

Abstract
The aim of this study was to generate a specific aptamer against human jaw periosteal cells (JPCs) for tissue engineering applications in oral and maxillofacial surgery. This aptamer should serve as a capture molecule to enrich or even purify osteogenic progenitor cells from JPCs or from adult stem cells of other sources. Using systematic evolution of ligands by exponential enrichment (SELEX), we generated the first aptamer to specifically bind to human osteogenically induced JPCs. We did not detect any binding of the aptamer to undifferentiated JPCs, adipogenically and chondrogenically induced JPCs, or to any other cell line tested. However, similar binding patterns of the identified aptamer 74 were detected with mesenchymal stromal cells (MSCs) derived from placental tissue and bone marrow. After cell sorting, we analyzed the expression of osteogenic marker genes in the aptamer 74-positive and aptamer 74-negative fractions and detected no significant differences. Additionally, the analysis of the mineralization capacity revealed a slight tendency for the aptamer positive fraction to have a higher osteogenic potential. In terms of proliferation, JPCs growing in aptamer-coated wells showed increased proliferation rates compared with the controls. Herein, we report the development of an innovative approach for tissue engineering applications. Further studies should be conducted to modify and improve the specificity of the generated aptamer.
Introduction
The development and application of targeting ligands such as aptamers are promising goals in biotechnology and regenerative medicine. Upon selection, aptamers bind specifically to cell surface molecules that are differentially expressed in different tissues or cells (i.e., adult stem cells or tumor cells) (Cerchia et al., 2005; Guo et al., 2006). The spectrum of aptamer applications ranges from drug delivery approaches to tissue engineering purposes as attractors for specific cell types. One important application of aptamers can be to separate subpopulations from the whole cell collective (Mayer et al., 2010). Nevertheless, some cell lines or proteins are not feasible for aptamers, and it is not possible to predict whether a target molecule is aptamerogenic (MAYER, 2009). Aptamers can be conjugated to well-known drugs or small interfering RNA (siRNA) and immobilized on carrier materials. In this context, aptamers have a high potential for use in diagnostics and therapeutics (Bagalkot et al., 2006; Dhar et al., 2008) and imaging (Famulok and Mayer, 2011). Different areas of operation are described in detail in several reviews (MAYER, 2009; Esposito et al., 2011).
For the generation and amplification of aptamers, the process called SELEX (systematic evolution of ligands by exponential enrichment) is often used (Ellington and Szostak, 1990; Tuerk and Gold, 1990). The SELEX method is based on repeated incubations of a random DNA library with the target cells, followed by repeated amplifications of the target-bound nucleic acids by polymerase chain reaction (PCR). Through the iteration loops, generated aptamers with higher specificities to the target can be enriched (Wendel et al., 2010).
Aptamers are single-stranded DNA or RNA molecules that are typically 40–120 bases in length that fold into well-defined tertiary structures and bind their targets with levels of affinity and specificity similar to those of antibodies. The advantages of aptamers in comparison with antibodies are their small size (∼10–30 kDa), low immunogenicity, and the facile production process with a low batch-to-batch variability (Bunka and Stockley, 2006). Chemical modifications of aptamers to increase their serum stability and half-life are easy to perform.
For tissue engineering, many different methods for attracting cells or binding cells to a carrier matrix have been developed. One technique includes (arginine-glycine-aspartic acid) peptides (Hersel et al., 2003) or growth factors such as bone morphogenetic proteins (BMPs) (He et al., 2008; Schofer et al., 2008). However, these strategies lack a distinct cell specificity. Therefore, the generation of aptamers as cell-specific attractors for the biofunctionalization of matrices could be a feasible approach. Mesenchymal stromal cells (MSCs) provide a well-established cell source for tissue engineering purposes. These cells can differentiate into all mesodermal lineages and into osteocytes, adipocytes and chondrocytes (Dominici et al., 2006). The best established source for MSCs is bone marrow, but MSCs can also be isolated with high frequency from adipose tissue (Zuk et al., 2001), umbilical cord blood (Bieback et al., 2008), dental pulp (Demarco et al., 2011), periosteum (De Bari et al., 2001; Ringe et al., 2008), and placenta (Chan et al., 2007).
The jaw periosteum is a promising niche for adult MSCs that can be used for tissue engineering purposes in oral and maxillofacial surgeries. Jaw periosteal cells (JPCs) possess a higher bone formation capacity than bone marrow-derived MSCs (Zhu et al., 2006; Agata et al., 2007). Further studies have been undertaken to characterize this cell source in detail (Hutmacher and Sittinger, 2003; De Bari et al., 2006; Zhu et al., 2006; Alexander et al., 2008; Ringe et al., 2008; Alexander et al., 2009; Alexander et al., 2010; Alexander et al., 2011).
