterol oxidase (6), which is an indirect, enzyme-based method that may also disrupt cell membranes (7); (ii) fluorescence microscopy of fluorescent analogs, such as dehydroergosterol and dansyl cholesterol, which may not be faithful mimics of cholesterol in terms of membrane organization (8); (iii) binding of fluorescent compounds, such as filipin, which form complexes with cholesterol but also disrupt cell membranes (8); and (iv) cholesterol-dependent binding of cytolysins, such as Perfringolysin O (PFO), which binds selectively to cholesterol-rich membranes but also forms pores (9, 10).
Although the pore-forming drawback of PFO limits its use in measuring cholesterol in the plasma membrane of living cells, it does not interfere with its use as a probe for cholesterol in isolated membranes. In this regard, PFO binding to purified ER membranes was observed only after the molar concentration of cholesterol exceeded a threshold of 4–6% of membrane lipids (11). This threshold coincided almost exactly with the threshold concentration above which cholesterol blocks ER-to-Golgi transport of sterol-regulatory element binding proteins (SREBPs) (11, 12), which are transcription factors that stimulate lipid synthesis (13). SREBP transport is mediated by Scap, an ER protein that binds SREBPs and escorts them to the Golgi, where the SREBPs are processed to their transcriptionally active forms. When the cholesterol content of ER membranes exceeds 4–6%, Scap binds cholesterol and this triggers a conformational change that causes the Scap/SREBP complex to be retained in the ER (14, 15). The common ∼5% threshold for Scap and PFO binding to cholesterol suggests that both proteins recognize a pool of ER cholesterol that is accessible for protein binding only when the cholesterol content rises above ∼5%. Unfortunately, PFO binding cannot be used to measure cholesterol in plasma membranes of intact cells because PFO disrupts the membrane by forming pores. Two groups have described an approach to overcoming PFO’s lytic properties by using a fluorescently labeled truncated fragment that does not form pores (16, 17). However, neither group used their PFO fragment to quantify the cholesterol concentration in the plasma membrane.
In the present study, we take advantage of a mutated form of PFO that retains its normal cholesterol-binding properties but was shown not to form pores, at least in model liposomes (18, 19). We show that this modified water-soluble PFO, designated as PFO*, can be readily purified in large quantities and does not lyse cultured mammalian cells at 4 °C. PFO* can be labeled with radioactive 125I and can be used to directly measure the amount of accessible cholesterol in the plasma membrane of intact cells. We also use a surface biotinylation method to isolate plasma membranes and quantify their cholesterol content. Our studies show that cholesterol in plasma membranes is not accessible to 125I-PFO* until the content of cholesterol exceeds 35 mol% of membrane lipids. These data suggest that mammalian cells maintain a considerable fraction of cholesterol in a pool that is inaccessible to a cholesterol-binding protein like PFO*.
Results
To prepare a modified version of PFO that would not permeabilize cell membranes at 4 °C, we produced a plasmid that encodes PFO with a tyrosine to alanine mutation at position 181. This mutant PFO retains the ability to bind to cholesterol in model liposomes, but it loses the ability to form pores (18, 19). We hereafter refer to this mutant version of PFO as PFO*. Fig. 1A shows a Coomassie-stained gel illustrating the homogeneity of PFO* after its purification from transformed Escherichia coli. Fig. S1 compares the binding of purified PFO and PFO* to cholesterol-containing liposomes. For both PFO and PFO*, binding does not occur until the cholesterol concentration of the liposomes exceeds a threshold of ∼40 mol%.
Fig. 1.

Biochemical characterization of PFO*. (A) SDS/PAGE of purified recombinant PFO*. Protein was purified as described in Materials and Methods. An aliquot (1 μg) was subjected to 8% SDS/PAGE, and proteins were visualized with Coomassie Brilliant blue R-250. (B) Lysis of SV-589 cells by PFO, but not by PFO*, as measured by release of LDH. On day 0, cells were set up in 12-well plates as described in SI Materials and Methods. On day 3, each monolayer received 0.5 mL PBS containing varying amounts of the indicated protein. After incubation for 1 h at 4 °C, the PBS was removed and assayed for LDH as described in Materials and Methods. (C) Immunoblot analysis of two cytosolic proteins released from SV-589 cells after incubation with varying amounts of purified PFO, but not PFO*. Aliquots of the cell-free PBS solution from B (lanes 1–7) and from the total cell lysate (lane 8) were immunoblotted with antibodies directed against the ubiquitin activating enzyme E1 and LDH. Before immunoblot analysis, the total cell lysate was adjusted to the same volume as each cell-free PBS solution. Blots were exposed to film at room temperature for 30–60 s.
