

Abstract
The present work aimed at the development and application of a lipid-based nanocarrier for targeted delivery of nucleic acids to glioblastoma (GBM). For this purpose, chlorotoxin (CTX), a peptide reported to bind selectively to glioma cells while showing no affinity for non-neoplastic cells, was covalently coupled to liposomes encapsulating antisense oligonucleotides (asOs) or small interfering RNAs (siRNAs). The resulting targeted nanoparticles, designated CTX-coupled stable nucleic acid lipid particles (SNALPs), exhibited excellent features for in vivo application, namely small size (<180 nm) and neutral surface charge. Cellular association and internalization studies revealed that attachment of CTX onto the liposomal surface enhanced particle internalization into glioma cells, whereas no significant internalization was observed in noncancer cells. Moreover, nanoparticle-mediated miR-21 silencing in U87 human GBM and GL261 mouse glioma cells resulted in increased levels of the tumor suppressors PTEN and PDCD4, caspase 3/7 activation and decreased tumor cell proliferation. Preliminary in vivo studies revealed that CTX enhances particle internalization into established intracranial tumors. Overall, our results indicate that the developed targeted nanoparticles represent a valuable tool for targeted nucleic acid delivery to cancer cells. Combined with a drug-based therapy, nanoparticle-mediated miR-21 silencing constitutes a promising multimodal therapeutic approach towards GBM.
Keywords: chlorotoxin, glioblastoma, liposome, miR-21, stable nucleic acid lipid particle
Introduction
Glioblastoma (GBM) is the most common and lethal primary brain tumor in humans.1 Despite the ongoing research efforts, current treatment options for GBM are largely unsatisfactory and the prognosis is usually poor with a 9- to 12-month median survival time (following diagnosis) that has not improved significantly over the last decade.2,3 It is therefore important to develop new therapeutical strategies that could encompass both high specificity for tumor cells and complete tumor eradication.
Driven by the tremendous advances in molecular biology, gene therapy constitutes an attractive approach for modulation of the cell genetic background. Regulators of gene transcription and translation operate at multiple levels in order to fine-tune the genome end products. MicroRNAs (miRNAs) are elements of this complex modulatory network that play a pivotal role in cell fate. Dysregulation of several miRNAs (such as miR-21) has indeed been associated with development and progression of several cancers, including GBM.4,5 Therefore, these small post-transcriptional regulators constitute novel and highly promising targets for antitumoral strategies.
Due to their unique characteristics (low size, low immunogenicity, high target affinity), antisense oligonucleotides (asOs) constitute an important tool for the manipulation of miRNA function in biological systems. In this regard, recent studies (including those reported in our previous manuscript) have shown that miRNA modulation in GBM cells results in decreased tumor cell migration and proliferation, as well as increased cytotoxic effect of antineoplastic drugs.6,7,8 Nevertheless, the successful therapeutic application of oligonucleotide-based therapies to brain cancer requires novel strategies to overcome the barriers imposed by this complex organ. The presence of the blood–brain barrier, which restricts entry of therapeutic molecules into the brain,9 and the possible degradation of nucleic acids by nucleases present in the blood constitute major obstacles associated with nucleic acid delivery in vivo. It is, therefore, crucial that oligonucleotides are properly delivered by vehicles that are not only reliable and effective in overcoming cellular and physiological barriers, but are also highly target specific. Carrier systems, such as viruses or liposomes, have been developed to ensure protection and improvement of nucleic acid delivery into target cells.10,11,12 Recently, a new class of nucleic acid lipid particles, designated stable nucleic acid lipid particles (SNALPs), were shown (by us and other groups) to be very efficient in delivering small interfering RNAs (siRNAs), both in vitro and in vivo.13,14,15 Targeted therapy using peptides coupled to liposomal systems towards overexpressed tumoral receptors enables tumor-specific delivery, while minimizing side effects to normal cells. In this regard, chlorotoxin (CTX), a scorpion-derived peptide, was reported as a specific marker for gliomas16 and is currently used as a targeting agent in imaging studies (as well as in delivery of RNA interference therapeutics).17,18 Moreover, CTX was reported to bind to matrix metalloproteinase-2 that is specifically upregulated in gliomas and related cancers, but poorly expressed in brain and normal tissues.19
In this work, we employed CTX as a ligand to design targeted SNALPs for the delivery of asOs and siRNAs to GBM. Our results show that CTX-coupled SNALPs are more effective (both in vitro and in vivo) in mediating nucleic acid delivery to tumor cells than their nontargeted (NT) counterpart. Moreover, we demonstrate that SNALP-mediated miR-21 silencing in GBM/glioma cells increases the expression of the tumor suppressors PTEN and PDCD4, enhances caspase 3/7 activity, and, importantly, enhances the cytotoxic effect of the antiangiogenic drug sunitinib.
Results
Preparation and physicochemical characterization of targeted (CTX-coupled) and NT SNALPs
We have previously developed a lipid formulation composed of DODAP/DSPC/Chol/C16 mPEG2000 Ceramide (25:22:45:8, mol%), which was shown to efficiently encapsulate both siRNAs and asOs.15 Here, locked nucleic acid (LNA)-modified asOs or siRNAs were encapsulated into this lipid formulation. Aiming at achieving specific tumor-targeting and increasing intracellular delivery, the ligand CTX was attached to the liposomal surface. In this regard, when liposomes, encapsulating anti-miR-21 oligonucleotides or siRNAs, were incubated with 4 mol% of micelles, values of 5.4 ± 2.8 and 5.0 ± 3.0 nmol CTX/µmol total lipid (respectively) and ~500 molecules of CTX per liposome were obtained. As shown in Table 1, the postinsertion step did not interfere with the loading of the encapsulated nucleic acids, since high encapsulation yields were obtained for anti-miR-21 oligonucleotides and siRNA in both targeted (89.3 ± 19.8 and 87.5 ± 11.1%, respectively) and NT (82.2 ± 15.8 and 85.5 ± 17.1%) formulations. Moreover, the SNALPs exhibited a net surface charge close to neutrality, with lower values of ζ potential for NT formulations (encapsulating siRNAs or anti-miR-21 oligonucleotides) as compared with CTX-coupled formulations (Table 1).
Table 1. Physicochemical characterization of CTX-coupled and NT liposomes encapsulating anti-miR-21 oligonucleotides (anti-miR-21) or anti-survivin siRNAs.
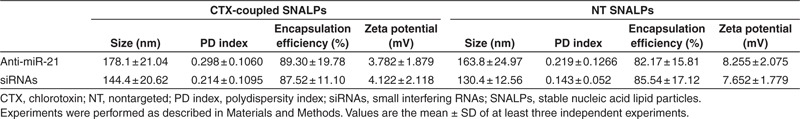
The developed formulations also revealed capacity to protect the nucleic acid molecules from nuclease degradation, since in the absence of the detergent C12E8, the intercalation of the probe SYBR Safe with the encapsulated anti-miR-21 oligonucleotides/siRNAs was reduced by ~87 and 92%, respectively. Results obtained from photon correlation spectroscopy revealed that all formulations exhibited a size under 180 nm, nanoparticles encapsulating siRNAs being generally smaller than those encapsulating anti-miR-21 oligonucleotides with a narrow distribution (polydispersity index <0.3) (Table 1). The insertion of protein conjugates onto the liposome surface resulted in a small increase of liposomal size: NT liposomes encapsulating anti-miR-21 oligonucleotides or siRNAs exhibited 163.8 ± 25.0 and 130.4 ± 12.6 nm, respectively, whereas CTX-coupled liposomes encapsulating anti-miR-21 oligonucleotides or siRNAs exhibited 178.1 ± 21.0 and 144.4 ± 20.6 nm, respectively. Nevertheless, particle aggregation (and consequent increase in size) was observed for CTX-coupled SNALPs 3 months after their preparation, whereas particle aggregation was observed to a lesser extent in the NT formulations (data not shown), thus indicating that the presence of CTX may decrease the stability of the formulation over time.