In the present study, we aimed to generate aptamers that are specific to the cell surface molecules of the osteogenic-induced progenitor cells derived from the jaw periosteum (JPCs) for cell isolation or capturing approaches. In future studies, generated aptamers could serve as molecules for the biofunctionalization of scaffolding materials used in tissue engineering applications.
Materials and Methods
Isolation of jaw periosteal cells and other progenitor cells
The human periosteal tissue was obtained during routine interventions in our department after informed patient consent. The 1 cm2 pieces of tissue were minced in phosphate buffered saline (PBS) using a scalpel and digested in collagenase XI (1,500 U/mL; Sigma Aldrich) for 90 minutes. The cells were then plated in 75 cm2 culture flasks and cultivated in Dulbecco's modified Eagle's medium (DMEM):F12 (Invitrogen) containing 10% fetal calf serum (FCS, Sigma Aldrich), 1% fungizone, and 1% streptomycin. For these studies, JPCs from the fifth passage were used. The placental and bone marrow MSCs were kindly provided from the Center for Regenerative Medicine, University Hospital Tübingen and were isolated, cultured, and expanded as described recently (Pilz et al., 2011). The prostate fibroblasts were kindly provided by the group of Prof. H.P. Wendel. These fibroblasts were isolated by the passive outgrowth of the cells in culture dishes out of small pieces of prostate and cultured in high glucose DMEM containing 10% FCS (Sigma Aldrich), 1% penicillin, 1% streptomycin, and 2 mM l-glutamine. The cell line BICR56 (oral squamous carcinoma cell line, commercially available from Health Protection Agency Culture Collections) was cultured in RPMI 1640 medium containing 10% FCS, 1% fungizone, and 1% streptomycin. The human lung carcinoma cell lines A594 and SK-MES and the endothelial cell line EAHy926 (ATCC) were cultured in high glucose DMEM (PAA) containing 10% FCS and 1% streptomycin. The primary endothelial cells and the endothelial cells derived from umbilical cord blood were cultured in the VascuLife EnGS Medium Complete Kit (Lifeline Cell Technology). These cells were isolated by digestion with collagenase II (0.05%) (PAA) solution for 30 minutes at 37°C and then flushed out with basal medium (Lifeline Cell Technology). The cell lines Cal72 and MG63 (ATCC) were kindly provided by the group of Prof. G. Klein. The Cal72 cells were cultured in RPMI 1640 containing 10% FCS, 1% antibiotic/antimycotic solution (100 U/mL penicillin, 100 μg/mL streptomycin sulfate, 0.25 μg/mL amphotericin B), and the MG73 cells were cultured in RPMI 1640 containing 10% FCS and 1% streptomycin.
Isolation of peripheral blood mononuclear cells from whole blood
The blood was diluted 1:2 with PBS and carefully layered over 15 mL of Histopaque (Sigma Aldrich). Plasma and red blood cells were eliminated from the peripheral blood mononuclear cell (PBMC) by centrifugation for 30 min, 810 g, room temperature. The thin interphase containing the PBMCs was carefully aspirated and transferred into another tube. The PBMCs were washed 3 times with PBS and cultivated in x-vivo medium (Invitrogen) without any additional supplements.
Induction of JPC and MSC osteogenesis
To differentiate the progenitor cells into the osteogenic lineage, the cells were treated with DMEM:F12 containing 10% FCS, 4 μM dexamethasone, 10 mM ß-glycerophosphate, and 100 μM ascorbic acid (OB medium). For the induction of the early phase of osteogenesis, the cells were stimulated for 5 days with the OB medium before performing the binding assays with aptamers. Cells treated with normal culture medium served as untreated controls.
Induction of JPC and MSC adipogenesis
To differentiate the progenitor cells into the adipogenic lineage, the cells were treated with culture medium containing 1 μM dexamethasone, 0.2 mM indomethacine, 0.5 mM 3-isobutylxanthine, and 0.01 mg/mL insulin for 5 days. Afterwards, the binding assays were performed with aptamer 74.
Induction of JPC and MSC chondrogenesis
For the induction of chondrogenesis, the cells were treated with DMEM:F12 containing 1% FCS, 10 ng/mL TGFβ-1, 0.5 μg/mL insulin, 2 mM l-glutamine, and 50 mM ascorbic acid for 5 days before incubation with aptamer 74.
Detection of calcium phosphate precipitates by Alizarin red staining
The cell monolayers were washed with PBS and fixed with ice-cold methanol for 1 minute. The following steps included the staining of precipitates with a 40 mM Alizarin red S solution (Sigma Aldrich) for 20 minutes and intensive washing steps with distilled water and PBS. Finally, the monolayers were dehydrated and air-dried. The calcium phosphate precipitates appeared bright red after staining.
