

Abstract
Fetal brain-directed gene addition represents an under-appreciated tool for investigating novel therapeutic approaches in animal models of central nervous system diseases with early prenatal onset. Choroid plexuses (CPs) are specialized neuroectoderm-derived structures that project into the brain's ventricles, produce cerebrospinal fluid (CSF), and regulate CSF biochemical composition. Targeting the CP may be advantageous for adeno-associated viral (AAV) gene therapy for central nervous system disorders due to its immunoprivileged location and slow rate of epithelial turnover. Yet the capacity of AAV vectors to transduce CP has not been delineated precisely. We performed intracerebroventricular injections of recombinant AAV serotype 5-green fluorescent protein (rAAV5-GFP) or rAAV9-GFP in embryonic day 15 (E15) embryos of CD-1 and C57BL/6 pregnant mice and quantified the percentages of GFP expression in CP epithelia (CPE) from lateral and fourth ventricles on E17, postnatal day 2 (P2), and P22. AAV5 was selective for CPE and showed significantly higher transduction efficiency in C57BL/6 mice (P = 0.0128). AAV9 transduced neurons and glial cells in both the mouse strains, in addition to CPE. We documented GFP expression in CPE on E17, within just 48 hours of rAAV administration to the fetal lateral ventricle, and expression by both the serotypes persisted at P130. Our results indicate that prenatal administration of rAAV5 and rAAV9 enables rapid, robust, and sustained transduction of mouse CPE and buttress the rationale for experimental therapeutics targeting the CP.
Keywords: blood-cerebrospinal fluid barrier, choroid plexus, in utero gene therapy, rAAV5, rAAV9, survival surgery
Introduction
Fetal gene transfer represents an under-appreciated tool for investigating novel therapeutic approaches in mouse models of human neurometabolic diseases.1 Advantages of this approach include the short murine gestation time, large litter sizes that permit near-simultaneous vector administration to multiple animals, the existence of monogenic diseases comparable to conditions in humans, and the ability to target embryonic- or perinatal-lethal alleles for therapeutic rescue. For brain-directed treatments in which entry of the therapeutic molecular entity to cerebrospinal fluid (CSF) is desired, the space targeted is considerably larger in mouse fetuses than in postnatal mice, since the ratio of ventricular volume:brain volume is higher in early development.2,3
The choroid plexuses (CPs) are highly vascularized neuroectoderm-derived structures continuous with the ependymal cells lining the brain ventricles, and which project into the ventricular cavities.4 The polarized epithelia of the CP produce CSF by transporting water and ions into the ventricles from the blood and secreting a large number of proteins. CP development begins by embryonic day 10 (E10) in rodents.5 The fourth ventricle CP develops first, followed by the CPs of both the lateral ventricles and finally the third ventricle CP,3 a sequence consistent across mammalian species. CP epithelia (CPE) are contiguous with, though functionally differentiated from, the ependymal cells that line the walls of the ventricles. The early morphology of CPE is single-layered cuboidal to columnar. By E12.5, differentiation into a simple cuboidal pattern occurs at all sites of CPE formation in mice and is accompanied by activation of the CPE-specific gene, transthyretin.2
We recently reported that neonatal intracerebroventricular administration of recombinant adeno-associated virus serotype 5 (rAAV5) and copper chloride resulted in efficient gene transfer to mouse CPE, and rescued mottled-brindled (mo-br), a lethal mouse model of the copper metabolism disorder, Menkes disease.6 Untreated mo-br mice died by 2 weeks of age. Fetal gene transfer to CPE with rAAV might also rescue an embryonic lethal model of Menkes disease, mottled-dappled (mo-dp), that usually does not survive beyond E17.7 Rescue of this complete loss-of-function allele would provide compelling additional proof-of-concept in support of human applications of rAAV gene therapy for this illness. In addition to rAAV5, the capacity of rAAV serotypes 1, 4, 8, and rh10 to transduce neonatal mouse CPE has been described.8,9,10,11,12 However, rAAV9 tropism for CPE has not been clearly established.
