

Abstract
Activating mutations in the epidermal growth factor receptor (EGFR) tyrosine kinase are frequently found in many cancers. It has been suggested that changes in the equilibrium between its active and inactive conformations are linked to its oncogenic potential. Here, we quantify the effects of some of the most common single (L858R and T790M) and double (T790M-L858R) oncogenic mutations on the conformational free-energy landscape of the EGFR kinase domain by using massive molecular dynamics simulations together with parallel tempering, metadynamics, and one of the best force-fields available. Whereas the wild-type EGFR catalytic domain monomer is mostly found in an inactive conformation, our results show a clear shift toward the active conformation for all of the mutants. The L858R mutation stabilizes the active conformation at the expense of the inactive conformation and rigidifies the αC-helix. The T790M gatekeeper mutant favors activation by stabilizing a hydrophobic cluster. Finally, T790M with L858R shows a significant positive epistasis effect. This combination not only stabilizes the active conformation, but in nontrivial ways changes the free-energy landscape lowering the transition barriers.
Keywords: conformational changes, resistance-causing mutations
The epidermal growth factor receptor (EGFR) is one of the most studied members of the receptor tyrosine kinases family due to its central role in many cellular pathways and its importance as a drug target (1). Activating mutations of EGFR are common in human malignancies (2), where they are implicated in the cellular survival, proliferation, and migration, and are the oncogenic drivers in glioblastoma and nonsmall-cell lung cancer (NSCLC) (3, 4).
Structurally, EGFR is composed of a cell-surface receptor, a transmembrane region, and an intracellular catalytic domain. The activation of the enzyme is triggered by binding of the EGF ligand on the extracellular domain, which induces dimerization of the receptors (5). During activation, an asymmetric dimer is formed in which the C lobe of the catalytic domain playing the “activator” interacts with the N lobe of the “receiver” domain, promoting and stabilizing the active conformation of the latter (6–8). The mechanism of activation bears some resemblance to that of cyclin-dependent kinase 2 (CDK2), which is activated by cyclin A (9). The catalytic domain (CD) of EGFR assumes a fold that is common to most protein kinases (10). It consists of a smaller N-terminal lobe, containing mostly β-sheets, and an α-helical C-terminal lobe. The ATP binding cleft is at the interface between the lobes. Kinase activation is mainly controlled by conformational changes in three conserved structural motifs close to the active site: the activation loop (A loop), the Asp-Phe-Gly (DFG) motif, and the αC-helix (Fig. 1). The active state conformation is characterized by an extended conformation of the A loop and the presence of a hairpin in its N-terminal region. The αC-helix assumes an “in” conformation, which is kept in place by a salt bridge formed by E762 and K745 (Fig. 1). Crystal structures of EGFR have revealed an inactive conformation similar to the one observed in Src tyrosine kinase (“Src-like” inactive conformation), in which a short α-helix is formed in the A loop, making an autoinhibitory interaction with a shifted αC-helix (6, 11).
Fig. 1.

Comparison of active (Left) and Src-like inactive (Right) structures of EGFR CD. Key structural elements are colored in red (αC helix), green (P-loop) and yellow (A-loop). Also shown as sticks is the DFG motif and the residues either mutated (L858, T790) or participating in salt-bridge interactions and used to define a collective variable (K745, D855, E762).
Many somatic mutations of the CD of EGFR have clinical importance due to their activating or drug-resistance effects. The L858R mutation (also referred to as L834R; see Materials and Methods for residue numbering conventions) is the most common oncogenic mutation in the EGFR kinase domain (12, 13) and one of the most observed kinase mutations in human cancers (2, 14). It is found in a significant subset of patients with NSCLC (4, 15–17). L858R is activating; the mutant shows a 20–50-fold increase in the catalytic efficiency with respect to wild-type (WT) EGFR (6, 18). Two contrasting activation mechanisms have been proposed for this mutation. The first, based on the comparison of WT and mutant crystal structures, posits that the L858R mutation locks the kinase in the active state because it prevents residue 858, and flanking residues, from forming the helical conformation typical of the inactive state (18). The second mechanism, based on observations of microsecond-long molecular dynamics trajectories, proposes that L858R acts by reducing the intrinsic disorder in the αC-helix located in the N lobe of the “receiver” kinase. This in turn would favor dimerization and indirectly stabilize the active conformation (19).