Both PFO and PFO* form pores in animal cells at 37 °C. To test whether PFO* would form pores at 4 °C in mammalian cells, we added native PFO or PFO* to cultured SV-589 fibroblasts at 4 °C. Native PFO permeabilized the plasma membrane as revealed by enzymatic assays of cytosolic lactate dehydrogenase (LDH) activity that was released into the culture medium (Fig. 1B). PFO* had no such effect. This result was confirmed by immunoblotting of the released cytosol for LDH and for another cytosolic protein, the ubiquitin-activating enzyme E1 (Fig. 1C).
When cultured SV-589 fibroblasts were incubated with 125I-labeled PFO* at 4 °C, we observed a time-dependent binding of the iodinated protein to the cell surface (Fig. 2A). Binding was markedly reduced when the cells had been pretreated with 2-hydroxypropyl-β-cyclodextrin (HPCD), which is known to remove cholesterol from cell membranes (20). When 125I-PFO* was incubated with cells for 2 h at 4 °C, the binding reaction showed saturation kinetics with an apparent half-maximal 125I-PFO* concentration of 70 μg/mL (Fig. 2B). Again, 125I-PFO* binding was markedly reduced when the cells were pretreated with HPCD.
Fig. 2.

Binding of 125I-PFO* to SV-589 cells. On day 0, cells were set up in medium A at 1 × 105 cells per 60-mm dish. On day 3, cells were refed with medium A. On day 4, cells were treated with fresh medium B with or without 2% (wt/vol) HPCD for 1 h at 37 °C, after which the cells were washed five times as described in Materials and Methods and then incubated at 4 °C for the indicated time with 2 mL of ice-cold buffer E containing either 100 μg/mL 125I-PFO* (6.5 × 103 cpm/μg protein) (A) or for 2 h with the indicated concentration of 125I-PFO* (10 × 103cpm/μg) (B). The total amount of 125I- PFO* bound to the cells was determined as described in Materials and Methods. Each value represents the average of duplicate incubations. The values (mean ± SEM) for total cellular protein content did not differ significantly in cells treated with or without HPCD (0.59 ± 0.02 and 0.56 ± 0.02 mg per dish, respectively).
To test the sterol specificity of the 125I-PFO* binding reaction, we depleted cells of cholesterol by incubating them in lipoprotein-deficient serum together with compactin, a 3-hydroxy-3-methyl-glutaryl-CoA (HMG CoA) reductase inhibitor that blocks cholesterol synthesis. We included a small amount of mevalonate to maintain the supply of nonsterol products of HMG CoA reductase (21). The cells were then incubated for 3 h with a variety of sterols (complexed with methyl-β-cyclodextrin, MCD), after which the cells were chilled to 4 °C and 125I-PFO* binding was measured (Fig. 3). In the absence of sterols, 125I-PFO* binding was low; it rose progressively when increasing concentrations of cholesterol or desmosterol were added. There was no increase when epicholesterol or 25-hydroxycholesterol was added. This binding specificity matches the known specificity of PFO (11, 19).
Fig. 3.

125I-PFO* binding to SV-589 cells after prior treatment with different sterols. On day 0, cells were set up in medium A at 1 × 105 cells per 60-mm dish. On day 2, cells were switched to lipoprotein-deficient medium C. On day 3, cells were refed with medium C containing 10 μM compactin and 50 μM sodium mevalonate and then incubated for 16 h at 37 °C. On day 4, each dish received fresh medium C containing 10 μM compactin, 50 μM sodium mevalonate, and varying concentrations of the indicated sterol complexed with MCD (sterol/MCD molar ratio of 1:12). After incubation for 3 h at 37 °C, the cells were washed five times as described in Materials and Methods and then incubated with 2 mL ice-cold buffer E containing 5 μg/mL 125I-PFO* (30 × 103 cpm/μg) for 30 min at 4 °C. The total amount of cell surface 125I-PFO* binding was determined as described in Materials and Methods. Each value represents the average of duplicate incubations.