Viral-mediated transduction of CPE might also provide a safe and efficacious treatment approach for a broader category of pediatric neurometabolic disease, lysosomal storage disorders (LSDs).9,12,13,14,15,16 These conditions are caused by inherited deficiencies of lysosomal enzyme activity and more than 60 such human diseases have been identified.17 Enzymes within lysosomes normally break down specific nutrients, including glycoproteins, mucopolysaccharides, oligosaccharides, and sphingolipids. Patients with LSDs have greatly diminished lifespans and reduced quality of life, particularly those with neurological manifestations. There are few current therapeutic options available to treat the neurological signs and symptoms of LSDs. Bone marrow transplantation has been carried out in some LSDs in which there is neurological involvement.18 Such therapy is based on the concept of metabolic cross-correction in which the lysosomal enzyme of one cell is taken up by the enzyme-deficient adjacent cell.19 However, bone marrow transplantation itself is associated with significant morbidity and mortality, and many LSD patients who receive this treatment do not exhibit improvements in cognitive signs and symptoms.20
Enzyme-replacement therapy for LSDs administered intravenously has been ineffective in achieving adequate delivery of enzyme to the brain. However, a growing body of research in animal models supports the potential efficacy of direct injection into the CSF.21 Studies in dogs with mucopolysaccharidosis type I showed that this intrathecal approach enabled widespread distribution of the recombinant protein within the brain, and reduced neuropathology.22 Studies in mouse and dog models of mucopolysaccharidosis IIIA also revealed that the strategy was effective in ameliorating neuropathology and improving clinical signs in affected animals.23,24 Other studies in mice with the LSDs Krabbe disease and late infantile neuronal ceroid lipofuscinosis demonstrated that delivery of recombinant lysosomal enzymes into the CSF was efficacious in reducing disease pathology and neurological signs and symptoms.25,26 Thus, the principle that provision of enzyme into the CSF is beneficial for LSDs is gaining considerable traction. The short half-lives of recombinant lysosomal enzymes, which would necessitate weekly or monthly administration, complicates translation to clinical applications however.
Viral vector-mediated gene therapy targeting the CPE represents a potential alternative method for provision of enzyme into the CSF without the need for chronic intrathecal injections. We speculate that patients with LSDs could benefit from a CP-targeted gene therapy approach that results in steady production of the deficient enzyme and secretion into the CSF. The CSF circulation normally carries molecules throughout the ventricular system into the subarachnoid space, from which molecules ultimately reach the entire brain.27
To better characterize mammalian CP transduction for these important potential applications, we conducted experiments to specifically evaluate rAAV-mediated transgene expression in CPE in two strains of wild-type mice.
Results
Survival of mouse fetuses following early intracranial gene transfer
Fetal intracranial administration of rAAV5-green fluorescent protein (GFP), rAAV9-GFP, or lactated Ringer's at 15 days of gestation (Figure 1) was performed on a total of 141 CD-1 and 117 C57BL/6 fetuses. The choice of the CD-1 outbred strain was grounded in the knowledge that this background has been used successfully for fetal gene surgery by others.28,29 The choice of C57BL6 inbred strain reflected our related work on a Menkes disease mouse model that has the C57BL6 background.6 There was no maternal mortality or morbidity related to fetal surgery in either mouse background. Overall survival to birth was higher in CD-1 fetuses (70.2%) compared with C57BL/6 fetuses (42.7%; P < 0.0001, Tables 1 and 2). The increased prenatal mortality in C57BL/6 was accounted for by the results of rAAV9 administration, after which only 29% of E15 C57BL/6 embryos survived to birth. rAAV5-treated C57BL/6 embryos survived at a twofold higher rate (57.8%) than rAAV9-treated C57BL/6 (P = 0.0192). In contrast, rAAV5 administration appeared to lower survival in CD-1 animals compared with mock-treated CD-1 controls (P = 0.0097).
Figure 1.

Surgical approach for intracerebroventricular injection of E15 fetal mouse brains. Following exposure of the uterine horns, individual fetal brains were injected on the left side with 5 μl of lactated Ringer's solution containing 5 × 109 viral particles of either rAAV5-GFP or rAAV9-GFP, or lactated Ringer's alone (mock). GFP, green fluorescent protein; rAAV, recombinant adeno-associated virus.
Table 1. Survival to birth by strain.
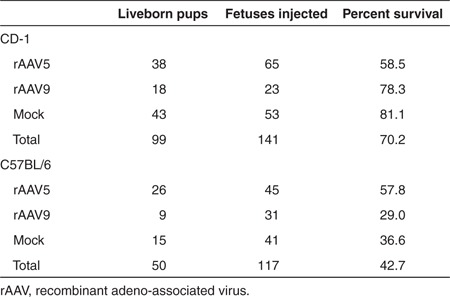
Table 2. Survival to birth by AAV serotype.

All liveborn pups that had been treated at E15 with either rAAV5 or rAAV9 or lactated Ringer's survived into late adulthood unless harvested for analysis. Fetuses harvested at E17 were not included in survival rates.