Two L858R-selective, ATP-competitive tyrosine kinase inhibitors (TKIs), erlotinib and gefitinib (16, 20), are in clinical use (18). In the case of gefitinib, its selectivity for the mutant might be the consequence of increased affinity of the drug, which binds to an active-like conformation, combined with decreased affinity of ATP (21, 22). However, the conformational selectivity of EGFR inhibitors has been recently questioned, when erlotinib was found to also bind to the inactive EGFR CD conformation (23).
An additional problem is the emergence of drug-resistance mutations. One of the most frequent resistance mutations is the T790M, in which the so-called “gatekeeper” threonine at position 790 is mutated to methionine (17, 24). This mutation was first thought to reduce the affinity to the drugs due to a sterical clash in the drug binding site (17). However, Yun et al. showed that both the single-mutant T790M and the double-mutant L858R–T790M maintain the same low nanomolar affinity to gefitinib as the L858R mutant does (21). In contrast, the T790M mutation confers a higher affinity to ATP than L858R mutant so that the combined mutation L858R–T790M results in an activated enzyme resistant to ATP-competitive TKIs (25). What is more, there are several lines of evidence showing that L858R and T790M have a positive epistasis effect (14).
Although the biological effects of these important mutations are clear, a mechanistic explanation linking the mutations to changes in the conformational free-energy landscape is still missing. Thanks to the advances in force fields (26) and the use of specialized computer architectures (27) or enhanced sampling methods (28, 29), it is now possible to use all-atom molecular dynamics (MD) simulations to accurately describe the complex conformational transitions involved in kinase activation. With standard MD and specialized computer hardware, events on the microsecond (at most millisecond) timescale can be sampled, without the need of any a priori knowledge of the relevant reaction coordinates (19). Thus, they are useful to observe unexpected events, but cannot be used to obtain statistically converged populations of high-in-energy secondary conformations. With enhanced sampling methods, such as metadynamics (29, 30), there is no limit on the timescales that can be sampled, but some a priori knowledge of the reaction coordinates is usually needed (30). This need is less stringent when enhanced sampling methods are combined with a multiple-replica approach, as in the case of the parallel-tempering metadynamics simulations approach (PT-metaD) (28, 31). Indeed, it has been shown that PT-metaD is able to converge very complex conformational free-energy surfaces of kinases as a function of a few relevant variables (32, 33).
To elucidate the structural and dynamical consequences of the L858R and T790M mutations on the catalytic domain of EGFR and their synergistic interaction, here we performed extensive PT-metaD simulations of WT-EGFR, three oncogenic mutants: L858R, T790M, and T790M–L858R, and a control mutant, L858I. The total sampling time for each system was at least 3.7 μs, leading to well-converged free-energy surfaces.
Results
We have obtained converged free-energy surfaces (FESs) of the activation of the EGFR catalytic domain and of three EGFR oncogenic mutants (Fig. 2 and Fig. S1). Converged conformational FESs have been obtained for these systems with a fully atomistic explicit-solvent force field. In Fig. 2 we show the FES projected along the first two collective variables (CVs), CV1 and CV2, for the four systems. CV1 is defined as the difference between the distances of the two salt-bridge–forming couples: K745:E762 and K745:D855. When both salt bridges are formed, CV1 is close to 0. This CV is able to characterize the displacement of the functionally important glutamic acid E762 located in the αC-helix of the N lobe in its transition from the so-called “αC-in” to the “αC-out” conformation. The use of CV2 allows for a broad characterization of the A-loop conformations (Materials and Methods). It measures the displacement of the heavy atoms of the A loop from the Src-like inactive conformation (Fig. 1, Right). When its value is close to 0, the A loop assumes a conformation similar to that found in the Src-like inactive structure. Values larger than 0.7 correspond to open A-loop conformations as in the active state. In-between values of 0.4–0.5 are also observed and will be referred to as “semiclosed” conformations.
Fig. 2.