Fig. 4 shows an experiment designed to test the ability of 125I-PFO* binding to monitor the receptor-mediated delivery of LDL-derived cholesterol from lysosomes to the plasma membrane. Fibroblasts were depleted of cholesterol by culturing the cells with compactin in lipoprotein-deficient serum and were then incubated with LDL, after which 125I-PFO* binding was measured. In the absence of LDL, 125I-PFO* binding was low. Binding rose progressively when the cells had been incubated with LDL for varying times (Fig. 4A). This increase was blocked when LDL was added in the presence of chloroquine, which is known to block LDL degradation in lysosomes, leading to a buildup of cholesteryl esters within this organelle (22). The delivery of LDL-derived cholesterol to the plasma membrane was dependent on the concentration of LDL with saturation kinetics that mimic the saturation curve for LDL binding to the LDL receptor (Fig. 4B) (3). Again, this delivery was blocked by chloroquine. As a control for the specificity of the chloroquine effect, we added cholesterol delivered in MCD, which enters the plasma membrane without a requirement for endocytosis or lysosomal degradation. Chloroquine did not block the increase in 125I-PFO* binding when cholesterol was delivered in MCD (Fig. 4C). Moreover, we observed that compounds known to block lysosomal processing of LDL-derived cholesterol, such as U18666A and imipramine (23), abolished the cell surface binding of 125I-PFO* to SV-589 cells incubated with LDL (Fig. S2).
Fig. 4.

Movement of LDL-derived cholesterol from lysosomes to plasma membrane as determined by cell surface 125I-PFO* binding. On day 0, SV-589 cells were set up in medium A at 1 × 105 cells per 60-mm dish. On day 2, cells were switched to lipoprotein-deficient medium C. On day 3, cells were refed with medium C containing 50 μM compactin and 50 μM sodium mevalonate and incubated for 16 h at 37 °C. On day 4, cells received fresh medium E containing 50 μM compactin, 50 μM mevalonate, and one of the following treatments in the absence or presence of 100 μM chloroquine: (A) 50 μg protein/mL LDL for the indicated time; (B) the indicated concentration of LDL for 5 h; or (C) the indicated concentration of cholesterol/MCD complex. After incubation for 5 h at 37 °C, the cells were washed five times as described in Materials and Methods and then incubated with 2 mL of ice-cold buffer E containing 25 μg/mL 125I-PFO* (6 × 103 cpm/μg). After 2 h at 4 °C, the total amount of cell surface binding of 125I-PFO* was determined. Each value represents the average of duplicate incubations.
As a further test for the role of the LDL receptor pathway in delivery of cholesterol to the plasma membrane, we used cells from subjects with mutations that are known to disrupt this pathway. Mutations in the LDL receptor, as seen in homozygous familial hypercholesterolemia (FH), prevent receptor-mediated LDL uptake into cells (3). Mutations in lysosomal acid lipase, as seen in Wolman syndrome, permit LDL uptake but lead to the accumulation of LDL-derived cholesteryl esters in lysosomes (24). Mutations in Niemann-Pick C1 (NPC1) permit the lysosomal hydrolysis of LDL-derived cholesteryl esters, but they block the egress of unesterified cholesterol from lysosomes (25, 26). Compared with fibroblasts from a normal subject, fibroblasts from subjects with any of these three mutations showed a marked reduction in 125I-PFO* binding after incubation with LDL (Fig. 5A). None of these mutant fibroblasts showed a reduction in 125I-PFO* binding when cholesterol was added in complex with MCD (Fig. 5B).
Fig. 5.

Reduced delivery of LDL-derived cholesterol to the cell surface of fibroblasts from subjects with NPC1 disease, Wolman disease, and homozygous FH. On day 0, diploid fibroblasts from the indicated subject were set up in medium A at 2 × 104 (control), 4 × 104 (NPC1 and Wolman), and 8 × 104 (FH) cells per 60-mm dish, respectively. On day 3, cells were refed with the same medium. On day 5, cells were switched to lipoprotein-deficient medium D. On day 6, cells were refed with medium D containing 50 μM compactin and 50 μM sodium mevalonate. On day 7, each dish was refed with medium F containing 50 μM compactin, 50 μM mevalonate, and the indicated amount of LDL (A) or 50 μM cholesterol/MCD complex (B). After 5 h at 37 °C, each monolayer was washed 5 times as described in Materials and Methods and then incubated with 2 mL ice-cold buffer E containing 5 μg/mL 125I-PFO* (23 × 103 cpm/μg). After 30 min at 4 °C, the total amount of cell surface 125I-PFO* binding was determined. (A and B) Each value represents the average of duplicate incubations.