Detection of calcium phosphate precipitates by van Kossa staining
Cells were fixed by adding 100% ice-cold ethanol for 15 minutes and were washed with distilled water afterwards. The next step included the incubation with 5% silver nitrate in the dark for 1 hour, followed by further washing steps with distilled water. The reduction was done by adding sodium carbonate and several washing steps. For the final fixation, sodium thiosulfate was added for 2 minutes.
Scanning electron microscopy and energy dispersive x-ray analysis of cell-seeded 3D constructs
In order to visualize the mineralization capacity of aptamer 74-positive and –negative cells, scanning electron microscopy (SEM) was performed using the electron microscope LEO 1430 (Zeiss, Oberkochen). Fixation of cells on scaffolds followed with glutaraldehyde (2%) in 1 mM cacodylic acid sodium salt (pH 7.4) for 90 minutes, and thereafter, constructs were lyophilized. Cell-seeded constructs were sputter coated with gold/palladium. Energy dispersive x-ray (EDX) analyses were performed using the Roentec UHV-Si(Li)-Detector (Roentec).
Nano-computed tomography analyses of cell-seeded polylactic acid scaffolds
Aptamer 74-sorted cells were seeded onto polylactic acid scaffolds and cultured for 40 days under untreated and osteogenic induced conditions. Lyophilized cell-seeded scaffolds were then used for high-resolution x-ray analyses (phoenix nanotom s, GE Healthcare).
Quantification of calcium phosphate precipitates
After Alizarin red staining, the monolayers were treated with 10% acetic acid to dissolve the dye from the monolayers, and the cells were detached from the well bottom using a cell scraper. The samples were vortexed and heated at 85°C for 5 minutes and then cooled down on ice for further 5 minutes, followed by centrifugation at 16,000 g for 20 minutes. The supernatants were transferred into fresh tubes and neutralized with 400 μL of ammonium hydroxide. A standard dilution series using the Alizarin red solution (40 mM) was freshly prepared following the manufacturer's instructions for the osteogenesis quantitation kit (Millipore). The photometric measurements of the optical densities were determined in triplicate at 405 nm using an ELISA reader (ELX-800; Biotek)
RNA extraction
The aptamer 74-positive and -negative cell fractions were separated using a fluorescence activated cell sorting (FACS) technique. After centrifuging each subpopulation, the cell pellets were resuspended in lysis buffer containing 0.01% ß-mercaptoethanol. The RNA was isolated using the RNeasy Micro Kit (Qiagen) following the manufacturer's instructions. The amount of isolated RNA was photometrically measured and quantified (GeneQuant Pro; GE Healthcare).
cDNA synthesis for quantitative PCR analysis
For cDNA synthesis, 1 μg of RNA was used following the manufacturer's instructions for the Clonetech Advantage for RT-PCR Kit. Briefly, to isolate the mRNA, 2 μL of the oligo dT primer was hybridized with 1 μg of RNA for 2 minutes at 70°C. After cooling the probes, 4 μL of the 5×reaction buffer, 0.5 μL of the recombinant inhibitor, 1 μL of the dNTP mix and 1 μL of the reverse transcriptase were added, and diethylpyrocarbonate (DEPC)-treated water was added to give a final volume of 20 μL. The cDNA synthesis was carried out at 42°C for 60 minutes in a personal thermocycler (Eppendorf). The samples were then heated at 95°C and diluted to a final volume of 100 μL. For the quantitative PCR analysis, 0.02 μg of DNA was used for each sample per reaction.
Quantification of mRNA expression by real-time PCR
The quantification of mRNA expression was performed using the real-time LightCycler System (Roche Diagnostics). For the PCR reactions, 2 μL of each sample was added to the master mix containing 2 μL of the primer mix (commercial primer kits from Search LC), 2 μL of the DNA Master Sybr Green 1 mix (Roche Molecular Biochemicals) and 14 μL of DEPC-treated water. The amplification was performed with 35 cycles of denaturation for 10 seconds at 95°C, primer annealing for 10 seconds at 68°C, and amplification for 16 seconds at 72°C. The transcript levels were normalized to those of the housekeeping gene glycerinaldehyde 3-phosphate dehydrogenase, and the induction factors of the aptamer 74-positive samples in relation to those of aptamer 74-negative samples were calculated.