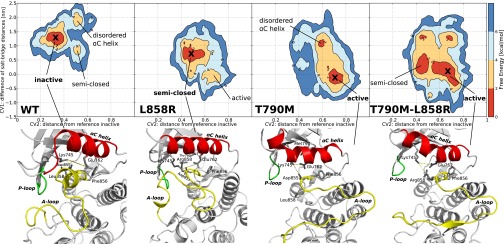
Free energy surface of wild-type EGFR and the three mutants as a function of CV1 (y axis) in nm and CV2 (x axis) in arbitrary units. A cross indicates the global free energy minimum for which a representative structure is shown below. Each minimum is also named according to the corresponding main feature. The contour lines are drawn every 2 kcal/mol. In the structures below the free energy surfaces the αC-helix is shown in red, the A-loop in yellow and the P-loop in green. The distances between K745:E762 and K745:D855, whose difference constitute CV1, are displayed with a dashed yellow line.
WT.
The deepest free-energy minimum of WT-EGFR (Fig. 2) corresponds to the autoinhibited conformation where the A loop is folded and forms a short helix at its N-terminal end and the E762 residue of the αC-helix is rotated out of the catalytic site (αC-out) as in the inactive Src-like state (Fig. 1). This inactive conformation is found both in Src-family kinases (34) and cyclin-dependent kinases (CDKs) (35). Notably, there are other less-populated states, with higher free energy (2–4 kcal/mol), corresponding to inactive structures (αC-out) where the αC-helix is deformed and the A loop is semiclosed. The disordered αC-helix basin is characterized by a large deformation of the αC-helix in which only two turns are maintained, whereas the others are disordered (Fig. S2). In the semiclosed basin the αG-helix is surprisingly far from its usual position and interacts both with the P-loop and the C-terminal region of the A loop (Fig. S2). The ensemble of states found in this basin seems to correspond to the intermediate “extended” structures described in ref. 36. The transition from the semiclosed to the inactive state proceeds by local unfolding or “cracking” (37). Finally, in agreement with previous proposals (19, 36), we find that the active conformation of WT-EGFR, in the absence of ATP, is energetically unfavorable and infrequently sampled.
In contrast to the WT, all of the mutants populate to some extent the active conformation. Moreover, the investigated oncogenic mutations alter the free-energy landscape in a global way. Surprisingly, they generate new minima in different locations of the FES and alter the barriers between the minima.
L858R Mutant.
The L858R mutation changes the conformational free-energy landscape. The disordered states seen in the case of WT-EGFR have disappeared. The most stable state of L858R corresponds to an ensemble of structures which are in between the active and inactive conformation (Fig. 2). The glutamic acid 762 is on average 0.5 nm closer to the catalytic site compared with WT-EGFR, but the salt bridge with lysine 745 is never formed as in the fully active conformation. The A loop assumes a semiclosed conformation where neither the short helix nor the hairpin at the N terminal end is stable. The observed changes are mainly due to the interactions that the positively charged R858 engages with the three negatively charged residues of the αC-helix. As shown in Fig. 3, R858 makes a salt bridge with the negatively charged glutamic acids 758 and 762 or with the aspartic acid 761, strongly stabilizing the αC-helix. At the same time, these favorable interactions prevent the formation of the short helical turn at the N-terminal region of the A loop (residues 858–862) that is typical of the inactive conformations (18).
Fig. 3.

Close-up of two possible conformations of the L858R mutant in its main free energy minimum. The R858 forms salt bridge interactions with either A) D761 and E762 or B) E758 and E762.
In contrast with WT-EGFR, the fully active state is populated (L858R, Fig. 2), albeit it has a thermodynamic penalty (2–4 kcal/mol) with respect to the inactive state. At variance with the structures of the active L858R mutant crystallized in the presence of different inhibitors (18), the side chain of arginine 858 is found either completely exposed to the solvent or interacting with the aspartate 831 of the DFG motif. The electrostatic interactions involving R858 have also been observed in microsecond-long MD simulations (19).
Altogether, the effect of the L858R mutation is twofold: on one hand it destabilizes the inactive conformation; on the other it stabilizes the αC-helix structure. The stabilization of the α- helix structure is expected to favor the formation of the dimer by decreasing the entropic penalty associated with the ordering of the disordered interface observed in the WT-EGFR monomer (7, 19).
L858I Mutant.