Viral-mediated expression of GFP in CPE of CD-1 mice
We quantified CPE in the lateral and fourth ventricles (Supplementary Figure S1) because the third ventricle CP was smaller and not consistently visualized in the brain sections. Constructs used in these experiments contained the cDNA for the reporter gene, GFP. Transgene expression was driven by the chicken β-actin promoter and human cytomegalovirus enhancer combination (Figure 2a).
Figure 2.

Recombinant adeno-associated virus (rAAV)-mediated gene expression in CD-1 fetal mouse choroid plexus. (a) Elements of the rAAV construct. Flanked by inverted terminal repeat (ITR) motifs, the rAAV construct includes a cytomegalovirus (CMV) enhancer, chicken β-actin (CBA) promoter, intronic sequence (triangle), complementary DNA (cDNA) for green fluorescent protein (GFP), and a poly-adenylation (poly-A) tail. (b) Representative immunohistochemistry images of GFP transgene expression at E17 in CD-1 mouse brains, after rAAV serotype 5 or 9, or mock injection on E15. (c) Representative images of P2 and P22 CD-1 mouse brains after rAAV serotype 5 or 9, or mock injection on E15. Brown stain indicates GFP expression. Arrows indicate AAV9-GFP–mediated expression in adjacent brain parenchyma on P22. CPL: choroid plexus-lateral ventricle; CP4: choroid plexus-fourth ventricle. Bars, 100 μm. Of the three timepoints evaluated, densitometric quantitation showed peak transgene expression on P2 in the (d) lateral and (e) fourth cerebral ventricles. Statistically significant differences between rAAV5- and rAAV9-mediated GFP expression were evident only in the fourth ventricle choroid plexus epithelia, as shown.
In CD-1 mice, transgene expression was easily detectable on E17 (2 days after rAAV administration) in the CPE of the lateral and fourth ventricles and not in mock-injected controls (Figure 2b). CPE expression of GFP mediated by rAAV5 and rAAV9 was robust on postnatal day 2 (P2) and P22 (Figure 2c). Mock-injected controls showed no discernible GFP staining (data not shown). No statistically significant differences were evident in head-to-head comparisons of the percent of CPE expressing GFP between rAAV5 and rAAV9, with two exceptions (Figure 2d,e). Expression mediated by rAAV5 proved significantly higher in fourth ventricle CPE at P2 compared with rAAV9-GFP (P = 0.02, Figure 2e). By P22, rAAV9-mediated expression exceeded that by rAAV5 in the fourth ventricle (P = 0.03, Figure 2e). GFP expression was also detected in CPE of the third ventricle (data not shown).
In addition to expression in CPE, E15 administration of rAAV9 led to transgene expression throughout the CD-1 mouse brains, including neuronal cells in olfactory bulb, cerebral cortex, cerebellum, and glial cells in the brainstem on P2 (Figure 3a). In contrast, rAAV5 administered to CD-1 mouse embryos on E15 did not result in detectable transgene expression in neuronal or glial cells (data not shown) and expression was confined to the CPE (Figure 2c). Quantitation of rAAV9 viral genome copy number by CD-1 brain region on P2 confirmed a pattern of transduction consistent with the P2 expression data (Figure 3b). By P22, rAAV9-mediated expression in neuronal cells remained robust (Figure 4a).
Figure 3.

Broad transduction of CD-1 mouse brain on P2 after E15 intracerebroventricular administration of rAAV9-GFP. (a) Representative images of GFP transgene expression in CD-1 mouse brains on P2 after rAAV9 injection on E15 shows robust expression of GFP (brown stain) in olfactory bulb, cerebral cortex, cerebellum, and brainstem. Bar, 100 μm. (b) Results of real-time PCR quantitation of rAAV9 genome copy number per mouse diploid genome in CD-1 mouse brain on P2 (n = 3). Consistent with the expression data in a, transduction appeared highest in olfactory bulb followed by brainstem, cortex, and cerebellum. Error bars represent SD corrected for log scale. GFP, green fluorescent protein; rAAV, recombinant adeno-associated virus.
Figure 4.

Neuronal transduction of CD-1 mouse brain on P22 after intracerebroventricular administration of rAAV9-GFP on E15. (a) Hippocampal, cortical, olfactory, and some cerebellar neurons demonstrate sustained GFP expression ~1 month after vector administration. (b) Immunohistochemical staining at P130 shows sustained transgene expression in the choroid plexus in the fourth ventricle (CP4) of CD-1 mouse brains, more than 4 months after intracerebroventricular administration on E15 with either rAAV5-GFP or rAAV9-GFP. Bars, 100 μm. GFP, green fluorescent protein; rAAV, recombinant adeno-associated virus.