Evaluation of cellular association of SNALPs by flow cytometry
Extensive association was observed 4 hours after exposure of U87 cells (at 37 °C) to CTX-coupled liposomes encapsulating 0.5 µmol/l of oligonucleotides, which was further enhanced when cells were exposed to 1 µmol/l of oligonucleotides encapsulated in targeted SNALPs (~75% fluorescent cells, 9.6 ± 4.2-fold increase in fluorescence intensity) when compared with that observed in cells exposed to 1 µmol/l of oligonucleotides encapsulated in NT liposomes (~5% fluorescent cells, 1.6 ± 0.4) (Figure 1a,b), as assessed by flow cytometry. Similar results were obtained when GL261 mouse glioma cells were incubated with CTX-coupled or NT liposomes encapsulating 0.5 or 1 µmol/l of oligonucleotides (Figure 1c,d). In contrast, following cell incubation with SNALPs at 4 °C (Figure 1b) or with 1 µmol/l of oligonucleotides, either per se or encapsulated in liposomes associated with a smaller amount of CTX (1 mol% of micelles instead of 4 mol%) (data not shown), no significant cellular association was detected.
Figure 1.

Association of stable nucleic acid lipid particles (SNALPs) with U87 human glioblastoma, GL261 mouse glioma and HEK293T human embryonic kidney cells. Cells were incubated with chlorotoxin (CTX)-coupled or nontargeted (NT) liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides (for 4 hours), rinsed with phosphate-buffered saline and prepared for flow cytometry analysis (as described in Materials and Methods). The extent of cell association was assessed only in viable cells, these being gated on the basis of morphological features (including cell volume and complexity). (a,c) Cellular association and (b,d) fluorescence intensity plots of (a,b) U87 and (c,d) GL261 cells incubated with SNALPs at 4 (U87) and 37 °C (U87, GL261). (e) Fluorescence intensity plot of U87 cells exposed to CTX-coupled or NT SNALPs either per se (without free CTX) or following preincubation with 20 µmol/l of free CTX (20 µmol/l free CTX), or incubated with bovine serum albumin-coupled liposomes. (f) Cellular association and (g) fluorescence intensity plots of U87 and HEK293T cells incubated with CTX-coupled or NT SNALPs at 37 °C. The percentage of cellular association in a, c, and f was normalized to control cells (untreated). Relative fluorescence units to control cells (untreated) are indicated for b, d, e, and g. Values are presented as means ± SD (n = 3). ***P < 0.001 compared to cells exposed to a similar amount of NT SNALP-formulated oligonucleotides. @P < 0.05 compared to cells exposed to 0.5 µmol/l of CTX-coupled SNALP-formulated oligonucleotides. ##P < 0.01, ###P < 0.001 compared to U87 cells exposed to 1 µmol/l of CTX-coupled SNALP-formulated oligonucleotides.
To demonstrate that cellular association of CTX-coupled SNALPs was mediated by specific interaction with cellular receptors, U87 cells were preincubated with 20 µmol/l of free CTX to block the CTX receptors. A moderate decrease in cellular association (reflected in the decrease in fluorescence intensity) was observed when cells were exposed to free CTX before the addition of CTX-coupled liposomes encapsulating 1 µmol/l of oligonucleotides (7.4 ± 1.9) when compared with that detected in cells exposed to 1 µmol/l of targeted SNALPs (11.3 ± 3.0). Reduced extent of association was also observed in cells exposed to bovine serum albumin-coupled liposomes encapsulating 0.5 or 1 µmol/l of oligonucleotides (Figure 1e).
Aiming at evaluating whether CTX-coupled SNALPs would specifically target tumor cells, experiments were performed to determine the extent of their association with the nonmalignant cell line HEK293T (human embryonic kidney). As demonstrated in Figure 1f,g, a significant decrease in the extent of cellular association was observed following incubation with CTX-coupled SNALPs when compared with that determined in U87 GBM cells exposed to similar amounts of targeted SNALP-formulated oligonucleotides. Similar results were obtained from parallel experiments performed with primary cultures of mouse astrocytes (Supplementary Figure S1).
Evaluation of cellular internalization by confocal microscopy
In order to confirm the results obtained on targeting specificity of CTX-coupled SNALPs by flow cytometry, cell internalization studies were performed using confocal microscopy. The results shown in Figure 2 reveal that following incubation of U87 cells, at 37 °C, with rhodamine-labeled CTX-coupled liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides, intensive red (lipid) and moderate green (oligonucleotide) fluorescence was detected throughout the cell cytoplasm (Figure 2b,d), whereas residual fluorescence was detected in the cytoplasm of cells exposed to NT liposomes (Figure 2a,c). A similar pattern of internalization was observed in GL261 mouse and F98 rat glioma cells exposed to liposomes encapsulating FAM-labeled oligonucleotides (Supplementary Figure S2), while only residual fluorescence was detected in mouse primary astrocytes (Figure 2e,f) or HEK293T cells (Figure 2g,h) incubated under the same conditions. Moreover, reduced internalization was observed upon cell incubation with CTX-coupled liposomes at 4 °C (Figure 3c,f) or presaturation of the CTX receptor with excess of free CTX (20 µmol/l) (Figure 3b,e) when compared with that observed in cells exposed to CTX-coupled liposomes at 37 °C (Figure 3a,d).
Figure 2.
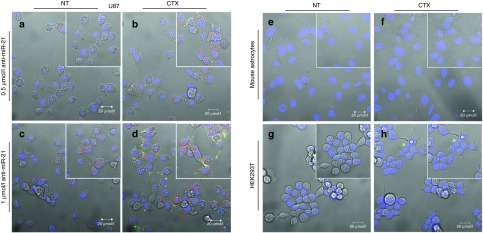
Stable nucleic acid lipid particle (SNALP) internalization in U87 glioblastoma cells, HEK293T human embryonic kidney cells and mouse primary astrocytes. Cells were incubated with chlorotoxin (CTX)-coupled or nontargeted (NT) liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides (for 4 hours at 37 °C), rinsed twice with phosphate-buffered saline, stained with DNA-specific Hoechst 33342 (blue) and then observed by confocal microscopy. The panel shows representative images at ×40 magnification of (a–d) U87 cells incubated with either (a,c) rhodamine-labeled NT or (b,d) CTX-coupled liposomes at a final oligonucleotide concentration of (a,b) 0.5 or (c,d) 1 µmol/l. The yellow dots are most likely due to the cellular colocalization of lipid and nucleic acid. Representative images of (e,f) mouse astrocytes and (g,h) HEK293T cells incubated with 1 µmol/l of SNALP-formulated oligonucleotides. Results are representative of two independent experiments. Bars corresponds to 20 µm.
Figure 3.

Stable nucleic acid lipid particle (SNALP) internalization in U87 glioblastoma cells and effect of cell preincubation with free chlorotoxin (CTX). Cells were incubated with CTX-coupled liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides (for 4 hours), rinsed twice with phosphate-buffered saline, stained with DNA-specific Hoechst 33342 (blue) and then observed by confocal microscopy. The panel shows representative images at ×40 magnification of U87 cells exposed to targeted SNALP-formulated oligonucleotides at 37 °C (a,d) either per se (37 °C) or (b,e) following preincubation with 20 µmol/l of free CTX for 1 hour (37 °C + CTX). (c,f) Cells exposed to targeted SNALP-formulated oligonucleotides at 4 °C. Results are representative of two independent experiments. Bars corresponds to 20 µm.
Although aggregation has been observed for CTX-coupled SNALPs 3 months after their preparation, images obtained by confocal microscopy (Supplementary Figure S3) and data from quantitative PCR showing a decrease in miR-21 expression levels in cells exposed to these SNALPs (data not shown) suggest that this formulation was internalized by the cells.
Cellular association and internalization studies of liposomes encapsulating siRNAs also revealed enhanced particle uptake in cells exposed to increased concentrations of siRNAs encapsulated in CTX-coupled liposomes when compared with their NT counterpart (Supplementary Figure S4).