Given the significant effect of the L858R mutation on the conformational free-energy landscape, we wondered what would be the effect of a “neutral” mutation at the same position. We have chosen L858I for two reasons. First, from a physico-chemical viewpoint the properties of isoleucine are similar to those of leucine. Second, this specific mutation is not reported among the cancer samples in the Catalogue of Somatic Mutations (COSMIC) database (13). Of the 5,556 EGFR L858 mutations currently reported in the database, L858R, with 5,541 instances, is the most common. However, many other mutations such as L858M, L858P, L858A, L858Q, L858K, L858W, L858V, and L858G are found in at least one case. Because L858I is not found in the database, we expect it either to be neutral or inactivating. The L858I FES is reported in Fig. S3. As expected, it is very similar to the WT FES. The most populated energy basin corresponds to the inactive structure, in agreement with the expected inactivating effect.
T790M Mutant.
In the case of the gatekeeper T790M mutant, the change in the FES is more dramatic and the equilibrium is shifted toward the active conformation (Fig. 2). The substitution of the threonine with a methionine stabilizes the active conformation by favoring the formation of a hydrophobic cluster around the phenylalanine F856 of the DFG motif. The cluster involves M790, F856, the methionine M766 in the αC-helix, and other residues of the regulatory spine (38). The M790–M766 distance is on average much shorter in the mutant than the corresponding T790–M766 distance in the WT-EGFR (0.5 0.1 nm vs. 0.9
0.1 nm vs. 0.9 0.2 nm, respectively). Interestingly, a basin where the αC-helix is disordered is also populated. Conformations in this basin show a deformed αC-helix and the broken salt bridge E762:K745 (Fig. S2). We do not observe the aspartate D858 of the DFG motif pointing outside of the catalytic site (DFG-out) as proposed in ref. 21. However, we have not explored the effect of protonation of the Asp of the DFG, which might stabilize the DFG-out form (36, 39).
0.2 nm, respectively). Interestingly, a basin where the αC-helix is disordered is also populated. Conformations in this basin show a deformed αC-helix and the broken salt bridge E762:K745 (Fig. S2). We do not observe the aspartate D858 of the DFG motif pointing outside of the catalytic site (DFG-out) as proposed in ref. 21. However, we have not explored the effect of protonation of the Asp of the DFG, which might stabilize the DFG-out form (36, 39).
T790M-L858R Mutant.
The double-mutant T790M–L858R retains some of the features of the single mutants L858R and T790M, but shows also some characteristics hinting at a synergistic effect of the combined mutations. In particular, the αC-in conformation is very stable (CV1 in the interval 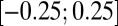 nm) and only a small barrier (
nm) and only a small barrier ( 2 kcal/mol) is present for the opening of the A loop, which thus exhibits a greater flexibility populating both the active and the semiclosed basins (Fig. 2). The global effect is to significantly stabilize the fully active state and to lower the free-energy barriers for the A-loop transition between the semiclosed and the fully extended structure. Some inactive conformations are still sampled and correspond to structures in which the αC-helix is distorted (Fig. S2).
2 kcal/mol) is present for the opening of the A loop, which thus exhibits a greater flexibility populating both the active and the semiclosed basins (Fig. 2). The global effect is to significantly stabilize the fully active state and to lower the free-energy barriers for the A-loop transition between the semiclosed and the fully extended structure. Some inactive conformations are still sampled and correspond to structures in which the αC-helix is distorted (Fig. S2).
Discussion
In contrast with other tyrosine kinases, the active state of the WT-EGFR CD monomer is rarely visited (40). This is in agreement with both its low catalytic activity (6) and the behavior observed in very long MD simulations (19, 36). Conformations in which the αC-helix is partially disrupted correspond to local minima. Because the αC-helix is at the dimer interface, its disorder could affect the stability of the dimer (19). In the L858R mutant, the substitution of the hydrophobic leucine residue in the A loop, with the positively charged arginine, creates new possibilities for the charged residues of the αC helix to interact. This mutation was first proposed to act by destabilizing the inactive conformation of the A loop, favoring the active conformation (6, 18). More recently, Shan et al. proposed an indirect mechanism whereby the mutation reduces the disorder in the αC-helix region, thus favoring dimerization (19). Our observations reconcile both views. We observe a dual effect. On one hand, the inactive state is disfavored due to the destabilization of the inactive-like short α-helix of the A loop. The fully active conformation that, in the absence of ATP, is unfavorable for the WT EGFR, appears as a stable secondary minimum in L858R. On the other hand, the αC-helix is stabilized by a cluster of salt bridges, suppressing the disordered states of WT-EGFR. Both the change in the relative stability of the active vs. the inactive state and the suppression of disorder at the interface, which favors the formation of the dimer, seem to play a role in the activation of L858R. The global effect is to shift the balance toward the active state and favor the formation of the asymmetric dimer.