We also documented persistence of GFP expression in CD-1 mouse CPE at 130 days, the endpoint of this study (Figure 4b), consistent with the slow rate of CPE turnover.30,31
Viral-mediated expression of GFP in CPE of C57BL/6 mice
In a second mouse strain, C57BL/6, viral-mediated expression in CPE at E17 was minimal following E15 administration (Figure 5a), whereas expression at P2 and P22 was robust with either rAAV5 or rAAV9 (Figure 5b). Similar to the pattern in mice with the CD-1 background (Figure 2d,e), expression mediated by rAAV5-GFP in C57BL/6 was highest on P2, of the three timepoints evaluated (Figure 5c,d). Expression in third ventricle CPE was also detected while all mock-injected controls were negative for GFP staining (data not shown). P2 expression mediated by rAAV9 proved significantly higher in fourth ventricle CPE compared with rAAV5-GFP (P = 0.03, Figure 5d).
Figure 5.
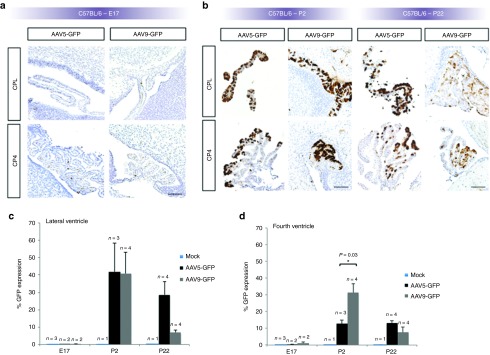
GFP transgene expression in C57BL/6 mice. (a) Minimal transduction of choroid plexus (CP) epithelia was evident in E17 C57BL/6 mouse fetuses after intracerebroventricular administration on E15 of either rAAV5-GFP or rAAV9-GFP. (b) Intense expression of GFP in CP epithelia was evident by P2 and P22 compared with E17 and to CD-1 mice at P2 and P22 (Figure 2c). CPL: CP-lateral ventricle; CP4: CP-fourth ventricle. Bars, 100 μm. Densitometric quantitation of GFP expression in C57BL/6 mice on E17, P2, and P22 in (c) lateral and (d) fourth cerebral ventricles. As in CD-1 mice (Figure 2d,e), expression in C57BL/6 appears generally the highest on P2 (of the timepoints evaluated). GFP expression mediated by rAAV9 was statistically higher than by rAAV5 in fourth ventricle CP epithelia on P2 (in d). GFP, green fluorescent protein; rAAV, recombinant adeno-associated virus.
Mouse background influences rAAV transduction
As suggested by an overall comparison of Figures 2 and 5, a higher percentage of CP cells expressed GFP in C57BL/6 than in CD-1 mice. This difference was statistically significant for rAAV5 (P = 0.01), and nearly significant for rAAV9 (P = 0.06). GFP expression in C57BL/6 compared with CD-1 was strikingly higher with rAAV5 at P22 in the lateral and fourth ventricles (Figure 5c,d compared with Figure 2d,e).
Discussion
In this work, we utilized an in utero approach to characterize rAAV transduction and transgene expression in mouse brain CPE, a unique target tissue for gene transfer not previously studied in detail. Potential advantages of CPE as a target for viral gene therapy include (i) an immunoprivileged location, (ii) access to the CSF for secretion of transgene products, and (iii) a slow CPE turnover rate30,31 that favors sustained transgene expression.
In the present study, we compared rAAV5 to a more recently identified AAV serotype, rAAV9, to assess the relative efficiency of transgene expression in CPE for two wild-type mouse strains. Limited knowledge exists concerning the capacity of prenatally administered rAAV9 to target the developing CPs.28,29 Here, we show that intracerebroventricular administration of rAAV5 and rAAV9 to fetal mouse brain results in rapid (within 48 hours) and robust transgene expression in CPE. The relative decline in transgene expression from P2 to P22 that we document (Figures 2 and 5) may reflect growth and division of CPE in neonatal mice, with loss of episomes harboring the GFP transgene. Nonetheless, we continued to detect GFP in the CP of animals for more than 4 months (P130) post-AAV administration (Figure 4b), confirming the capacity for persistent transgene expression.