MiR-21 silencing mediated by CTX-coupled SNALPs and its effect on the expression of the target proteins PTEN and PDCD4
Having shown that CTX-coupled SNALPs efficiently deliver oligonucleotides to GBM/glioma cells, we evaluated whether intracellularly delivered anti-miR-21 oligonucleotides could modulate the expression of mature miR-21. As illustrated in Figure 4a,b, incubation of U87 and GL261 cells with 0.25 µmol/l of SNALP-formulated anti-miR-21 oligonucleotides resulted in a significant decrease in miR-21 levels (0.17 ± 0.22 and 0.21 ± 0.09, respectively), which was further enhanced with increasing concentrations of anti-miR-21 oligonucleotides (0.5 and 1 µmol/l). Parallel experiments demonstrated that cell exposure to NT liposomes encapsulating anti-miR-21 oligonucleotides did not considerably affect the levels of miR-21 (Supplementary Figure S5).
Figure 4.
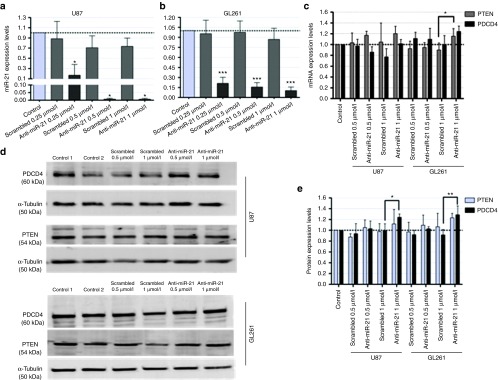
MiR-21 and PTEN/PDCD4 expression in U87 glioblastoma and GL261 glioma cells following incubation with chlorotoxin (CTX)-coupled liposomes encapsulating anti-miR-21 oligonucleotides. (a,b) MiR-21 and (c) PTEN/PDCD4 mRNA expression levels in (a) U87 and (b) GL261 cells, 48 hours after cell incubation with CTX-coupled liposomes encapsulating anti-miR-21 or scrambled oligonucleotides (n = 3). MiR-21 expression levels, normalized to the reference snord44 (human) or snord110 (mouse), and PTEN and PDCD4 expression levels, normalized to the reference HPRT1, are presented as relative expression values to control untreated cells. (d) Representative gel showing PTEN and PDCD4 protein levels in U87 (upper panel) and GL261 (lower panel) cells 48 hours after cell incubation with CTX-coupled liposomes encapsulating anti-miR-21 or scrambled oligonucleotides (n = 3). (e) Quantification of PTEN and PDCD4 bands observed in d, corrected for individual α-tubulin signal intensity. Results are presented as PTEN and PDCD4 expression levels relative to control. Values are presented as means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 to cells incubated with a similar amount of CTX-coupled liposomes encapsulating scrambled oligonucleotides.
MiR-21 silencing was also reflected on the expression of two of its targets, the tumor suppressors PDCD4 and PTEN.20,21 As shown in Figure 4c, a moderate increase in PTEN mRNA levels was observed in both U87 (~15%, P > 0.05) and GL261 (25%, P < 0.05) cells incubated with 1 µmol/l anti-miR-21 oligonucleotides as compared with those observed when cells were exposed to a similar amount of scrambled oligonucleotides. Although no significant changes were observed in U87 cells, a small increase in PDCD4 mRNA was obtained in GL261 cells under the same experimental conditions (~20%, P > 0.05). More importantly, a considerable and significant increase in PDCD4 protein expression was observed in both U87 (25%, P < 0.05) and GL261 (30%, P < 0.01) cells incubated with 1 µmol/l of anti-miR-21 oligonucleotides when compared with that observed in cells transfected with a scrambled sequence (Figure 4d,e).
Similarly to what was observed with the intracellularly delivered anti-miR-21 oligonucleotides, CTX-coupled liposome-mediated anti-survivin-siRNA delivery resulted in decreased levels of survivin mRNA (Supplementary Figure S4).
Evaluation of caspase activation and apoptosis in tumor cell lines with reduced miR-21 expression
Since miR-21 has been proposed to play an antiapoptotic role in GBM, we investigated whether miR-21 silencing mediated by CTX-coupled SNALPs would affect the activity of the effector caspases 3 and 7, crucial components of the apoptotic cell death. As shown in Figure 5a, incubation of U87 and GL261 cells with 1 µmol/l of SNALP-formulated anti-miR-21 oligonucleotides resulted in a twofold increase (~1.76 and 1.66, respectively) in caspase 3/7 activity (P > 0.05) as compared with that observed upon incubation to SNALP-formulated scrambled oligonucleotides. More importantly, silencing of miR-21 followed by cell exposure to 15 (U87) and 5 (GL261) µmol/l of the tyrosine kinase inhibitor sunitinib resulted in a considerable increase in caspase 3/7 activity (5.3 ± 2.3 and 4.8 ± 1.9, respectively) when compared with that observed for cells exposed to sunitinib, either per se (1.6 ± 0.7 and 2.1 ± 0.8) or in combination scrambled oligonucleotides (2.1 ± 1.1, P < 0.01 and 2.9 ± 0.8, P > 0.05).
Figure 5.
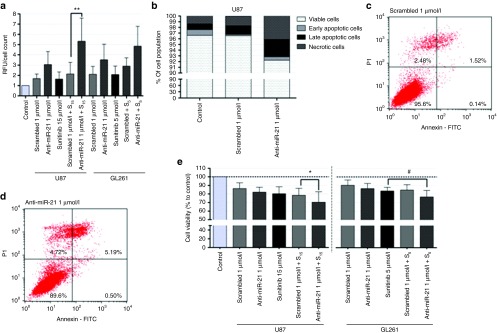
Evaluation of caspase activation, apoptosis and tumor cell proliferation in U87 glioblastoma and GL261 glioma cells. Cells were incubated with chlorotoxin-coupled liposomes encapsulating anti-miR-21 or scrambled oligonucleotides for 4 hours, washed with phosphate-buffered saline (PBS) and further incubated for 24 hours with fresh medium. Cells were subsequently exposed to 15 (U87) or 5 µmol/l (GL261) of sunitinib for 24 hours, rinsed with PBS, after which caspase/cell death detection and cell viability assays were performed. (a) Caspase 3/7 activity in U87 and GL261 cells incubated with 1 µmol/l of stable nucleic acid lipid particle (SNALP)-formulated anti-miR-21 or scrambled oligonucleotides, either per se or in combination with sunitinib. Results, presented as relative fluorescence units with respect to control untreated cells, were normalized for the number of cells in each condition. (b) Cell death detection in U87 cells exposed to 1 µmol/l of SNALP-formulated anti-miR-21 or scrambled oligonucleotides. For each condition (control, scrambled/anti-miR-21 1 µmol/l), results are presented as percentage of viable, early/late apoptotic and necrotic cells. Representative cell death plots for U87 cells incubated with (c) SNALP-formulated scrambled and (d) anti-miR-21 oligonucleotides. The percentage of viable (lower left), early apoptotic (lower right), late apoptotic (upper right) and necrotic (upper left) cells in the cell population is indicated in the plots. (e) Cell viability, evaluated by the Alamar Blue assay (as described in Materials and Methods) immediately after cell incubation with sunitinib. Values are presented as means ± SD (n = 3). Scrambled/anti-miR-21 1 µmol/l + S15/5: cells transfected with scrambled or anti-miR-21 oligonucleotides and further incubated with 15 or 5 µmol/l sunitinib. **P < 0.01 compared to cells incubated with SNALP-formulated scrambled oligonucleotides and further treated with 15 µmol/l sunitinib.
Furthermore, an increase in the percentage of late apoptotic (P > 0.05) and necrotic cells (P < 0.05) was observed in U87 cells incubated with 1 µmol/l of SNALP-formulated anti-miR-21 oligonucleotides (Figure 5b,d), compared to that observed for cells incubated to 1 µmol/l of SNALP-formulated scrambled oligonucleotides (Figure 5b,c). The presence of sunitinib, a fluorescently active molecule, interfered with the cytometric detection of FAM-labeled annexin V and propidium iodide and, therefore, no information could be obtained about apoptotic cell death in the presence of sunitinib.