In agreement with recent proposals (21), the gatekeeper T790M mutant does not seem to act by steric hindrance with the ATP-competitive inhibitors, but rather by stabilizing the active conformation. In this case the methionine participates in the hydrophobic core surrounding the active site. In particular, interacting with M766 of the αC-helix it promotes a more compact active site, with a closer helix, favoring the αC-in conformation. These results agree with the enhanced stabilization of the catalytic site observed when comparing the collective motions of the WT and mutant kinase domain (41). However, the T790M mutant does not suppress the disorder at the dimerization interface, and it is not expected to favor formation of the dimer. This observation might explain the greater frequency of L858R with respect to the T790M mutation in cancer patients.
In light of the present results, we can fully rationalize and quantify the epistatic effect due to the combination of the two mutations. As proposed in ref. 14, the total stabilization of the active state by the double mutations is more than that expected from a simple combination of the stabilization by the two single mutations. However, the effect is not limited to the relative stability of the active and inactive states, but it reshapes the free-energy landscape in a complex manner. Indeed, the combination of the two mutations in the T790M-L858R double mutant not only has the activating and rigidifying effects of the two single mutations, but it also lowers the free-energy barrier of the active–inactive transition of the A loop and stabilizes the correct helical structure of the αC-helix. In the T790M–L858R mutant the αC-helix and the E762:K745 salt bridge are stable. Thus, the conformation adopted by the A loop determines the balance between the active and the inactive form.
The complex changes seen in the conformational free-energy landscapes of the mutants are due both to local changes in the network of electrostatic and hydrophobic interactions and to long-range allosteric couplings. This nontrivial knowledge not only reconciles the observed differences of the mutants with the WT, but can also be used to devise new strategies for the development of TKIs less susceptible to resistance.
Materials and Methods
Protein Structures and Protein Data Bank.
The Protein Data Bank (PDB) codes used for the active and inactive WT-EGFR structures are 2GS2 and 2GS7, respectively, and the used sequence comprises the interval L703–Q976 in our notation, L679–Q952 in the PDB notation. The amino acid numbering convention adopted here includes the 24-residue membrane targeting signaling sequence. Mutants and missing residues were modeled with MODELER (42).
Simulation Details.
The MD simulations were performed using GROMACS 4.5 (43) with the PLUMED plug-in (44) for Metadynamics calculations. Each system is described by the Amber99SB*-ILDN force field (45) with the dihedral corrections of Best and Hummer (46), solvated with tip3p water molecules (47) and enclosed in a dodecahedron box with periodic boundary conditions. The van der Waals interactions were smoothly shifted to zero between 0.8 and 1.0 nm; the long-range electrostatic interactions were calculated by the particle mesh Ewald algorithm (48), with mesh spaced 0.12 nm, combined with a switch function for the direct space between 0.8 and 1.0 nm for better energy conservation. The system evolves in the canonical ensemble, coupled with a velocity-rescale thermostat (49) and a time step of 2 fs. Each solvated system was prepared as detailed in SI Text.
Enhanced Sampling.