Generation of aptamers using the SELEX procedure
Incubation of the starting oligonucleotide library with JPCs
The JPCs were thawed, and 2×105 cells were seeded in a 6-well plate and cultured for 2 days. The starting library consisted of 2 primer-binding regions on both ends of the DNA and a wobble region of 40 bases in between, generating random 40-mer DNA oligonucleotides with a variability of 1015 different sequences possible [see (a) in list of oligonucleotides used, Table 1]. For the initial de- and renaturation of the oligonucleotide pool, 200 pmol of oligonucleotides was heated in SELEX buffer (DMEM:F12, 0.1% bovine serum albumin, 3 mM MgCl2 and 100 μg/mL yeast tRNA) at 85°C and incubated on ice for 5 minutes. In the first rounds of SELEX, the libraries were incubated with JPCs only. From the eighth SELEX round on, the library was preincubated with other cells, such as BICR56 cells or PBMCs, for 30 minutes on ice with slight agitation for negative counterselection. Then, the supernatants containing the unbound aptamers were transferred to the cultured and/or induced JPCs or other MSCs for positive selection by incubation for 30 minutes on ice. Because PBMCs are suspension cells, a centrifugation step at 400g was performed to pellet the cells so that the oligonucleotide cocktail could be removed without removing the PBMCs. The JPCs were then washed 5 times with SELEX buffer, and the cells with the aptamers were scraped from the well bottom in 200 μL of DEPC-treated water. To remove the DNA from the cell debris, the cell/aptamer solution was heated at 95°C and centrifuged at 16,000g at room temperature for 5 minutes. The supernatants containing the selected DNA aptamers were amplified by PCR.
Table 1.
List of Oligonucleotides Used
| Application | Sequence | |
|---|---|---|
| Starting library | (a) | 5′-GGGAGACAAGAATAAACGCTCAA-N40-TTCGACAGGAGGCTCACAACAG GC-3′ |
| Modified PCR primers | (b) | 5′phosphate-GCCTGTTGTGAGCCTCCTGTCGAA-3′ |
| (c) | FITC-C18 - GGGAGACAAGAATAAACGCTCAA-3′ | |
| Gel elution | (d) | A20 - C18 -5′ GCCTGTTGTGAGCCTCCTGTCGAA-3 |
| Coupling of aptamer 74 to Dynabeads or western blotting | (e) | 5′-biotin-C18- GGGAGACAAGAATAAACGCTCAACAAATGGGTGGGTGTGGTGGGTGTGAAGGTGCGAGTTGATTCGACAGGAGGCTCACAACAGGC |
| Coupling aptamer 74 to gold electrodes | (f) | 5′-thiol-C18- GGGAGACAAGAATAAACGCTCAACAAATGGGTGGGTGTGGTGGGTGTGAAGGTGCGAGTTGATTCGACAGGAGGCTCACAACAGGC |
FITC, fluorescein isothiocyanate; PCR, polymerase chain reaction.
Polymerase chain reaction
For the PCR amplifications of the bound sequences, 2 different PCR protocols were applied (see Table 2). For the initial qualitative amplification step (PCR 1), we used 10 cycles, and for the quantitative amplification step (PCR 2), we used 20 cycles. The amplification reactions were carried out using the Fast Start PCR Master Kit (Roche) and primers synthesized by Ella Biotech at a concentration of 100 μM. Therefore, the template DNA was diluted 1:2 with the master/primer mix. For the PCR 1, 12 probes with a final volume of 100 μL/sample were amplified by PCR and then pooled. The second amplification step (PCR 2) was performed with a doubled amount of samples (24 samples with a final volume of 100 μL) and pooled together.
Table 2.
Protocols Used for Polymerase Chain Reactions
| Step | Temperature | Duration |
|---|---|---|
| PCR 1 (10 cycles) | ||
| Initial Denaturation | 95°C | 5 minutes |
| Denaturation | 95°C | 30 seconds |
| Annealing | 55°C | 30 seconds |
| Amplification | 72°C | 30 seconds |
| PCR 2 (20 cycles) | ||
| Initial Denaturation | 95°C | 5 minutes |
| Denaturation | 95°C | 30 seconds |
| Annealing | 55°C | 30 seconds |
| Amplification | 72°C | 30 seconds |
PCR, polymerase chain reaction.
Table 1 identifies the oligonucleotides (a)–(f) used in the amplification reactions. The starting library consisted of 2 primer regions on both ends of the DNA and a wobble region of 40 bases between the primer regions (a). The normal SELEX amplification was performed using the fluorescein isothiocyanate (FITC)–labeled forward primer (c) for flow cytometry analysis and a phosphate-labeled reverse primer (b) for the lambda exonuclease digestion of this synthesized strand.
To exclude misamplifications resulting in a ladder of different fragment lengths, bands with the desired length of 87 bp were extracted and eluted from the gel in regular intervals. Additionally, we used a reverse primer (d) with a poly-A tail at the 5′ end. For aptamers that were used for immobilization to Dynabeads (Invitrogen) or for western blotting analysis, binding to streptavidin was necessary, so biotin-labeled aptamers at the 5′ ends were synthesized (e). After the cloning and sequencing of the generated oligonucleotides, different aptamers were synthesized for binding assays (for details, see Table 3). For target identification, biotinylated aptamer 74 was synthesized (e), and the aptamers were labeled with a thiol group (f) for the immobilization of the aptamers on gold electrodes.