Evaluation of tumor cell death following miR-21 silencing
We further evaluated whether the increase in tumor suppressor expression and caspase activity observed following SNALP-mediated miR-21 silencing would correlate with changes in tumor cell proliferation. Initial experiments were performed by exposing GL261 cells to different concentrations of sunitinib for 24 hours (Supplementary Figure S6) to determine the optimal concentration of drug to be used in the assay. As demonstrated in Figure 5e, a small decrease in cell viability was observed when U87 cells were incubated with 1 µmol/l of SNALP-formulated anti-miR-21 oligonucleotides (82.1 ± 5.7) as compared with that observed for cells incubated with the same formulation encapsulating 1 µmol/l of scrambled oligonucleotides (86.3 ± 6.7, P > 0.05). Remarkably, a considerable decrease in the percentage of viable cells was observed when cells were exposed to 1 µmol/l of anti-miR-21 oligonucleotides and further exposed to sunitinib (70.3 ± 12.3) when compared to that observed upon exposure to sunitinib, either per se (80.2 ± 8.1, P > 0.05) or in combination with 1 µmol/l of scrambled oligonucleotides (78.5 ± 8.1, P < 0.05).
A significant decrease in the percentage of viable cells was also observed when GL261 cells were incubated with 1 µmol/l of anti-miR-21 oligonucleotides and further exposed to sunitinib when compared to that observed upon exposure to sunitinib per se (~8% decrease, Figure 5e).
Characterization of the glioma mouse model and intravenous injection of SNALP-formulated siRNAs
Following the demonstration of the efficacy of CTX-coupled SNALPs to deliver LNA-modified asOs (and siRNAs) to glioma cells, studies were addressed to investigate whether the developed nanoparticles would be efficient in delivering their encapsulated contents to intracranial tumors when administered via systemic route. For this purpose, we employed a previously developed (and characterized) mouse glioma model that displays molecular and histopathological features of human GBM.22 The stereotactic injection of 1.25 × 105 GL261 cells resulted in the formation of tumors, microscopically visible after 10 days, with an average tumor size of 75.5 ± 18.9 mm3 20 days after tumor implantation (Figure 6a,b).
Figure 6.

Tumor characterization and biodistribution analysis of systemically-administered liposome-formulated FAM-labeled small interfering RNAs (siRNAs). Following tumor implantation, animals (n = 3) were killed at defined time points (10, 14, and 20 days), the brains were removed and fixed in 4% paraformaldehyde, frozen and further processed for histological evaluation, as described in Materials and Methods. (a) Average tumor volume (calculated as described in Materials and Methods) on the day of animal killing. Unpaired t-test with Welch's correction was used to calculate the statistical significance. (b) Representative photographs of GL261 tumors 20 days after implantation and (c) representative image of a 20-µm thick tumor section (14 days after tumor implantation), stained with cresyl violet and observed under a light microscope. The scale corresponds to 5,000 µm. Flow cytometry analysis (fluorescence intensity plots) of (d) tumor, brain and (e) liver, kidney, and lung homogenates from animals injected intravenously with chlorotoxin-coupled/nontargeted (NT) liposomes encapsulating FAM-labeled siRNAs or saline solution (phosphate-buffered saline (PBS)). The protocol for intravenous administration and tissue processing is described in Materials and Methods, as well as in Supplementary Materials and Methods. The extent of cellular association was assessed only in viable cells, these being gated on the basis of morphological features (including cell volume and complexity). Relative fluorescence units are indicated for d and e. Values are presented as means ± SD. @P < 0.05 compared to the tumor volume of animals killed 14 days after tumor implantation. #P < 0.05 compared to animals injected with a similar amount of NT stable nucleic acid lipid particle-formulated siRNAs. *P < 0.05 compared to animals injected with PBS.
Intravenous administration of SNALP-formulated siRNAs was performed 14 days after tumor cell implantation (Figure 6c), so that the tumor could reach a volume which would allow achieving a therapeutic effect with potential clinical impact. The detection of FAM in tissue homogenates through indirect immunofluorescence revealed an increase in fluorescent intensity in tumors of animals injected with CTX-coupled SNALPs (9.2 ± 2.9) when compared to that detected in tumors of animals injected with NT SNALPs (5.7 ± 2.2, P < 0.05) or saline solution (Figure 6d). Increased fluorescence was also detected in the liver of animals injected with CTX-coupled or NT SNALPs when compared to that detected in animals injected with phosphate-buffered saline (PBS) (Figure 6e).
Discussion
Despite the potential of nucleic acid-based strategies to generate highly specific and biocompatible drugs, these molecules have intrinsic limitations that restrain their in vivo administration, namely poor pharmacokinetics, fast blood clearance and inability to target specific tissues or cells. Nonviral vectors, namely cationic liposomes, were developed to improve both nucleic acid protection and the biodistribution profile, while enhancing uptake by the target cells. Despite being extensively utilized in vitro and in vivo, clinical application of cationic liposomes is limited by their large size and impaired ability to reach tissues beyond the vasculature, unless directly injected into the tissue.23 Advances in lipid chemistry and liposome preparation enabled the development of a new class of lipid-based carriers, designated SNALPs, which was found to be very efficient for in vivo delivery of siRNAs and asOs.15,24,25
In this work, we generated tumor-targeted stabilized liposomes encapsulating either LNA-modified anti-miR-21 oligonucleotides or siRNAs, and evaluated their biological activity in different glioma cell lines. In accordance with previously reported studies,24,26 high encapsulation yields were obtained for both asOs and siRNA (Table 1), which can be attributed to the inclusion in the formulation of DODAP, an ionizable lipid that is positively charged at pH 4 and was shown to improve nucleic acid entrapment.24 Moreover, the developed liposome formulation exhibited a net surface charge close to neutrality (Table 1), which is extremely important for successful in vivo application, as it reduces their ability to interact with serum proteins involved in the early clearance from the blood stream,27 thus increasing particle bioavailability. Although low yields of peptide-conjugate insertion were obtained upon attachment of CTX onto the liposomal surface, the obtained values are in agreement with those reported in studies involving transferrin-targeted liposomes,15 which may be due to the similar temperature used in the postinsertion step (39 °C) and the similar amount of CerC16 mPEG incorporated in the preformed liposomes (8 mol%).
Taken together, our observations indicate that CTX-coupled SNALPs display optimal physicochemical properties for an in vivo application, including high encapsulation efficiency, low size, electrical neutrality, and high protection against enzymatic degradation.
The stability of the generated nanoparticles was also reflected in their capacity to interact with the target cells. Cellular association and internalization studies demonstrated that the attachment of CTX to the liposomal surface (at 4 mol%) strongly enhanced uptake of the targeted SNALPs in U87 GBM and GL261 glioma cells when compared to their NT counterpart, this effect being dependent on the concentration of CTX (Figures 1 and 2). Furthermore, a moderate decrease in cellular association and internalization was observed in cells either incubated with 20 µmol/l of free CTX (to saturate the CTX receptor) before the addition of CTX-coupled SNALPs, or exposed to bovine serum albumin-coupled SNALPs (Figure 1), thus suggesting that the cellular uptake was peptide-specific, although unspecific binding may also be responsible for the internalization of a small fraction of the nanoparticles. Cellular association of CTX-coupled SNALPs was strongly inhibited when incubations were performed at 4 °C (Figures 1 and 3), which indicates that an energy-dependent process, most likely receptor-mediated endocytosis, is involved in the uptake of the targeted nanoparticles. In accordance with previously reported studies involving CTX,17,18 reduced extent of cellular association was observed in noncancer HEK293T human kidney cells and mouse astrocytes (Figures 1 and 2), which indicates that the interaction of CTX-coupled liposomes with the target cells was mostly tumor cell-specific. This observation may be of great relevance for clinical application, as it strongly indicates that the developed CTX-coupled formulation is internalized by tumor cells while sparing normal tissues, thus reducing the toxicity associated with its systemic administration.