PT-metaD in the Well-Tempered Ensemble has been performed for each system using six replicas at increasing temperatures (295, 300, 310, 325, 341, and 358 K) (see SI Text and Fig. S4 for further details). All six replicas are subject to the well-tempered metadynamics prescription in which a Gaussian is deposited in the collective variable space every 2 ps with height 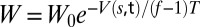 , where
, where 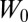 =5 kJ/mol is the initial height, T is the temperature of the replica, f=10 is the bias factor, and V(s,t) is the bias potential at time t and CV value s. The following three collective variables are used: the difference between atomic distances
=5 kJ/mol is the initial height, T is the temperature of the replica, f=10 is the bias factor, and V(s,t) is the bias potential at time t and CV value s. The following three collective variables are used: the difference between atomic distances 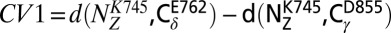 ; the distance in contact map space to the inactive A-loop conformation
; the distance in contact map space to the inactive A-loop conformation 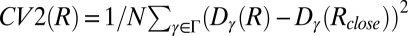 ; the distance in contact map space to the active A-loop conformation
; the distance in contact map space to the active A-loop conformation 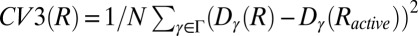 .
. 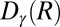 is a sigmoidal function that measures the degree of formation of the contact γ in the structure R and is defined as
is a sigmoidal function that measures the degree of formation of the contact γ in the structure R and is defined as 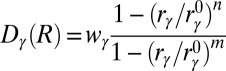 , where
, where 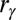 is the contact distance in the structure R,
is the contact distance in the structure R, 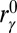 is the contact distance in either the reference inactive or active conformation depending if γ is a contact specific to the former or the latter conformation,
is the contact distance in either the reference inactive or active conformation depending if γ is a contact specific to the former or the latter conformation, 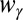 is the weight of the contact and is set to 1 for regular contacts and 3 for salt bridges, N is a normalization constant, n = 6, and m = 10. The set of contacts
is the weight of the contact and is set to 1 for regular contacts and 3 for salt bridges, N is a normalization constant, n = 6, and m = 10. The set of contacts 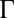 defining the contact map is defined in SI Text. The widths of the Gaussians in the three CV dimensions are
defining the contact map is defined in SI Text. The widths of the Gaussians in the three CV dimensions are 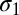 =0.015 nm and
=0.015 nm and 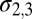 =0.0083. Note that CV2 and CV3 are adimensional.
=0.0083. Note that CV2 and CV3 are adimensional.
Convergence and Self-Correcting Properties of PT-metaD.
The simulations are run until the free energy in the bidimensional projections and in the monodimensional projections does not change more than 2 kcal/mol in the last 100 ns. This convergence criterion led to 680 ns long simulations for WT-EGFR and T790M-L858R and to 610 ns long simulations for L858R and T790M. To further check the convergence of the reconstructed free energy landscapes and to illustrate the self-correcting properties of PT-metaD calculations we performed two sets of calculations. Starting with a “wrong” free energy surface (the unconverged FES obtained for the WT kinase after 300ns) we run further 500ns of PT-metaD on the L858R and on the new L858I mutant (the total sampling time for each simulation was 3 μs). As shown in Fig. S5, the two free energy surfaces rapidly converge to the original FES, in the case of the L858R mutant and to a FES similar to that of the WT, in the case of the L858I mutant.
Analysis.
The free energy surfaces are obtained by integrating the deposited bias during the simulation, as required by the metadynamics algorithm. For convenience, they are shown as a function of two CVs at a time (CV1, CV2) and (CV2, CV3) in the Supplementary Materials. To obtain a representative structure of each free energy basin, a clustering of the set of structures falling within a small (CV1, CV2) patch surrounding each basin has been performed. The single-linkage clustering algorithm of g_cluster program from the GROMACS package has been used with the RMSD on the CA atoms as the distance with a cutoff of 0.2 nm. In the most populated cluster of each basin, the central structure, i.e., the structure with smallest distance to all of the other members of the cluster, has been picked as representative of the basin.