The data thus far indicate that 125I-PFO* binding bears some relation to the concentration of cholesterol in the plasma membrane. Previous studies have shown that PFO binding to native and artificial membranes does not occur unless a critical concentration of cholesterol is reached (11, 19). To determine whether 125I-PFO* binding to intact cells also shows a threshold-like response, we treated cells with varying concentrations of HPCD to deplete membrane cholesterol to varying extents. We measured 125I-PFO* binding to one set of intact cells. A parallel set of cells was incubated with a chemical reagent that biotinylates plasma membrane proteins. The cells were then homogenized, and plasma membranes were purified by adherence to streptavidin beads. The content of cholesterol and phospholipid in the purified plasma membranes was determined as described in Materials and Methods and compared with the binding of 125I-PFO* to the intact cells. Fig. 6A shows that the biotinylated plasma membrane fraction was pure as judged by the absence of immunoblottable markers for lysosomes, the ER, Golgi, mitochondria, peroxisomes, or endosomes. We noted that the plasma membrane markers Na+/K+ATPase and flotillin 1 showed altered mobility after elution from the streptavidin beads. This finding is attributable to the effects of the harsh urea-containing buffer that was necessary for elution (see the control experiment in Fig. S3).
Fig. 6.
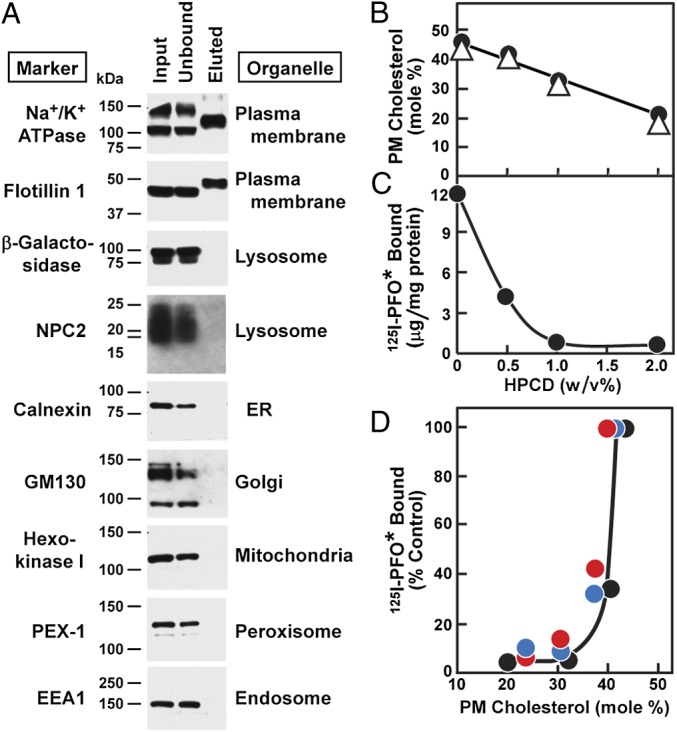
Relation between cholesterol content of plasma membrane and amount of 125I-PFO* bound to cell surface of SV-589 cells. (A) On day 0, cells were set up in medium A at 4 × 105 cells per 100-mm dish. On day 3, cells were refed with medium B. On day 4, plasma membranes were purified as described in Materials and Methods. Equal volumes of the input, unbound fraction, and bound (eluted) fraction were subjected to 8 or 15% SDS/PAGE and immunoblotted with antibodies against the indicated organelle marker. Filters were exposed to film at room temperature for 5–60 s. (B) On day 0, cells were set up in medium A at 1 × 105 cells per 60-mm dish. On day 3, cells were refed with medium B. On day 4, groups of 12 dishes of cells were each treated with the indicated concentration of HPCD in medium B for 1 h at 37 °C, after which the cells were washed five times as described in Materials and Methods. Each group of 12 dishes was then divided into two sets of six dishes. The cells from each six-dish set were pooled together and processed for purification of plasma membranes. Lipids were extracted from the plasma membranes, and the content of unesterified cholesterol and choline-containing phospholipids was measured. Values for total phospholipids were calculated from measurements of choline-containing phospholipids as described in Materials and Methods. Each value represents the average of duplicate measurements for each of the two pooled sets, each denoted by a different symbol (●, △). (C) Cells were cultured under identical condition as described in B. On day 4, after treatment with HPCD and washing, each cell monolayer was incubated with 2 mL of ice-cold buffer E containing 25 μg/mL 125I-PFO* (19 × 103 cpm/μg). After 2 h at 4 °C, the total amount of cell surface 125I-PFO* binding was determined. Each value represents the average of duplicate incubations. (D) Graph showing the amount of cell surface 125I-PFO* binding plotted as a function of the unesterified cholesterol content of the plasma membrane for three independent experiments (denoted by red, blue, and black circles), one of which (●) was done in parallel with B and C.
Fig. 6B shows that HPCD treatment over the entire concentration range of 0–2% lowered the concentration of plasma membrane cholesterol in a linear fashion as measured in isolated plasma membranes. In stark contrast, 125I-PFO* binding to the plasma membranes of intact cells declined nearly to zero at 1% HPCD, and there was no further decline at 2% HPCD (Fig. 6C). Fig. 6D plots 125I-PFO* binding to intact cells as a function of the plasma membrane cholesterol concentration (expressed as mole% of total plasma membrane lipids). Binding was undetectable until the plasma membrane cholesterol exceeded a threshold of ∼35 mol%.