Increased cellular association and internalization, as well as a significant decrease in the mRNA levels of survivin, were also obtained in cells exposed to CTX-coupled liposomes encapsulating anti-survivin siRNAs when compared to their targeted counterpart (Supplementary Figure S4), which provides evidence that the targeted nanoparticles are not only very efficient in delivering LNA-modified asOs to GBM/glioma cells, but also viable vehicles for the delivery of siRNAs to GBM/glioma cells.
While cellular association and internalization studies demonstrated that CTX enhances tumor cell uptake of liposomes encapsulating anti-miR-21 oligonucleotides or siRNAs, it is important to demonstrate that increased nanoparticle internalization results in alterations in the cell's biological functions. Several reports suggested that the incorporation of high percentages of CerC16-PEG2000 in stabilized liposomes may result in the loss of activity of the encapsulated siRNAs or asOs.13,28,29 The steric barrier imposed by PEG inhibits the interaction of liposomes with the endosomal membrane, which is essential for endosomal membrane destabilization and the subsequent release of the entrapped nucleic acids. In this regard, our results indicate that incorporation of 8 mol% of CerC16-PEG2000 in the developed formulation allows liposomal size stability without compromising the release of anti-miR-21 oligonucleotides into the cell cytoplasm, where the miRNA processing machinery is located. In accordance with previous studies,30,31 we demonstrate that targeted liposome-mediated anti-miR-21 delivery efficiently reduces miR-21 expression levels, thus increasing the expression of the tumor suppressor PTEN and PDCD4 (Figure 4), whose loss of expression (frequently observed in glioma) results in dysregulation of important signaling pathways that control cell proliferation, growth, differentiation, and survival.32,33,34 In contrast, cell incubation with NT liposomes encapsulating anti-muR-21 oligonucleotides did not significantly affect the levels of miR-21, which indicates that the presence of CTX in the formulation is crucial to achieve a biological effect.
The increased tumor suppressor expression and caspase 3/7 activity detected in U87 and GL261 cells with decreasing miR-21 levels not only provides evidence that CTX-coupled SNALPs are biologically active, but may also render the cells susceptible to drugs targeting other signaling pathways governing GBM tumorigenesis. In this regard, several in vitro studies (including our own) have already shown that miR-21 modulation potentiates the cytotoxic effect of antineoplastic drugs,6,35,36 which may be of great importance to overcome treatment resistance, one of the major unsolved problems in clinical oncology. The observation of increased cytotoxic effect of sunitinib, a drug currently being tested in several phase II clinical trials for GBM,37 following miR-21 silencing in U87 GBM and GL261 glioma cells via the generated CTX-coupled SNALPs (Figure 5), not only confirms the results obtained in our previously published studies, but enforces the huge potential of combining targeted liposome-mediated miR-21 silencing and antiangiogenic therapy, which may translate into meaningful therapeutics that benefit cancer patients.
Due to limitations inherent to in vitro models, the results from cell culture experiments should be validated in a reliable animal model of disease. Several reports have shown that the efficacy of in vivo tumor internalization of liposomes may be independent of the presence of a targeting ligand. Studies by Moreira and colleagues38 revealed that while antagonist G-targeted liposomes enhanced in vitro uptake, their tumor accumulation was similar to that observed for the NT liposomes. Similarly, Bartlett et al. demonstrated that transferrin-targeted and NT siRNA nanoparticles exhibited similar biodistribution and tumor accumulation with increased biological activity (reduction in tumor luciferase activity by 50%) being observed for the targeted formulation.39 In opposition, our results from experiments on the intravenous administration of SNALP-formulated siRNAs indicated that CTX enhances the tumor internalization of the nanoparticles when compared to their NT counterpart (Figure 6). Although the observed increase in particle internalization by the tumors was significantly smaller than that obtained in the cell-based assays, such increase was within the range of the values reported in the literature for other studies involving intravenous administration of nucleic acids formulations to the brain.40 Further studies involving SNALP-mediated anti-miR-21 intravenous administration, either per se or in combination with sunitinib, should ascertain the biological effect and tumor cell-killing potential of both targeted and NT formulations.
In accordance with other reported in vivo studies involving SNALPs,14,26,41 increased uptake of both CTX-coupled and NT SNALPs was also detected in the liver of animals following intravenous injection when compared to that detected in animals injected with PBS (Figure 6), thus suggesting that nonspecific particle retention occurs in organs involved in blood clearance (liver).
Overall, the results presented in this study demonstrate that the developed CTX-coupled SNALPs not only exhibit adequate physicochemical properties for intravenous administration, but also enhance the delivery of anti-miR-21 oligonucleotides to different glioma cell lines and intracranial tumors, while revealing reduced affinity for noncancer cells. Moreover, the molecular and cellular alterations that resulted from SNALP-mediated miR-21 silencing, including increased tumor suppressor expression and caspase activity, as well as increased cytotoxic activity of sunitinib, indicate that a multimodal SNALP-mediated therapeutic approach, combining miRNA silencing with antiangiogenic chemotherapy deserves to be explored in preclinical and clinical applications.
Materials and methods
Materials. Sunitinib malate (Sutent) was kindly offered by Pfizer (Basel, Switzerland). The lipids 1,2-dioleoyl-3-dimethylammonium-propane (DODAP), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), N-palmitoyl-sphingosine-1-[succinyl(methoxypolyethylene glycol) 2000] (C16 PEG2000 Ceramide), 1,2-distearoyl-sn-glycero-3-phospatidylethanolamine-N-[maleimide (polyethylene glycol)-2000] ammonium salt (DSPE-PEG-MAL), l-α-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) ammonium salt (NBD-PE) were acquired from Avanti Polar Lipids (Alabaster, AL). The LNA-modified anti-miR-21, FAM-labeled anti-miR-21 and noncoding (scrambled) oligonucleotides were acquired from Exiqon (Vedbaek, Denmark). The anti-survivin siRNA42 was obtained from Dharmacon (Lafayette, CO). All sequences are displayed in Supplementary Table S1. The 36 amino acid peptide CTX (MCMPCFTTDHQMARKCDDCCGGKGRGKCYGPQCLCR)43 was synthesized by Genecust (Dudelange, Luxembourg). The carboxyl terminus was modified by the addition of an amide group. All other reagents were obtained from Sigma (Munich, Germany) unless stated otherwise.
Cell lines and culturing conditions. The F98 rat and GL261 mouse glioma cell lines were kindly donated by Dr Hélène Elleaume (European Synchrotron Radiation Facility, Grenoble, France) and Dr Perez-Castillo (Universidad Autónoma de Madrid, Madrid, Spain), respectively; the U87 human GBM cell line was a kind gift from Dr Peter Canoll (Columbia University, New York, NY). HEK293T human embryonic kidney cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium containing 4.5 g/l glicose (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated FBS (Gibco, Paisley, Scotland), 100 U/ml penicillin (Sigma), 100 µg/ml streptomycin (Sigma), 10 mmol/l HEPES and cultured at 37 °C under a humidified atmosphere containing 5% CO2. Primary mouse cortical astrocyte cultures were prepared from the cerebral cortices of newborn pups according to established protocols.44 Cell plating densities that are not included in the Materials and Methods section are indicated in Supplementary Materials and Methods.