Table 3.
Sequences of the Synthesized Aptamers
| Aptamer | Sequence 5′ Fam-C18 |
|---|---|
| 4 | GGGAGACAAGAATAAACGCTCAAGGCCGCTAGTTCCCGGGTGCACAGCTTATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
| 5 | GGGAGACAAGAATAAACGCTCAATAGACAGTGGTTGGGGTAGGGGAAGGCGGGTGTCGGCGTATTCGACAGGAGGCTCACAACAGGC |
| 7 | GGGAGACAAGAATAAACGCTCAAGGCCGCCAGTTCCCGGGTTGCACAGCATATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
| 9 | GGGAGACAAGAATAAACGCTCAAACGGGGCTGCACATAAGTGGTTTCTACGCTCAGATTCGACAGGAGGCTCACAACAGGC |
| 13 | GGGAGACAAGAAtAAACGCTCAACGACGCTGATCTGGGACAGAGGTTTCTACGTTCAGATTCGACAGGAGGCTCACAACAGGC |
| 24 | GGGAGACAAGAATAAACGCTCAAGGCCGCCAGTTCCCGGGGTGCACAGCATATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
| 28 | GGGAGACAAGAATAAACGCTCAACGACGCTGATCTGGGACAGAGGTTTCTACGTTCAGATTCGACAGGAGGCTCACAACAGGC |
| 34 | GGGAGACAAGAATAAACGCTCAACAAATGGGTGGGTGTGGTGGGTGTGAAGGTGCGAGTTGATTCGACAGGAGGCTCACAACAGGC |
| 45 | GGGAGACAAGAACAACGCTCAACGGTCGAAGTGGCTGGGTAGGGGTAGGCGGGTTCGACAGGAGGCTCACAACAGGC |
| 46 | GGGAGACAAGAATAAACGCTCAATGGGGGGGAGGTCGGAGGGTGGATGGGGGGTAAGGATGAATTCGACAGGAGGCTCACAACAGGC |
| 54 | GGGAGACAAGAATAAACGCTCAACAAATGGGTGGGTGTGGTGGGTGTGAAGGTGCGAGTTGATTCGACAGGAGGCTCACAACAGGC |
| 57 | GGGAGACAAGAATAAACGCTCAAGGCCGCCAGTTCCCGGGTGCACAGCATATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
| 60 | GGGAGACAAGAATAAACGCTCAACGGTCGAAGTGGCTGGGTAGGGGTAGGCGGGTTCGACAGGAGGCTCACAACAGGC |
| 67 | GGGAGACAAGAATAAACGCTCAATAGACAGTGGTTGGGGTAGGGGAAGGCGGGTGTCGGTGCGTTCGACAGGAGGCTCACAACAGGC |
| 68 | GGGAGACAAGAATAAACGCTCAATGGGGGGGAGGTCGGAGGGTGGATGGGGGGTAAGGATGAATTCGACAGGAGGCTCACAACAGGC |
| 74 | GGGAGACAAGAATAAACGCTCAACAAATGGGTGGGTGTGGTGGGTGTGAAGGTGCGAGTTGATTCGACAGGAGGCTCACAACAGGC |
| 77 | GGGAGACAAGAATAAACGCTCAATAGACAGTGGTTGGGGTAGGGGAAGGCGGGTGTCGGTGCGTTCGACAGGAGGCTCACAACAGGC |
| 80 | GGGAGACAAGAAtAAACGCTCAAGGTACCCATCCGTCCGTACAGATTCACGTTCACTGGGTTATTCGACAGGAGGCTCACAACAGGC |
| 84 | GGGAGACAAGAATAAACGCTCAACGACGCTGATCTGGGACAGAGGTTTCTACGTTCAGATTCGACAGGAGGCTCACAACAGGC |
| 87 | GGGAGACAAGAATAAACGCTCAAGGCCGCCAGTTCCCGGGTGCACAGCATATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
| 88 | GGGAGACAAGAATAAACGCTCAATCGGTGGAGATATCCGCTAAGGAGTTGGCCCTTCGGTGTTTTCGACAGGAGGCTCACAACAGGC |
| 91 | GGGAGACAAGAATAAACGCTCAACGACGCTACTCCACACCATTAATGGGTTTCTACGCTCAGATTCGACAGGAGGCTCACAACAGGC |
| 92 | GGGAGACAAGAATAAACGCTCAAGGCCGCCAGTTCCCGGGTGCACAGCATATGTCCGGTTAATTCGACAGGAGGCTCACAACAGGC |
Purification of the generated DNA aptamers
The double-stranded DNA aptamers generated from the PCR reactions were purified using the MinElute Kit (Qiagen) following the manufacturer's instructions. Briefly, 100 μL aliquots of the samples were added to 500 μL (1:5 dilution) of PB buffer (components patented), pipetted to the MinElute columns and centrifuged at 16,000g for 1 minute. The flow-through was discarded, and the columns were washed with PE buffer. The DNA was then eluted from the membranes with 25 μL of EB buffer (10 mM Tris CL, pH 8.5), and the DNA concentrations were measured photometrically at a 1:50 dilution.