Overall, GFP expression was higher in C57BL/6 compared with CD-1 and the difference was especially striking with rAAV5 at P22 (Figure 5c,d). Differences between the levels of CP transduction with rAAV5 and rAAV9 for different ages and mouse backgrounds may reflect developmental differences in CPE cell membrane receptors, or differential rates of intracellular rAAV uncoating and nuclear translocation.32 In addition to CPE transduction, intracerebroventricular rAAV9 administration to fetal mouse brain resulted in broad neuronal and glial cell transduction, similar to patterns described by others using rAAV1 and rAAV8, as well as rAAV9.14,29
An unexpected finding of this study involved the higher prenatal losses in both the mouse strains compared with mock-treated controls following in utero administration of rAAV at E15 (Tables 1 and 2). While AAV and GFP are generally considered safe, we cannot formally exclude the possibility of inflammatory or other toxic cellular responses to the rAAV capsid, transgene product, or AAV preparation in these mice.33,34 While a contaminant such as lipopolysaccharide or other endotoxin(s) in the rAAV preparations remain possible explanations for the high mortality, we suspect that the mortality data are more related to the invasive nature of E15 fetal surgery. We also assume that the higher overall fetal survival in CD-1 compared with C57BL/6 (70.2 versus 42.7%), reflects differences in breeding robustness between these strains.
We previously showed that rAAV5 transduction of CPE in the neonatal period was associated with rescue of the mo-br mouse model of Menkes disease, an X-linked recessive neonatal lethal disorder of copper metabolism.6 This condition is caused by mutations in the highly conserved copper transporter, ATP7A.7 The successful gene therapy advanced the concept that CPE are critically important for central nervous system copper homeostasis. Untreated mo-br mice live only for 2 weeks due to markedly reduced copper transport activity (≈10% of wild-type) in the context of an in-frame deletion of two highly conserved amino acid residues.6,7 The mo-dp allele, in contrast, represents a complete loss-of-copper transport function due to a large genomic deletion in the ATP7A homolog and dies at E17.7 Viral-mediated addition of ATP7A to mo-dp prenatally may enable rescue of this embryonic lethal. While relevant to our investigations of copper transport as a proof-of-principle, in utero gene therapy is not however being considered for clinical application in Menkes disease.
Our findings reinforce the prospect that CPE, which form the blood–CSF barrier, represent a useful therapeutic avenue for amelioration of various inherited neurometabolic diseases.9,10,11,12,13,14,15,16,17 During early mammalian brain development, the ventricular lining is constituted by proliferating neuroectodermal cells among which both ependymal and CPE progenitor cells are prominent.4 It is unknown whether the CPE progenitors possess cell surface receptors necessary to mediate rAAV capsid binding and endocytosis, although this appears possible. Our data supports the presence of cell surface receptors for rAAV5 and rAAV9 in mouse CPE at least as early as E15.
Murine CPE have an extremely slow turnover rate and are not replaced during the entire mouse adult lifespan.30 Slow turnover of these cells is also considered the situation in larger mammals.31 We suspect that with a moderately high multiplicity of infection, sufficient numbers of episomal genomes would persist in CPE daughter cells. Thus, the neuroectoderm-derived epithelial cells of the CP may represent ideal targets for a non-integrating, episomal rAAV vector capable of sustaining long-term transgene expression.
Since the normal flow of CSF carries molecules secreted by CPE throughout the ventricular system into the subarachnoid space from which molecules ultimately reach the entire brain,27 rAAV-mediated gene transfer to remodel CPE could transform the treatment of lysosomal storage diseases. The prospect of single administration to an immunoprivileged site, in combination with the favorable overall safety profile of rAAV vectors, renders this an attractive prospective approach. Further preclinical investigations to test this hypothesis are warranted.
Materials and methods
Mice. The NICHD Animal Studies Committee approved all experimental procedures. CD-1 and C57BL/6J breeding pairs were obtained from Charles River Laboratories (Wilmington, MA). CD-1 and C57BL/6 matings were set up and the females monitored for vaginal plugs. The observed day of vaginal plug was considered E1, and the male was separated from the female.
AAV vectors. The Powell Gene Therapy Center Vector Core Lab, University of Florida-Gainesville (Gainesville, FL) produced rAAV5-GFP and rAAV9-GFP. A UF11 vector harboring the chicken β-actin promoter and the cytomegalovirus enhancer was used to drive transgene expression.