Preparation of liposomes encapsulating LNA-modified asOs or siRNAs. The preparation of liposomes encapsulating asOs or siRNAs was performed as described previously13,15 with a few modifications. Thirteen micromoles of a lipid mixture composed of DODAP:CHOL:DSPC:CerC16-PEG2000 (25:45:22:8, % molar ratio to total lipid) in 200 µl of absolute ethanol were slowly added, under strong vortex, to 0.086 µmol of anti-miR-21, 0.116 µmol of scrambled oligonucleotides or 0.041 µmol of anti-survivin siRNAs in 300 µl of 20 mmol/l citrate buffer (pH 4), previously heated at 60 °C. The final charge ratio of the preparation was 2:1 (cationic lipid:asOs). The resulting particles were extruded 21 times through 100-nm diameter polycarbonate membranes, using a LiposoFast basic extruder (Avestin, Toronto, Ontario, Canada). The removal of ethanol and nonencapsulated asOs or siRNAs was carried out by running extruded nanoparticles through a Sepharose CL-4B column equilibrated with HEPES-buffered saline (20 mmol/l HEPES, 145 mmol/l NaCl, pH 7.4). Subsequently, the total lipid concentration was assessed by cholesterol quantification, using the Liebermann–Burchard test.45 Briefly, 150 µl of Liebermann–Burchard reagent were added to 5 µl of sample (in a 96-well plate) followed by incubation at 37 °C for 30 minutes. Absorbance was measured at 625 nm in a spectrophotometer and the concentration was determined from a standard curve for cholesterol content.
Preparation and purification of targeted SNALPs. CTX-coupled (targeted) SNALPs were prepared by the postinsertion method.15,46 Briefly, CTX was modified with the addition of thiol groups upon reaction with freshly prepared 2-iminothiolane hydrochloride (2-IT, in HEPES-buffered saline pH 8) at a molar ratio of 1:10 (CTX:2-IT). The reaction occurred under gentle stirring for 1 hour in the dark at room temperature (RT). Thiolated CTX was then coupled to DSPE-PEG-MAL micelles, prepared in MES buffer pH 6.5,15 by a thioesther linkage (1:1, CTX:DSPE-PEG-MAL molar ratio). The coupling reaction was performed overnight (at RT) in the dark with gentle stirring. For the NT SNALPs, postinsertion was performed with plain micelles (without conjugated ligand), which were prepared by adding HEPES-buffered saline (pH 8.0) to the DSPE-PEG-MAL micelles. The neutralization of free maleimide groups in the micelles was carried out upon incubation with β-mercaptoethanol at a maleimide:β-mercaptoethanol molar ratio of 1:5 (0.52:2.6 µmol), under stirring for 30 minutes (at RT). The insertion of CTX-DSPE-PEG-MAL conjugates or plain DSPE-PEG-MAL micelles onto the preformed liposomes, at 1 or 4 mol% (relative to the total lipid concentration), was performed upon incubation in a water bath at 39 °C for 16 hours (in the dark). Targeted and NT SNALPs were purified by size exclusion chromatography on a Sepharose CL-4B column using HEPES-buffered saline (pH 7.4) as running buffer to remove nonconjugated micelles and chemical reagents (including unreacted 2-IT and β-mercaptoethanol) used during SNALPs preparation.
Characterization of the SNALPs. The final total lipid concentration was assessed by cholesterol quantification (using the Liebermann–Burchard test) as described above. The quantification of encapsulated asOs/siRNAs was performed with the DNA-intercalating probe SYBR Safe (Life Technologies, Carlsbad, CA) in the presence of the detergent octaethylene glycol monododecyl ether (C12E8) (Sigma). The encapsulation efficiency was calculated from the formula ((asO/total lipid) final molar ratio/(asO/total lipid) initial molar ratio) × 100. The extent of nucleic acid protection resulting from the encapsulation of the asOs/siRNAs into the liposomes was determined by evaluating the ability of the SYBR Safe probe to intercalate into the asOs/siRNAs in the absence of C12E8. The amount of CTX associated with the SNALPs determined using with the BCA Protein Assay Kit (Pierce, Rockford, IL) from a CTX standard curve (at 562 nm) in a microplate reader (SpectraMax Plus 384; Molecular Devices, Sunnyvale, CA). The insertion efficiency was calculated from the formula ((CTX/total lipid) final molar ratio/(CTX/total lipid) initial molar ratio) × 100. SNALPs size distribution was assessed by photon correlation spectroscopy using an N5 submicrometer particle size analyzer (Beckman Coulter, Miami, FL). Measurements were made at a 90° angle and at 20 °C. ζ-Potential measurements of targeted and NT SNALPs were performed at RT using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).
Assessment of cellular association by flow cytometry. To evaluate the extent of cellular association of the SNALPs, cells were plated onto 48-well plates at densities of 5.5 × 104 (HEK293T), 5 × 104 (U87), and 4.5 × 104 cells/well (GL261, mouse astrocytes). Twenty-four hours after plating, cells were incubated in OptiMEM (Gibco) with targeted (CTX-coupled or bovine serum albumin-coupled) or NT liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides for 4 hours at 4 or 37 °C. Subsequently, cells were washed twice with cold PBS (pH 7.4), detached by exposure to trypsin (5 minutes, 37 °C) and further washed twice with PBS. Cells were then resuspended in 350 µl of cold PBS and immediately analyzed in a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). FAM fluorescence was evaluated in the FL-2 channel and a total of 20,000 events were collected (unless otherwise stated). The data were analyzed by Cell Quest software (BD Biosciences).
Assessment of cellular association by confocal microscopy. To assess the extent of cellular internalization of the SNALPs, cells were plated onto ibiTreat 8-well slides (Ibidi, Munich, Germany) at densities of 3.5 × 104 (HEK293T), 3 × 104 (U87, GL261, F98), and 2.5 × 104 cells/well (mouse astrocytes). Twenty-four hours after plating, cells were incubated in OptiMEM with CTX-coupled or NT liposomes encapsulating FAM-labeled anti-miR-21 oligonucleotides for 4 hours at 4 or 37 °C. Cells were rinsed twice with PBS, stained with the DNA binding dye Hoechst 33342 (Molecular Probes, Eugene, OR) (1 µg/ml) for 5 minutes (in the dark), rinsed twice with PBS and maintained in this saline solution for image acquisition. Confocal images (single layer) were acquired in a point scanning confocal microscope Zeiss LSM 510 Meta (Carl Zeiss Microscopy, Goettingen, Germany), as described in Supplementary Materials and Methods.
Cell transfection with anti-survivin or scrambled siRNAs. For transfection with siRNAs, complexes of siRNAs with Lipofectamine RNAiMax (Invitrogen) were prepared, following the manufacturer's instructions, and added to cells, maintained in OptiMEM medium (Gibco), at a final concentration of 50 or 100 nmol/l siRNA. After incubation for 4 hours, cells were washed with PBS and further cultured in fresh Dulbecco's modified Eagle's medium for 48 hours.
RNA extraction and cDNA synthesis. RNA extraction and cDNA synthesis for miRNA and mRNA quantification were performed as described previously.6 The detailed protocol is provided in Supplementary Materials and Methods.
Quantitative PCR quantification of miRNA expression. miRNA quantification was performed in an iQ5 thermocycler using 96-well microtitre plates and the SYBR Green Master Mix (Exiqon). The primers for miR-21 and references snord110 (mouse) and snord44 (human) were also acquired from Exiqon. For each primer set, a master mix was prepared containing a fixed volume of SYBR Green master mix and the appropriate amount of each primer. For each reaction, performed in duplicate, 6 µl of master mix were added to 4 µl of template cDNA. Reaction conditions consisted of enzyme activation and well-factor determination at 95 °C for 10 minutes, followed by 40 cycles at 95 °C for 10 seconds (denaturation) and 60 seconds at 60 °C (annealing and elongation). The melting curve protocol started immediately after and consisted of 1 minute heating at 55 °C followed by 80 (10 seconds steps) with 0.5 °C increases in temperature at each step. Threshold values for threshold cycle determination (Ct) were generated automatically by the iQ5 Optical System Software. Relative miRNA levels were determined following the Pfaffl method for relative miRNA quantification in the presence of target and reference genes with different amplification efficiencies.47 The protocol for quantitative PCR quantification of mRNA is provided in Supplementary Materials and Methods.