Discussion
In the present studies, we report the use of a sensitive probe that measures the accessibility of cholesterol in the plasma membrane of living cells. This probe is a radioiodinated version of the bacterial cholesterol-binding protein PFO. In preparing this probe, we inserted a point mutation (tyrosine-181 to alanine) that was shown previously to retain cholesterol binding to liposomes, but to eliminate pore formation (18, 19). The probe, which we named 125I-PFO*, binds to the cell surface of human and animal cells at 4 °C in a cholesterol-dependent manner without detectable pore formation.
We found that 125I-PFO* binding was low when cells were depleted of cholesterol by incubation with HPCD or by incubation in lipoprotein-deficient serum together with compactin to block cholesterol synthesis. 125I-PFO* binding was restored by incubating the cells with cholesterol delivered with MCD or with LDL. The data of Fig. 5 show that in order for LDL-cholesterol to reach the plasma membrane, the LDL had to undergo receptor-mediated endocytosis, as indicated by the failure of this response in homozygous FH fibroblasts. LDL-cholesterol delivery also required lysosomal hydrolysis of its cholesteryl esters, as indicated by the delivery failure in cells from a patient with Wolman syndrome. Finally, the LDL-derived cholesterol had to exit the lysosomes, as revealed by the failure to see an increase in plasma membrane cholesterol when LDL was added to cells with a mutation in NPC1 protein. The role of the lysosomes was also supported by the failure of LDL to increase plasma membrane cholesterol normally when cells were treated with the lysosomal inhibitor chloroquine (Fig. 4).
A striking finding emerged when 125I-PFO* binding to intact cells was correlated with the chemically measured cholesterol content of isolated plasma membranes (Fig. 6). When the cells were treated with increasing concentrations of HPCD, the cholesterol content of plasma membranes declined linearly with the HPCD concentration (Fig. 6B). In contrast, 125I-PFO* binding declined precipitously, reaching its lowest point when cholesterol still represented 35 mol% of plasma membrane lipids (Fig. 6D). The ∼35% threshold concentration of PFO binding to plasma membranes is similar to that seen with simple binary liposomes containing phosphatidylcholine and cholesterol (11) (Fig. S1). The ∼35% threshold for the plasma membrane is in sharp contrast to the findings with ER membranes. When PFO was incubated with ER membranes, the threshold for cholesterol binding was in the range of 5% (11). This finding was similar to the ∼5% threshold that was seen when the cholesterol content of ER membranes was varied in intact cells and the activity of the intrinsic ER cholesterol-binding protein Scap was studied (11, 12). Scap activity, as reflected by ER-to-Golgi transport, was decreased sharply when the cholesterol content of ER membranes rose above 5%.
Considered together, these results indicate that cholesterol is much less accessible when it is contained in plasma membranes compared with ER membranes. It should be noted that PFO and PFO* are monomers in solution, but they form large oligomers upon binding to cholesterol-containing membranes (18). One could argue that positive cooperativity arising from this oligomerization could account for the threshold-like response to cell membranes. However, the striking difference between plasma membranes and ER membranes (35% vs. 5%) is more likely to be caused by differences in the phospholipid composition of the two membranes. Previous studies with model membranes have revealed that the accessibility of cholesterol to PFO depends on the type and structure of the acyl chain of the phospholipids (11, 19, 27). In general, saturated acyl chains make cholesterol less accessible. Whether the difference between ER and plasma membranes relates to acyl chains or to some other characteristic of the membrane will need to be the subject of further study.
Materials and Methods
Materials.
We obtained cholesterol and 25-hydroxycholesterol from Steraloids; MCD and HPCD from Cyclodextrin Technologies Development; Insulin-Transferrin-Selenium and Dulbecco’s PBS from Life Technologies; 125I-NaI from Perkin-Elmer; DMEM (Cat. No. D6064), l-glutamine-free DMEM (Cat. No. D5546), FCS, newborn calf serum, NADH, sodium pyruvate, BSA, and biotin from Sigma; Sulfo-NHS-S S-Biotin and Pierce Iodination Tubes from Thermo Scientific; PD-10 columns from GE Healthcare; NanoLink Streptavidin Magnetic Beads (0.8 μm) from Solulink. Human LDL (d < 1.019–1.063 g/mL), human or newborn calf lipoprotein-deficient serum (LPDS, d < 1.215 g/mL) were prepared by ultracentrifugation, as described previously (28). Solutions of compactin and sodium mevalonate were prepared as described previously (29). Stock solutions of various sterols/MCD complexes were prepared at a final sterol concentration of 2.5 mM and a sterol/MCD molar ratio of 1:12 (15).