In utero surgery. Pregnant CD-1 females underwent surgery on E15 under sterile conditions, using endotoxin-free material and autoclave-sterilized surgical instruments. Pregnant females were anesthetized with isoflurane (1–5%) inhalation and an analgesic (buprenorphine; 1 mg/kg) was injected subcutaneously before and after the surgery. The eyes were coated with ophthalmic ointment to keep the corneas moist. The abdominal area of the mouse was shaved and prepped sterilized. A 3-cm skin incision was made followed by a 2-cm midline laparotomy. The uterine horns were gently extracted and the number of embryos per horn was evaluated. The embryos were kept moist with phosphate-buffered saline. Using a 32-gauge Hamilton syringe, each fetus received an injection of 5 μl of either rAAV5-GFP (1 × 109 viral particles/μl) or rAAV9-GFP (1 × 109 viral particles/μl) in lactated Ringer's solution, or lactated Ringer's alone (mock control) into the left lateral ventricle of the fetal brain (Figure 1). We confirmed the accuracy of intraventricular administration by injecting several untreated fetuses with 5 μl of 0.6% trypan blue solution, which rapidly filled the ventricles of both the hemispheres on visual inspection. During the procedure, the pregnant mouse body temperature was monitored using a rectal probe thermometer and the animals were kept warm to prevent hypothermia. After all fetuses were injected, the uterine horns were gently reinserted into the abdomen. The incision in the muscle was closed using absorbable 6-0 sterile surgical sutures (Henry Schein, Melville, NY). The incision in the skin was closed with staples. Prophylaxis against postoperative infection was given with an intraperitoneal injection of cefazolin (dose 100 mg/kg). Post-surgery, acetaminophen (200 mg/kg/day) was administered via drinking water for 3 days. The pregnant dam was either killed 2 days post-injection to evaluate the E17 timepoint, or gave birth, typically at E21.
Pathology and immunohistochemistry. Mice were anesthetized, then decapitated, and E17, P2, and P22 brains were dissected and divided sagittally into right and left hemispheres, which were fixed in 10% formalin and dehydrated in 70% ethanol. The brain hemispheres were then embedded in paraffin (Histoserve, Germantown, MD), sectioned in 4 μm thick sections, placed on ProbeOn Plus slides (Fisher Scientific, Pittsburgh, PA), and de-paraffinized. All slides and negative controls were pre-treated in citrate buffer pH 6.0 (Cell Marque, Rocklin, CA) for 20 minutes in a steamer and allowed to cool to room temperature. Subsequently, all slides were blocked twice with Dual Endogenous Block (DAKO, Carpentaria, CA) for 5 minutes each, and with protein block (DAKO) for 10 minutes. A mouse monoclonal antibody against GFP (JL-8) (BD Biosciences Clontech, Palo Alto, CA; diluted 1:2,000) was applied for 60 minutes. Visualization was via the EnVisionTMG|2 Double stain system (DAKO), followed by CAT hematoxylin stain (Biocare, Concord, CA). For negative controls, mouse monoclonal IgG was used instead of antibody. The immunohistochemistry was performed in batches and for each batch, independent positive and negative controls were included. Brain hemispheres both ipsilateral and contralateral to the injection site were used for immunohistochemistry.
Densitometry. Photographs of microscopic brain sections were obtained using the Axioskop2 plus microscope and AxioVision 4.5 software (Carl Zeiss, Thornwood, NY), and visualized at ×200 magnification. The percentage of GFP expression was analyzed via color threshold adjustment and quantified densitometrically, using ImageJ software (Supplementary Figure S1).
Viral genome quantitation. Quantitative PCR, using primers specific for GFP, was used to quantitate rAAV9 viral genome copies in brain cortex, cerebellum, olfactory bulb, and brain stem. Uninjected animals were used as controls. Brains from P2 mice injected on E15 with AAV9-GFP, and from untreated mice, were dissected and divided sagittally. Olfactory bulb, cortex (and underlying brain tissue), cerebellum, and brainstem (Figure 3b) were separated, snap-frozen on dry ice, and stored at −80 °C. Genomic DNA was extracted from sections ipsilateral to the injection site using the Wizard Genomic DNA purification kit (Promega, Madison, WI). Purified DNA was used as template for genomic Real Time PCR on a DNA engine Opticon thermocycler (MJ Research, Waltham, MA). Amplification was performed using the SYBR Green Jumpstart Taq ReadyMix from Sigma (St Louis, MO), following the manufacturer's instructions. The biodistribution of AAV9-GFP was assessed in the four brain regions and corresponding tissues from untreated mice served as negative controls. Primers, forward: CAGCACGACTTCTTCAAGTCC and reverse: GCTGGAGTACAACTACAACAGC were used to amplify a region of the GFP transgene at a melting temperature of 60 °C. For quantitation, we used a control plasmid harboring the GFP transgene at a known concentration. We used serial dilutions of the plasmid to construct a linear curve of log quantity per cycle threshold (Ct). Based on this standard curve, we calculated the DNA quantities (pg) in our samples and divided them by the mass (pg) of one AAV genome to obtain vector genome copy. The final results were expressed as AAV vector genomes per diploid mouse genome, as described by others.35
Statistics. To evaluate for significant differences between AAV5- and AAV9-mediated GFP expression, we used two-tailed independent groups t-tests with equal variance. Fisher's exact test of independence was used to compare survival frequencies.36 Two-tailed P values ≤0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Densitometry to quantitate GFP transgene expression in mouse choroid plexus.