Western blot analysis. The preparation of protein extracts and protein quantification were performed as described previously6 and shown in Supplementary Materials and Methods. Twenty-five microgram of total protein were resuspended in loading buffer (20% glycerol, 10% sodium dodecyl sulfate, 0.1% bromophenol blue), incubated for 5 minutes at 95 °C and loaded onto a 10% polyacrylamide gel for electrophoretic separation. After electrophoresis, the proteins were blotted onto a polyvinylidene difluoride membrane, which was blocked in 5% nonfat milk and further incubated with an anti-PTEN or anti-PDCD4 (1:1,000) (Cell Signaling, Beverly, MA) overnight at 4 °C, and with the appropriate alkaline phosphatase-labeled secondary antibody (1:20,000) (Amersham, Uppsala, Sweden) for 2 hours at RT. Equal protein loading was shown by reprobing the membrane with an anti-α-tubulin antibody (1:10,000) (Sigma) and with the same secondary antibody. The blots were washed several times with TBS/T (25 mmol/l Tris–HCl, 150 mmol/l NaCl, 0.1% Tween-20), incubated with ECF (alkaline phosphatase substrate) for 5 minutes (at RT) and then submitted to fluorescence detection at 570 nm using a VersaDoc Imaging System Model 3000 (Bio-Rad, Hercules, CA). For each membrane, the band intensity was analyzed using the ImageJ software.48
Evaluation of caspase 3/7 activity. Caspase-3/7 activity was assessed using the SensoLyte homogenous AMC caspase-3/7 assay (AnaSpec, San Jose, CA), as described previously.6 The detailed protocol is provided in Supplementary Materials and Methods.
Evaluation of apoptotic cell death. The detection of apoptosis was performed in U87 cells using the FITC Annexin V Apoptosis Detection Kit II (BD Pharmingen, San Diego, CA). Briefly, 48 hours after SNALP-mediated oligonucleotide transfection or immediately after exposure to sunitinib, cells were detached using trypsin, washed twice with cold PBS and resuspended in 1X binding buffer at a concentration of 1 × 106 cells/ml. From this suspension, 1 × 105 cells were transferred to 5 ml polystyrene tubes, followed by the addition 5 µl of FITC Annexin V and 5 µl propidium iodide and cells were then incubated for 15 minutes (at RT) in the dark. Four-hundred microliter of 1X binding buffer were added to each tube and the samples were immediately analyzed in a FACSCalibur flow cytometer. FITC fluorescence was evaluated in the FL-2 channel, propidium iodide was evaluated in the FL-3 and a total of 20,000 events were collected. The data were analyzed by Cell Quest software.
Evaluation of cell viability. Cell viability was evaluated by a modified Alamar blue assay, as described previously.49 The detailed protocol is provided in Supplementary Materials and Methods.
Establishment of an orthotopic glioma mouse model and histological analysis. Primary tumors were induced in the right hemisphere of adult male C57BL/6 mice, obtained from Charles River Laboratories (Wilmington, MA) as described by Aguilar-Morante et al.22 with a few modifications. Briefly, 8-week-old mice anesthetized by intraperitoneal administration of ketamine/xylazine (100 and 10 mg/kg, respectively) were injected stereotactically with 1.25 × 105 GL261 glioma cells into the right hemisphere (stereotactic coordinates relative to bregma: −1.06 mm anterior, 3 mm lateral, 3 mm deep) using a Hamilton syringe with a 22-gauge needle (3 µl at 0.2 µl/minute). Following surgery, animals were monitored daily and killed as soon as they displayed neurological deficits or lost >20% of their body weight. For determination of the tumor growth rate, animals were killed at 11, 14, or 20 days after tumor implantation by intracardiac perfusion with 15 ml ice-cold PBS, followed by 15 ml cold 4% paraformaldehyde before tissue harvesting. Brains were collected into polystyrene tubes and fixed overnight at 4 °C in 4% paraformaldehyde, placed in 25% sucrose for additional 48 hours and frozen (after drying) at −80 °C. Sequential cryosections (20 µm) were obtained by microtome sectioning and processed for cresyl violet staining (as described in Supplementary Materials and Methods). Tumor volume was calculated from cresyl violet-stained sections using the software 3D Doctor (Able Software, Lexington, MA). All the procedures with animals were carried out in accordance with the International Recommendations for the Use of Animals in Scientific Research (normative 86/609 from the European Communities Council).
Systemic administration of SNALP-formulated siRNAs and biodistribution analysis. Fourteen days after tumor implantation, mice were randomly assigned to target (injected with CTX-coupled and NT SNALPs) or control (injected with PBS) groups (n = 3). SNALP-formulated FAM-labeled siRNAs (2.5 mg/kg) were administered via standard intravenous injection into the lateral tail vein. Four hours after injection, animals were killed by intracardiac perfusion with 30 ml of ice-cold PBS and brain, lungs, liver, and kidneys were harvested into polystyrene tubes containing PBS supplemented with 2% fetal bovine serum. The brains were then dissected to separate tumor from healthy tissue. Tissues were homogenized for flow cytometry analysis, as described in Supplementary Materials and Methods. FAM detection was performed by incubating ~1 × 106 cells with an anti-FAM antibody (Sigma; clone FL-D6, 1:100) for 2 hours at 4 °C, followed by cell incubation with an Alexa488-conjugated anti-mouse secondary antibody (1:200; Molecular Probes, Life Technologies, Paisley, UK), for 1 hour at RT. All samples were immediately analyzed in a FACSCalibur flow cytometer; Alexa488 fluorescence was evaluated in the FL-2 channel and a total of 30,000 events were collected. The data were analyzed by Cell Quest software.
Statistical analysis. All data are presented as means ± SD of at least three independent experiments, each performed in triplicate (unless stated otherwise). One way analysis of variance combined with the Tukey post hoc test was used for multiple comparisons (unless stated otherwise) and considered significant when P < 0.05. Statistical differences are presented at probability levels of P < 0.05, P < 0.01 and P < 0.001. Calculations were performed with Prism 5 (GraphPad, San Diego, CA).
SUPPLEMENTARY MATERIAL Figure S1. Association of SNALPs with U87 GBM cells and mouse primary astrocytes. Figure S2. Internalization of SNALPs in GL261 mouse and F98 rat glioma cells. Figure S3. Tumor cell internalization of SNALPs 3 months after their preparation. Figure S4. Association and internalization of liposomesencapsulating anti-survivin siRNAs in survivin-expressing U87 glioma cells, and effect on survivin mRNA expression. Figure S5. MiR-21 expression in U87 GBM cells following incubation with CTX-coupled or nontargeted liposomes encapsulating anti-miR-21 oligonucleotides. Figure S6. Effect of sunitinib in the viability of GL261 glioma cells. Table S1. Oligonucleotide, siRNA, and primer sequences. Materials and Methods.
Acknowledgments
The authors thank Maria da Graça Rasteiro (University of Coimbra) for making available the equipment used in the measurement of zeta potentials; Luisa Cortes, Isabel Nunes, and Ana Maria Cardoso (Center for Neuroscience and Cell Biology) for the assistance with the confocal microscopy imaging, flow cytometry, and zeta potential measurement (respectively). This work was supported by the Portuguese Foundation for Science and Technology (grants PTDC/DTP-FTO/0265/2012, PTDC/SAU-FAR/116535/2010 and PEst-C/SAU/LA0001/20119). The drug sunitinib was kindly provided by Pfizer. P.M.C. is recipient of a fellowship from the Portuguese Foundation for Science and Technology (SFRH/BD/45902/2008). The authors declared no conflict of interest.