Buffers.
Buffer A contains 50 mM Tris⋅HCl (pH 7.5) and 150 mM NaCl. Buffer B contains 25 mM Hepes-KOH (pH 7.4), 150 mM NaCl, and 2 mM CaCl2. Buffer C is buffer A supplemented with 100 mM glycine. Buffer D contains 50 mM Tris⋅HCl (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, and 250 mM sucrose. Buffer E contains 25 mM Hepes-KOH (pH 7.4), 150 mM NaCl, and 0.2% (wt/vol) BSA. Buffer F contains 50 mM Tris⋅HCl (pH 7.5) and 5 µM biotin. Buffer G contains 50 mM Tris⋅HCl (pH 7.5), 150 mM NaCl, 3 mM biotin, 6 M urea, 2 M thiourea, and 2% (wt/vol) SDS. Where indicated, buffers were supplemented with a protease inhibitor mixture containing 50 μg/mL leupeptin, 25 μg/mL pepstatin A, 10 μg/mL aprotinin, and 25 μg/mL N-acetyl-l-leucyl-l-norleucinal.
Culture Media.
Medium A is DMEM containing 100 units/mL of penicillin, 100 μg/mL streptomycin sulfate, and 10% (vol/vol) FCS. Medium B is DMEM containing 100 units/mL penicillin, 100 μg/mL streptomycin sulfate, and 1% (vol/vol) Insulin-Transferrin-Selenium. Medium C is DMEM containing 100 units/mL penicillin, 100 μg/mL streptomycin sulfate, and 5% (vol/vol) newborn calf LPDS. Medium D is DMEM containing 100 units/mL penicillin, and 100 μg/mL streptomycin sulfate, and 10% (vol/vol) human LPDS. Medium E and medium F are identical to medium C and medium D, respectively, except that medium E and medium F contain l-glutamine-free DMEM.
Cell Culture.
Stock cultures of human SV-589 fibroblasts (30) and human diploid fibroblasts were grown in monolayer at 37 °C in a 5% CO2 incubator and maintained in medium A. FH fibroblasts were derived from a patient with receptor-negative homozygous FH (31). NPC1 and Wolman fibroblasts were derived from a patient with NPC1 disease (American Type Culture Collection; GM3123) and Wolman disease (24), respectively.
Purification of PFO*.
The cDNA encoding a cysteine-less (C459A), fully active derivative of PFO with an NH2-terminal hexahistidine tag (10), hereafter referred to as PFO, was obtained from Arthur E. Johnson, Texas A&M University, College Station, TX. This PFO cDNA, cloned into a pRSET B vector (Life Technologies), was subjected to site-directed mutagenesis, changing tyrosine-181 to alanine. This version of PFO is not cytotoxic at 4 °C (Fig. 1) and is referred to as PFO*. The PFO* plasmid was expressed in E. coli, after which the cell pellet was lysed with a French pressure cell. The protein was then purified on a HisTrap HP column packed with nickel-Sepharose 6 beads (GE Healthcare) as described by Sokolov and Radhakrishnan (11). The fractions containing purified PFO* protein were pooled, concentrated, and stored at 4 °C until use. Typical yield was 8–10 mg protein/liter of culture medium.
Iodination of PFO*.
Protein iodination was performed using precoated iodination tubes according to the manufacturer’s protocol. Briefly, 2–2.5 mg of purified PFO* was added to an IODO-GEN precoated tube preequilibrated with buffer A, after which 2 mCi of 125I-NaI was added in a final volume of 500 µL. The tubes were incubated for 15 min at room temperature, after which 2 mL of buffer A was added and the solution was then passed through a PD-10 column preequilibrated with buffer A. Fractions containing 125I-labeled PFO*, were pooled and stored at 4 °C until use; cholesterol binding activity was maintained for at least 3 wk. The specific activity of 125I-PFO* ranged from 50 to 200 cpm/ng. More than 90% of the 125I-radioactivity in 125I-PFO* was precipitable after incubation with 10% (vol/vol) trichloroacetic acid.
Plasma Membrane Purification.