Acknowledgments
This work was done in Bethesda, MD, USA. We thank Daniel Abebe, Lynn Holtzclaw, and Matt Schech for assistance with mouse surgery, and Alan Flake, Ahad Rahim, Simon Waddington, and John Wolfe for helpful discussions. The authors declared no conflict of interest.
Supplementary Material
Densitometry to quantitate GFP transgene expression in mouse choroid plexus.
References
- Roybal JL, Endo M, Buckley SM, Herbert BR, Waddington SN, Flake AW. Animal models for prenatal gene therapy: rodent models for prenatal gene therapy. Methods Mol Biol. 2012;891:201–218. doi: 10.1007/978-1-61779-873-3_10. [DOI] [PubMed] [Google Scholar]
- Currle DS, Cheng X, Hsu CM, Monuki ES. Direct and indirect roles of CNS dorsal midline cells in choroid plexus epithelia formation. Development. 2005;132:3549–3559. doi: 10.1242/dev.01915. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Ek J, Habgood MD, Saunders NR. Development of the choroid plexus. Microsc Res Tech. 2001;52:5–20. doi: 10.1002/1097-0029(20010101)52:1<5::AID-JEMT3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr Top Dev Biol. 2005;71:1–52. doi: 10.1016/S0070-2153(05)71001-2. [DOI] [PubMed] [Google Scholar]
- Donsante A, Yi L, Zerfas PM, Brinster LR, Sullivan P, Goldstein DS, et al. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol Ther. 2011;19:2114–2123. doi: 10.1038/mt.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson DJ, Passini MA, Wolfe JH. Transduction of the choroid plexus and ependyma in neonatal mouse brain by vesicular stomatitis virus glycoprotein-pseudotyped lentivirus and adeno-associated virus type 5 vectors. Hum Gene Ther. 2005;16:49–56. doi: 10.1089/hum.2005.16.49. [DOI] [PubMed] [Google Scholar]
- Passini MA, Watson DJ, Vite CH, Landsburg DJ, Feigenbaum AL, Wolfe JH. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J Virol. 2003;77:7034–7040. doi: 10.1128/JVI.77.12.7034-7040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Fidler JA, Hester M, Haidet A, Handy C, et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol Ther. 2010;18:2075–2084. doi: 10.1038/mt.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Curtis MT, Wenger DA. Extended normal life after AAVrh10-mediated gene therapy in the mouse model of Krabbe disease. Mol Ther. 2012;20:2031–2042. doi: 10.1038/mt.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolewski BA, Wolfe JH. Genetic correction of the fetal brain increases the lifespan of mice with the severe multisystemic disease mucopolysaccharidosis type VII. Mol Ther. 2006;14:14–24. doi: 10.1016/j.ymthe.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, McNair R, Bradley J, Lunn D, Ziegler R, et al. Gene transfer corrects acute GM2 gangliosidosis–potential therapeutic contribution of perivascular enzyme flow. Mol Ther. 2012;20:1489–1500. doi: 10.1038/mt.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donsante A, Haddad MR, Kaler SG. Directed evolution to create choroid plexus-permissive AAV capsid variants. Mol Ther. 2012;20 suppl. 1:S229. [Google Scholar]
- Wolfe JH.2011Viral vector gene delivery to the brain to treat the disseminated lesions of neurogenetic diseases: focus on lysosomal storage diseases Glorioso J.ed.). Gene Vector Design and Application to Treat Nervous System Disorders Society for Neuroscience: Washington, DC; pp. 25–32. [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162:570–572. doi: 10.1126/science.162.3853.570. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Caviness VS, Jr, Erbe RW, Patterson MC, Schultz KR, Wenger DA, et al. The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet Med. 2009;11:450–454. doi: 10.1097/GIM.0b013e3181a16e04. [DOI] [PubMed] [Google Scholar]
- Hemsley KM, Hopwood JJ. Delivery of recombinant proteins via the cerebrospinal fluid as a therapy option for neurodegenerative lysosomal storage diseases. Int J Clin Pharmacol Ther. 2009;47 suppl. 1:S118–S123. doi: 10.5414/cpp47118. [DOI] [PubMed] [Google Scholar]