Supplementary Material
Acknowledgments
We thank M. Eck for useful discussions and suggestions concerning this work and K. Marino for proofreading the manuscript. We also acknowledge partial support from the Spanish Ministry of Science and Innovation (MICINN, Grant BIO2010-20166); the High End Computing Terascale Resource (HECToR) of the Partnership for Advanced Computing in Europe (PRACE) research infrastructure (Tier-1); (Tier-0) SuperMUC at the Leibniz-Rechenzentrum (LRZ), Germany (Seventh Framework Programme 2007-2013, Grant Agreement RI-283493); and the Emerald supercomputing facility at e-Infrastructure South for the computational resources.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221953110/-/DCSupplemental.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber AB, Libermann TA, Lax I, Yarden Y, Schlessinger J. Biological role of epidermal growth factor-receptor clustering. Investigation with monoclonal anti-receptor antibodies. J Biol Chem. 1983;258(2):846–853. [PubMed] [Google Scholar]
- 6.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Arkhipov A, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152(3):557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endres NF, et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell. 2013;152(3):543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffrey PD, et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376(6538):313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 10.Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303(5665):1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 11.Wood ER, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 12.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118(2):257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 13.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto K, Rogozin IB, Panchenko AR. Oncogenic potential is related to activating effect of cancer single and double somatic mutations in receptor tyrosine kinases. Hum Mutat. 2012;33(11):1566–1575. doi: 10.1002/humu.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 16.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 18.Yun CH, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan Y, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149(4):860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 20.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 21.Yun CH, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan S, Wright DW, Coveney PV. Mechanism of drug efficacy within the EGF receptor revealed by microsecond molecular dynamics simulation. Mol Cancer Ther. 2012;11(11):2394–2400. doi: 10.1158/1535-7163.MCT-12-0644-T. [DOI] [PubMed] [Google Scholar]
- 23.Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J. 2012;448(3):417–423. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam IYS, et al. Double EGFR mutants containing rare EGFR mutant types show reduced in vitro response to gefitinib compared with common activating missense mutations. Mol Cancer Ther. 2009;8(8):2142–2151. doi: 10.1158/1535-7163.MCT-08-1219. [DOI] [PubMed] [Google Scholar]
- 26.Lindorff-Larsen K, et al. Systematic validation of protein force fields against experimental data. PLoS ONE. 2012;7(2):e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw DE, et al. Millisecond-scale molecular dynamics simulations on Anton. New York: ACM Press; 2009. pp. 1–11. [Google Scholar]
- 28.Bussi G, Gervasio FL, Laio A, Parrinello M. Free-energy landscape for beta hairpin folding from combined parallel tempering and metadynamics. J Am Chem Soc. 2006;128(41):13435–13441. doi: 10.1021/ja062463w. [DOI] [PubMed] [Google Scholar]
- 29.Sutto L, Marsili S, Gervasio FL. New advances in metadynamics. WIREs Comput Mol Sci. 2012;2:771–779. [Google Scholar]
- 30.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonomi M, Parrinello M. Enhanced sampling in the well-tempered ensemble. Phys Rev Lett. 2010;104(19):190601–190604. doi: 10.1103/PhysRevLett.104.190601. [DOI] [PubMed] [Google Scholar]
- 32.Berteotti A, et al. Protein conformational transitions: the closure mechanism of a kinase explored by atomistic simulations. J Am Chem Soc. 2009;131(1):244–250. doi: 10.1021/ja806846q. [DOI] [PubMed] [Google Scholar]
- 33.Lovera S, et al. The different flexibility of c-Src and c-Abl kinases regulates the accessibility of a druggable inactive conformation. J Am Chem Soc. 2012;134(5):2496–2499. doi: 10.1021/ja210751t. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3(5):629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 35.De Bondt HL, et al. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363(6430):595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 36.Shan Y, Arkhipov A, Kim ET, Pan AC, Shaw DE. Transitions to catalytically inactive conformations in EGFR kinase. Proc Natl Acad Sci USA. 2013;110(18):7270–7275. doi: 10.1073/pnas.1220843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitford PC, Miyashita O, Levy Y, Onuchic JN. Conformational transitions of adenylate kinase: switching by cracking. J Mol Biol. 2007;366(5):1661–1671. doi: 10.1016/j.jmb.2006.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006;103(47):17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan Y, et al. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci USA. 2009;106(1):139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jura N, et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42(1):9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dixit A, Verkhivker GM. Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLOS Comput Biol. 2011;7(10):e1002179. doi: 10.1371/journal.pcbi.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 43.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 44.Bonomi M, et al. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput Phys Commun. 2009;180:1961–1972. [Google Scholar]
- 45.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113(26):9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahoney MW, Jorgensen WL. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys. 2000;112:8910. [Google Scholar]
- 48.Essmann U, et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577. [Google Scholar]
- 49.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.