Monolayers of SV-589 cells were washed twice at 4 °C with ice-cold PBS, followed by a 15-min incubation with PBS containing 1 mM MgCl2 and 0.1 mM CaCl2 after which buffer B containing 0.5 mg/mL sulfo-NHS-S S-biotin was added to each monolayer, and the dishes were shaken for 1 h at 4 °C. The biotinylation reaction was stopped by removing buffer B followed by addition of buffer C for 15 min at 4 °C. Cells were then scraped in buffer A and centrifuged at 1,000 × g for 10 min at 4 °C. The resulting pellet was resuspended in buffer D supplemented with protease inhibitor mixture. Cells were lysed by passage 13 times through a ball-bearing homogenizer with a 10-μm clearance (12). The homogenate was centrifuged at 3,000 × g for 10 min at 4 °C, after which the supernatant was incubated for 60 min at room temperature with streptavidin magnetic beads that were preequilibrated with buffer E. The beads were isolated by adherence to the tube wall in a magnetic field, after which the beads were washed for 10 min with buffer D, followed by seven 10-min washes with buffer F supplemented with protease inhibitor mixture, followed by one quick wash with PBS. The beads were then demagnetized, solubilized in PBS, and stored at 4 °C for up to 24 h before use. After removing the PBS, the proteins were eluted from the streptavidin beads by incubating the beads at room temperature with SDS-containing buffer G for 15 min followed by heating at 95 °C for an additional 15 min. Enrichment for plasma membrane proteins was assessed by immunoblotting.
Lipid Measurements.
Lipids were extracted with hexane/isopropanol (vol/vol, 3:2) from washed plasma membranes attached to streptavidin magnetic beads. The organic solvent containing the extracted lipids was evaporated under a gentle stream of nitrogen and used for measurement of unesterified cholesterol (Amplex Red Cholesterol Assay Kit; Life Technologies) and choline-containing phospholipids (EnzyChromTM Phospholipid Assay Kit; BioAssay Systems). In the experiments of Fig. 6, we estimated total plasma membrane phospholipids by measuring the content of choline, which is present on only some of the phospholipids (phosphatidylcholine and sphingomyelin). To convert the choline measurements to a measurement of total phospholipids, we conducted another experiment in which we treated SV-589 cells with and without 1% HPCD. We purified plasma membranes by surface biotinylation, extracted the lipids, and measured the mass of choline as described above. Another aliquot was treated with percholoric acid, and phosphate was measured (PiPer Phosphate Assay Kit, Invitrogen). We found that the ratio of total lipid phosphorus:choline was 1.47 in PMs without HPCD treatment and 1.59 in PMs from cells after HPCD treatment. Therefore, to convert choline content to total phospholipids we multiplied the choline content by the average of these two values, i.e., 1.53. The content of cholesterol is expressed on a molar basis as “percentage of total lipids” (i.e., moles of unesterified cholesterol divided by moles of total lipids multiplied by 100). Total lipids are defined as the measured moles of unesterified cholesterol plus phospholipid. We used 387 Da and 800 Da for the molecular masses of cholesterol and phospholipid, respectively.
125I-PFO* Binding to Surface of Cultured Cells.
Before addition of 125I-PFO*, cells were washed as follows to remove surface-bound lipoproteins or HPCD: three rapid washes with buffer E at room temperature, followed by two 10-min washes with the ice-cold buffer E in a 4 °C cold room. After these five washes, each 60-mm dish of cells was incubated at 4 °C with 2 mL of buffer E containing 125I-PFO* as described in the figure legends. After the indicated time, cell monolayers were washed rapidly three times with ice-cold PBS, dissolved with 1 mL of 0.1 N NaOH, and shaken on a rotary shaker for 15 min at room temperature. Aliquots of the dissolved cells were removed for scintillation counting in a gamma counter (500 μL) and for measurement of protein concentration (50 μL) (32). The data are expressed as μg 125I- PFO* bound per milligram of cell protein.
Reproducibility.
All results were confirmed in three to five independent experiments.
Other Methods.
Additional information for antibodies, cell viability assay, and immunoblot analysis can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank our colleagues Lina Abi-Mosleh and the late Y. K. Ho for invaluable suggestions and help in establishing the Perfringolysin O iodination protocol; Lauren Ziemian and Dorothy Williams for excellent technical assistance; Nimi Jacob and Ijeoma Onwuneme for invaluable help with tissue culture; Linda Donnelly and Angela Carroll for generating monoclonal Niemann-Pick C2 antibody; and Arthur Johnson for generously providing the parent Perfringolysin O plasmid. This work was supported by National Institutes of Health Grant HL20948 (to J.L.G. and M.S.B.), Welch Foundation Grant I-1793 (to A.R.), and American Heart Association Grant 12SDG12040267 (to A.R.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309273110/-/DCSupplemental.