Lambda-exonuclease digestion of double-stranded DNA
To obtain single-stranded DNA (ssDNA), the double-stranded DNA (dsDNA) from the PCR reactions was digested with lambda exonuclease by degrading the phosphate-labeled strand from the 5′ end. For each μg of DNA, 1 μL of lambda exonuclease was applied (5000 U/mL, New England BioLabs, Germany) and incubated for 4 hours with gentle agitation at 37°C. The enzyme was then heat inactivated at 70°C for 10 minutes (Wendel et al., 2010).
Phenol/chloroform purification of ssDNA
To purify the ssDNA, 0.3 volumes of phenol were added to the lambda exonuclease digestion. After vortexing the samples, the phases were mixed and then centrifuged at 16,000g for 5 minutes. The resulting upper layer was removed and added to 0.3 volumes of chloroform, mixed again and centrifuged under the same conditions. The aqueous phase was used for the precipitation of ssDNA by adding 3 volumes of absolute ethanol and 0.1 volumes of sodium acetate (3 M, pH 5.9) with an overnight incubation at −20°C. The precipitates were pelleted by centrifugation at 4°C and 16,000g for 20 minutes. The pellets were then washed with 70% ethanol. Afterwards, the ssDNA was diluted in 30 μL of DEPC-treated water, and the DNA concentration was determined photometrically. After amplification with primer (c), the ssDNA was FITC-labeled and used for the next SELEX round and for flow cytometry analysis.
Gel electrophoresis for confirmation of aptamer length
To confirm the amplicon length, the samples were mixed with the denaturing sequencing buffer (0.05% xylene cyanol, 0.05% bromophenol blue, 10 mM ethylenediaminetetraacetic acid (EDTA), 98% formamide) and denatured at 95°C for 10 minutes. The samples were then loaded onto polyacrylamide/urea gels (6.31 g urea, 3.75 mL Roti Phorese 40 (Roth, Germany), 125 μL ammonium persulfate, 10.5 μL tetramethyl-ethylenediamine, 0.75 mL 10×TBE buffer) and separated by electrophoresis at 120 V for 1 hour in Tris-borat-EDTA-buffer (89 mM boric acid, 89 mM Tris, 2 mM EDTA).
Gel elution of the ssDNA
To exclude the misamplifications of the polymerase, the DNA band with a length of approximately 87 bases (see explanation below) was cut from the gel after every third SELEX round instead of lambda exonuclease digestion. The PCR amplification was performed using the reverse primer with the poly-A tail (primer d). The poly-A tail was used for the elongation of the fragment to provide an exact visualization of the length of the two different strands. The desired fragment was cut from the gel using a scalpel, minced, and transferred into a tube in which the ssDNA extraction was performed passively by eluting with NH4Ac buffer (0.5 M NH4Ac, 1 mM EDTA) at 37°C overnight. The supernatants were collected, and the gel pieces were rinsed with NH4Ac buffer again. The supernatants containing the DNA were again collected. This step was repeated multiple times to achieve higher DNA yields. To dehydrate the ssDNA solution, the volume was concentrated using 0.5–1 volumes of isobutanol by inverting and discarding the butanol phase. This step was repeated until the volume of the ssDNA solution was approximately 200 μL. The solution was purified and desalted by adding the PN buffer (components patented) of the MinElute Kit (Qiagen, Germany) and using the kit according to the manufacturer's instructions.
The SELEX procedure including all the above-mentioned steps was performed for 17 rounds. To enrich the specificity of the aptamers, a negative counterselection was performed as mentioned above.
Cloning of generated aptamers
The dsDNA fragments were ligated into the pDrive vector (Qiagen PCR Cloning Kit, Germany). For each ligation, a 10 molar excess of the dsDNA (13 ng) was applied to 1 μL of the vector. Therefore, 1–4 μL DNA was added to 5 μL of the ligation master mix and diluted with DEPC-treated water to a final volume of 10 μL. The ligation was performed for 2 hours at 4°C.
The transformation was performed with the competent Escherichia coli strains provided by the cloning kit. The bacteria were thawed on ice, and the total volume of the ligation mix was added to the bacteria. After heating at 42°C for 30 seconds, the samples were cooled on ice for 2 minutes. Then, 250 μL of SOC medium was applied to the samples, and aliquots of 60 μL of the bacterial solution were plated on LB-agar plates that contained 100 μg/mL ampicillin, 40 mg/mL X-Gal, and 100 mM IPTG. After 16 hours of incubation at 37°C, the white colonies were picked and transferred into tubes containing 2 mL of LB medium.