- Dierenfeld AD, McEntee MF, Vogler CA, Vite CH, Chen AH, Passage M, et al. Replacing the enzyme alpha-L-iduronidase at birth ameliorates symptoms in the brain and periphery of dogs with mucopolysaccharidosis type I. Sci Transl Med. 2010;2:60ra89. doi: 10.1126/scitranslmed.3001380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemsley KM, King B, Hopwood JJ. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab. 2007;90:313–328. doi: 10.1016/j.ymgme.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Crawley AC, Marshall N, Beard H, Hassiotis S, Walsh V, King B, et al. Enzyme replacement reduces neuropathology in MPS IIIA dogs. Neurobiol Dis. 2011;43:422–434. doi: 10.1016/j.nbd.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Lee WC, Tsoi YK, Troendle FJ, DeLucia MW, Ahmed Z, Dicky CA, et al. Single-dose intracerebroventricular administration of galactocerebrosidase improves survival in a mouse model of globoid cell leukodystrophy. FASEB J. 2007;21:2520–2527. doi: 10.1096/fj.06-6169com. [DOI] [PubMed] [Google Scholar]
- Chang M, Cooper JD, Sleat DE, Cheng SH, Dodge JC, Passini MA, et al. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. doi: 10.1038/mt.2008.9. [DOI] [PubMed] [Google Scholar]
- Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA. Evidence for a ‘paravascular' fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 1985;326:47–63. doi: 10.1016/0006-8993(85)91383-6. [DOI] [PubMed] [Google Scholar]
- Rahim AA, Wong AM, Hoefer K, Buckley SM, Mattar CN, Cheng SH, et al. Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. FASEB J. 2011;25:3505–3518. doi: 10.1096/fj.11-182311. [DOI] [PubMed] [Google Scholar]
- Rahim AA, Wong AM, Ahmadi S, Hoefer K, Buckley SM, Hughes DA, et al. In utero administration of Ad5 and AAV pseudotypes to the fetal brain leads to efficient, widespread and long-term gene expression. Gene Ther. 2012;19:936–946. doi: 10.1038/gt.2011.157. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Green K. Cell turnover in ciliary epithelium compared to other slow renewing epithelia in the adult mouse. Curr Eye Res. 1988;7:247–252. doi: 10.3109/02713688809047029. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, Vandeberg JL, Saunders NR. Development of the lateral ventricular choroid plexus in a marsupial, Monodelphis domestica. Cerebrospinal Fluid Res. 2010;7:16. doi: 10.1186/1743-8454-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Bryant KD, Sun J, Brown SM, Troupes A, Pulicherla N, et al. Glycan binding avidity determines the systemic fate of adeno-associated virus type 9. J Virol. 2012;86:10408–10417. doi: 10.1128/JVI.01155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte TR, Conlon TJ, Poirier A, Campbell-Thompson M, Byrne BJ. Preclinical characterization of a recombinant adeno-associated virus type 1-pseudotyped vector demonstrates dose-dependent injection site inflammation and dissemination of vector genomes to distant sites. Hum Gene Ther. 2007;18:245–256. doi: 10.1089/hum.2006.113. [DOI] [PubMed] [Google Scholar]
- Salegio EA, Samaranch L, Jenkins RW, Clarke CJ, Lamarre C, Beyer J, et al. Safety study of adeno-associated virus serotype 2-mediated human acid sphingomyelinase expression in the nonhuman primate brain. Hum Gene Ther. 2012;23:891–902. doi: 10.1089/hum.2012.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar CN, Waddington SN, Biswas A, Johana N, Ng XW, Fisk AS, et al. Systemic delivery of scAAV9 in fetal macaques facilitates neuronal transduction of the central and peripheral nervous systems. Gene Ther. 2013;20:69–83. doi: 10.1038/gt.2011.216. [DOI] [PubMed] [Google Scholar]
- Dawson-Saunders B, Trapp RG.1994Basic and Clinical Biostatistics2nd edn. Appleton & Lange: Norwalk, CT; pp. 344 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Densitometry to quantitate GFP transgene expression in mouse choroid plexus.