References
- 1.De Duve C. Tissue fractionation. Past and present. J Cell Biol. 1971;50(1):20d–55d. [PubMed] [Google Scholar]
- 2.Lange Y, Swaisgood MH, Ramos BV, Steck TL. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J Biol Chem. 1989;264(7):3786–3793. [PubMed] [Google Scholar]
- 3.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 4.Lange Y, Ye J, Steck TL. Circulation of cholesterol between lysosomes and the plasma membrane. J Biol Chem. 1998;273(30):18915–18922. doi: 10.1074/jbc.273.30.18915. [DOI] [PubMed] [Google Scholar]
- 5.Liscum L, Munn NJ. Intracellular cholesterol transport. Biochim Biophys Acta. 1999;1438(1):19–37. doi: 10.1016/s1388-1981(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 6.Lange Y. Tracking cell cholesterol with cholesterol oxidase. J Lipid Res. 1992;33(3):315–321. [PubMed] [Google Scholar]
- 7.Underwood KW, Jacobs NL, Howley A, Liscum L. Evidence for a cholesterol transport pathway from lysosomes to endoplasmic reticulum that is independent of the plasma membrane. J Biol Chem. 1998;273(7):4266–4274. doi: 10.1074/jbc.273.7.4266. [DOI] [PubMed] [Google Scholar]
- 8.Gimpl G, Gehrig-Burger K. Probes for studying cholesterol binding and cell biology. Steroids. 2011;76(3):216–231. doi: 10.1016/j.steroids.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Ohno-Iwashita Y, et al. Cholesterol-binding toxins and anti-cholesterol antibodies as structural probes for cholesterol localization. Subcell Biochem. 2010;51:597–621. doi: 10.1007/978-90-481-8622-8_22. [DOI] [PubMed] [Google Scholar]
- 10.Flanagan JJ, Tweten RK, Johnson AE, Heuck AP. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry. 2009;48(18):3977–3987. doi: 10.1021/bi9002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sokolov A, Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J Biol Chem. 2010;285(38):29480–29490. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008;8(6):512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown MS, Goldstein JL. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 14.Yang T, et al. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 15.Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10(2):237–245. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- 16.Shimada Y, Maruya M, Iwashita S, Ohno-Iwashita Y. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur J Biochem. 2002;269(24):6195–6203. doi: 10.1046/j.1432-1033.2002.03338.x. [DOI] [PubMed] [Google Scholar]
- 17.Ishitsuka R, Saito T, Osada H, Ohno-Iwashita Y, Kobayashi T. Fluorescence image screening for chemical compounds modifying cholesterol metabolism and distribution. J Lipid Res. 2011;52(11):2084–2094. doi: 10.1194/jlr.D018184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotze EM, et al. Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J Biol Chem. 2002;277(13):11597–11605. doi: 10.1074/jbc.M111039200. [DOI] [PubMed] [Google Scholar]
- 19.Nelson LD, Johnson AE, London E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: Insights into the origin of perfringolysin O-lipid raft interaction. J Biol Chem. 2008;283(8):4632–4642. doi: 10.1074/jbc.M709483200. [DOI] [PubMed] [Google Scholar]
- 20.Kilsdonk EPC, et al. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270(29):17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein JL, Brunschede GY, Brown MS. Inhibition of proteolytic degradation of low density lipoprotein in human fibroblasts by chloroquine, concanavalin A, and Triton WR 1339. J Biol Chem. 1975;250(19):7854–7862. [PubMed] [Google Scholar]
- 23.Underwood KW, Andemariam B, McWilliams GL, Liscum L. Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J Lipid Res. 1996;37(7):1556–1568. [PubMed] [Google Scholar]
- 24.Brown MS, Sobhani MK, Brunschede GY, Goldstein JL. Restoration of a regulatory response to low density lipoprotein in acid lipase-deficient human fibroblasts. J Biol Chem. 1976;251(11):3277–3286. [PubMed] [Google Scholar]
- 25.Pentchev PG, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci USA. 1985;82(23):8247–8251. doi: 10.1073/pnas.82.23.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J Cell Biol. 1989;108(5):1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wattenberg BW, Silbert DF. Sterol partitioning among intracellular membranes. Testing a model for cellular sterol distribution. J Biol Chem. 1983;258(4):2284–2289. [PubMed] [Google Scholar]
- 28.Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 29.Brown MS, Faust JR, Goldstein JL, Kaneko I, Endo A. Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem. 1978;253(4):1121–1128. [PubMed] [Google Scholar]
- 30.Yamamoto T, et al. The human LDL receptor: A cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39(1):27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259–289. doi: 10.1146/annurev.ge.13.120179.001355. [DOI] [PubMed] [Google Scholar]
- 32.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.