Supplementary Material
References
- Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasraw M, Lassman AB. Advances in the treatment of malignant gliomas. Curr Oncol Rep. 2010;12:26–33. doi: 10.1007/s11912-009-0077-4. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Novakova J, Slaby O, Vyzula R, Michalek J. MicroRNA involvement in glioblastoma pathogenesis. Biochem Biophys Res Commun. 2009;386:1–5. doi: 10.1016/j.bbrc.2009.06.034. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PM, Cardoso AL, Nóbrega C, Pereira de Almeida LF, Bruce JN, Canoll P, et al. MicroRNA-21 silencing enhances the cytotoxic effect of the antiangiogenic drug sunitinib in glioblastoma. Hum Mol Genet. 2013;22:904–918. doi: 10.1093/hmg/dds496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CG, Wu WK, Feng SY, Wang XJ, Shao JF, Qiao J. Co-inhibition of microRNA-10b and microRNA-21 exerts synergistic inhibition on the proliferation and invasion of human glioma cells. Int J Oncol. 2012;41:1005–1012. doi: 10.3892/ijo.2012.1542. [DOI] [PubMed] [Google Scholar]
- Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J, et al. Sequence-dependent synergistic inhibition of human glioma cell lines by combined temozolomide and miR-21 inhibitor gene therapy. Mol Pharm. 2012;9:2636–2645. doi: 10.1021/mp3002039. [DOI] [PubMed] [Google Scholar]
- Catuogno S, Esposito CL, Quintavalle C, Condorelli G, de Franciscis V, Cerchia L. Nucleic acids in human glioma treatment: innovative approaches and recent results. J Signal Transduct. 2012;2012:735135. doi: 10.1155/2012/735135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AL, Simões S, de Almeida LP, Plesnila N, Pedroso de Lima MC, Wagner E, et al. Tf-lipoplexes for neuronal siRNA delivery: a promising system to mediate gene silencing in the CNS. J Control Release. 2008;132:113–123. doi: 10.1016/j.jconrel.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Hutterer M, Gunsilius E, Stockhammer G. Molecular therapies for malignant glioma. Wien Med Wochenschr. 2006;156:351–363. doi: 10.1007/s10354-006-0308-3. [DOI] [PubMed] [Google Scholar]
- Kanai R, Rabkin SD, Yip S, Sgubin D, Zaupa CM, Hirose Y, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104:42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-da-Silva LC, Santos AO, Bimbo LM, Moura V, Ramalho JS, Pedroso de Lima MC, et al. Toward a siRNA-containing nanoparticle targeted to breast cancer cells and the tumor microenvironment. Int J Pharm. 2012;434:9–19. doi: 10.1016/j.ijpharm.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A, et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest. 2009;119:661–673. doi: 10.1172/JCI37515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonça LS, Firmino F, Moreira JN, Pedroso de Lima MC, Simões S. Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug Chem. 2010;21:157–168. doi: 10.1021/bc9004365. [DOI] [PubMed] [Google Scholar]
- Mamelak AN, Jacoby DB. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin Drug Deliv. 2007;4:175–186. doi: 10.1517/17425247.4.2.175. [DOI] [PubMed] [Google Scholar]
- Veiseh O, Kievit FM, Fang C, Mu N, Jana S, Leung MC, et al. Chlorotoxin bound magnetic nanovector tailored for cancer cell targeting, imaging, and siRNA delivery. Biomaterials. 2010;31:8032–8042. doi: 10.1016/j.biomaterials.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, et al. Specific targeting of brain tumors with an optical/magnetic resonance imaging nanoprobe across the blood-brain barrier. Cancer Res. 2009;69:6200–6207. doi: 10.1158/0008-5472.CAN-09-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshane J, Garner CC, Sontheimer H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J Biol Chem. 2003;278:4135–4144. doi: 10.1074/jbc.M205662200. [DOI] [PubMed] [Google Scholar]
- Lu Z, Liu M, Stribinskis V, Klinge CM, Ramos KS, Colburn NH, et al. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene. 2008;27:4373–4379. doi: 10.1038/onc.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Morante D, Morales-Garcia JA, Sanz-SanCristobal M, Garcia-Cabezas MA, Santos A, Perez-Castillo A. Inhibition of glioblastoma growth by the thiadiazolidinone compound TDZD-8. PLoS ONE. 2010;5:e13879. doi: 10.1371/journal.pone.0013879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroso de Lima MC, Simoes S, Pires P, Faneca H, Duzgunes N. Cationic lipid-DNA complexes in gene delivery: from biophysics to biological applications. Adv Drug Deliv Rev. 2001;47:277–294. doi: 10.1016/s0169-409x(01)00110-7. [DOI] [PubMed] [Google Scholar]
- Semple SC, Klimuk SK, Harasym TO, Dos Santos N, Ansell SM, Wong KF, et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochim Biophys Acta. 2001;1510:152–166. doi: 10.1016/s0005-2736(00)00343-6. [DOI] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- Remaut K, Lucas B, Braeckmans K, Demeester J, De Smedt SC. Pegylation of liposomes favours the endosomal degradation of the delivered phosphodiester oligonucleotides. J Control Release. 2007;117:256–266. doi: 10.1016/j.jconrel.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Santos AO, da Silva LC, Bimbo LM, de Lima MC, Simões S, Moreira JN. Design of peptide-targeted liposomes containing nucleic acids. Biochim Biophys Acta. 2010;1798:433–441. doi: 10.1016/j.bbamem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- Gaur AB, Holbeck SL, Colburn NH, Israel MA. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro-oncology. 2011;13:580–590. doi: 10.1093/neuonc/nor033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1). Am J Physiol, Cell Physiol. 2004;287:C1541–C1546. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- Lino MM, Merlo A. PI3Kinase signaling in glioblastoma. J Neurooncol. 2011;103:417–427. doi: 10.1007/s11060-010-0442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, et al. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene. 2001;20:669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]
- Ren Y, Kang CS, Yuan XB, Zhou X, Xu P, Han L, et al. Co-delivery of as-miR-21 and 5-FU by poly(amidoamine) dendrimer attenuates human glioma cell growth in vitro. J Biomater Sci Polym Ed. 2010;21:303–314. doi: 10.1163/156856209X415828. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhou X, Mei M, Yuan XB, Han L, Wang GX, et al. MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer. 2010;10:27. doi: 10.1186/1471-2407-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol. 2011;103:491–501. doi: 10.1007/s11060-010-0402-7. [DOI] [PubMed] [Google Scholar]
- Moreira JN, Gaspar R, Allen TM. Targeting Stealth liposomes in a murine model of human small cell lung cancer. Biochim Biophys Acta. 2001;1515:167–176. doi: 10.1016/s0005-2736(01)00411-4. [DOI] [PubMed] [Google Scholar]
- Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- de Wolf HK, Snel CJ, Verbaan FJ, Schiffelers RM, Hennink WE, Storm G. Effect of cationic carriers on the pharmacokinetics and tumor localization of nucleic acids after intravenous administration. Int J Pharm. 2007;331:167–175. doi: 10.1016/j.ijpharm.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Trabulo S, Cardoso AM, Santos-Ferreira T, Cardoso AL, Simões S, Pedroso de Lima MC. Survivin silencing as a promising strategy to enhance the sensitivity of cancer cells to chemotherapeutic agents. Mol Pharm. 2011;8:1120–1131. doi: 10.1021/mp100426e. [DOI] [PubMed] [Google Scholar]
- Lyons SA, O'Neal J, Sontheimer H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. 2002;39:162–173. doi: 10.1002/glia.10083. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Culturing Nerve Cells (2nd Edition) The MIT: Cambridge, MA, USA; 1998. [Google Scholar]
- Xiong Q, Wilson WK, Pang J. The Liebermann-Burchard reaction: sulfonation, desaturation, and rearrangment of cholesterol in acid. Lipids. 2007;42:87–96. doi: 10.1007/s11745-006-3013-5. [DOI] [PubMed] [Google Scholar]
- Moreira JN, Ishida T, Gaspar R, Allen TM. Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharm Res. 2002;19:265–269. doi: 10.1023/a:1014434732752. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PM, Cardoso AL, Pereira de Almeida LF, Bruce JN, Canoll P, Pedroso de Lima MC. PDGF-B-mediated downregulation of miR-21: new insights into PDGF signaling in glioblastoma. Hum Mol Genet. 2012;21:5118–5130. doi: 10.1093/hmg/dds358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.