Purification of the plasmid DNA using the QIAprep Spin Miniprep Kit
The bacterial pellets were resuspended for the miniprep in 250 μL of the P1 buffer after an overnight incubation at 37°C and centrifugation at 16,000g. After adding 250 μL of buffer 2, the components were mixed by inverting. Then, 350 μL of buffer N3 was added, and the solution was mixed again by tube inversion. Following these steps, the solution was centrifuged for 10 minutes at 16,000g, and the supernatants were transferred to the QIAprep columns, which were then centrifuged at 16,000g for a further 60 seconds. For washing, 500 μL of the PB buffer was added to the columns followed by a centrifugation step for 60 seconds. To eliminate the remaining ethanol, the columns were centrifuged at maximum speed for 60 seconds and were then transferred into a clean collection tube before the plasmid DNA was eluted with 50 μL of the EB buffer. To confirm the correct insert length, the plasmid DNAs were digested for 1 hour at 37°C with EcoR1, which has restriction sites on both ends of the multiple cloning site of the pDrive vector, and were loaded onto agarose gels for visualization.
Sequencing of the generated aptamers
The plasmid DNA from the positive screened clones that included the insert length of 87 bp was purified using the QIAprep Spin Miniprep Kit. For the DNA sequencing analysis, 500 ng of the DNA was shipped to 4baselab (Germany). The identified oligonucleotides were synthesized by Ella-Biotech (Germany).
To generate the phylogenetic tree of the identified clones, the ClustalW2 software was used. To predict the secondary structures of the ssDNAs, the identified sequences were submitted to the RNAfold software (RNAfold webserver, University of Vienna).
Binding affinity analysis of generated aptamers by flow cytometry
To analyze the binding affinities of the aptamers to the analyzed cells, flow cytometry was performed using the FITC-labeled aptamers [see Table 1, oligonucleotide (c)]. The cells were detached from the culture dishes, washed with SELEX buffer and incubated with the aptamer/yeast tRNA/SELEX buffer cocktail (containing a total of 200 pmol of DNA after the de- and renaturation of the DNA by heating at 85°C for 5 minutes and cooling at 4°C for 15 minutes) for 1 hour with gentle agitation on ice. The FITC-labeled starting library served as a negative control in addition to the unlabeled control. The binding affinity was calculated by subtracting the percentage of the fluorescence intensity of the starting library from the percentage of the aptamer binding to the analyzed cells for the elimination of nonspecific binding affinities. Following the aptamer labeling step, the cells were pelleted by centrifuging at 300g, washed 3 times with SELEX buffer and once with FACS buffer (PBS, 0.1% bovine serum albumin, 0.1% sodium azide) and then analyzed in a flow cytometer (FACSScan, Becton Dickinson).
Fluorescence activated cell sorting of the aptamer 74-positive and aptamer 74-negative cell fractions
Out of all of the generated and analyzed aptamers, aptamer 74 exhibited the maximal binding affinity to osteogenic-induced JPCs and was, therefore, used for all further analyses. The cell labeling with aptamer 74 was performed under sterile conditions as described above for the flow cytometry analysis. However, the cells were resuspended in PBS instead of FACS buffer to maintain cell viability and physiological conditions for further cultivation after the cell sorting. The cells were sorted using a BD FACS Aria cell sorter (Becton Dickinson), and only the strongly aptamer 74-stained cells and the strongly negative fraction were plated for the analyses of the mineralizing capacity and gene expression.
Analysis of cell proliferation of the aptamer 74-positive and aptamer 74-negative cell population
Immediately after cell sorting, the aptamer 74-positive and -negative cells were washed with PBS, centrifuged and plated on special E-plates (Roche) at a cell density of 5×102 cells per well. The live monitoring of cell proliferation was performed using the xCELLigence System (Roche) for 4 weeks. The cell impedance (due to changes in the adherence/spreading of the cells) was automatically measured every 15 minutes.
Coating of the xCELLigence E-plates with aptamer 74 and analysis of cell proliferation
The sensor E-plates were cleaned with isopropanol and rinsed with sterile distilled water. To coat the E-plates, aptamer 74 was modified by adding a thiol group through which coupling to the sensor plates was possible. The plates were incubated with a 0.5 M NaHCO3 solution in a wet chamber in the dark for 15 minutes. The thiol aptamers were dissolved in distilled water and applied to the E-plates at a concentration of 1 nM. The immobilization of the thiol aptamers was performed for 4 hours in a wet chamber. Afterwards, the plates were washed with distilled water and were seeded with 5×102 cells. The live monitoring of cell proliferation was performed every 15 minutes over different time periods and for up to 4 weeks.
Results
Aptamer generation by SELEX
To generate an aptamer under physiological conditions, the SELEX procedure was performed in a modified culture medium containing 3 mM MgCl2 to enhance DNA binding to living cells. In the first round, the JPCs were incubated with the starting library. The unbound sequences were removed, and the bound oligonucleotides were amplified by PCR. After the third round of selection, we could detect an aptamer enrichment of 3.65% that increased up to 6.15% in the eighth round (Fig. 1A). As a control, we used the starting library. The percentage of bound cells was calculated by subtracting the percentage of binding of the unselected aptamers (i.e., starting library) from the total binding of the aptamer after SELEX. Depending on the inhomogeneity of the fragment lengths, the fragment of 87 bases was excised from the gel, and the DNA was eluted (indicated by the black arrow in Fig. 1A). To generate a more specific aptamer, we began the negative counterselection in round 8 using the cell line BICR56 (human squamous carcinoma cell line, indicated by the gray flash in Fig. 1A). The binding affinity did not increase exponentially as theoretically expected, but we detected a continuous increase in binding affinity during the SELEX process (Fig. 1A, B). After each negative counterselection, we obtained a decrease in the aptamer binding affinity (Fig. 1A; round 8, counterselection with BICR cells; round 16, counterselection with PBMCs, indicated by the white flash). After round 14, a maximal binding affinity of 12.63% was achieved, which decreased after the negative counterselection in rounds 16 and 17. To maintain the specificity to our target cells, we used the aptamers generated in round 17, although they had lower binding affinities than those from round 14.
FIG. 1.

Overview of the rounds performed and intermediate steps during the cell systematic evolution of ligands by exponential enrichment (SELEX) procedure. (A) The net aptamer binding affinities following the different cell SELEX rounds are illustrated. The intermediate steps were gel elution, indicated by the black arrows, negative counterselection with BICR56, indicated by the gray flash, and negative counterselection with PBMCs, indicated by the white flash. (B) Fluorescence activated cell sorting (FACS) histograms illustrating the obtained binding affinities during the different rounds of the SELEX process. PBMCs, peripheral blood mononuclear cells. Color images available online at www.liebertpub.com/nat
Cloning of generated aptamers
The generated aptamers were cloned into E. coli, and 100 positive clones were picked and further analyzed. After enzymatic digestion with EcoR1, for which restriction sites existed close to the insert ends, we identified 24 clones containing the insert of 103 bases. These clones were used for sequence analysis.
Sequence analysis of the generated aptamers
The sequence analysis of the 24 isolated clones was performed by the company 4baselab (Reutlingen). As shown in Fig. 2, multiple alignments were carried out. All sequences revealed the same flanking primer sequence at the 3′ and 5′ ends, whereas the differences in the sequences in the random region between bases 23 and 67 were noted. The respective bases are displayed in 4 different colors (orange: guanine; blue: adenine; pink: cytosine; green: thymine). When the bases appeared at a higher frequency, they were colored by the software. The primer sequence coincided in all of the clones with the exception of clone 45. The relative frequency of the bases at the corresponding position is denoted by black bars underneath the sequences. The maximal conformity was obtained at positions 25, 31–33, 38–40, 44–45, and 49–51.
FIG. 2.

Multiple sequence alignment of the 24 aptamer-positive clones using ClustalW2. Bases 1–23 and 66–89 represent the primer regions, as indicated below. Orange: guanine; blue: adenine; green: thymine; pink: cytosine; black: relative frequency of the bases. Color images available online at www.liebertpub.com/nat
Based on the identified sequences, a phylogenetic tree was generated to classify the 24 positive clones into 5 sequence families based on their degree of kinship (shown by the red boxes). Clones 80 and 88 had strongly deviating sequences (Fig. 3). The members of sequence family 4, which consisted of clones 34, 54, and 74, had the exact same insert sequence.
FIG. 3.

Phylogenetic sequence tree of the analyzed 24 aptamer-positive clones. Due to their degree of kinship, the identified aptamers could be classified into 5 sequence families. Aptamers 80 and 88 showed highly aberrant sequences. Color images available online at www.liebertpub.com/nat
Analysis of the secondary structures of sequenced aptamers
Because aptamers bind through their 3-dimensional secondary structures and not through their complementarity, an analysis of the DNA folding was of interest. We used the RNAfold software to predict the secondary structures. However, because the structure of the target was unknown, the calculation was quite imprecise and revealed the most likely folding pattern under normal conditions. In Fig. 4, it is apparent that the identified sequence families exhibited similar folded structures, confirming the previous analysis using the ClustalW2 software. Whereas sequence family 1 formed hairpin-like structures with small loops, sequence family 2 formed larger loops (with the exception of clone 45) and similar structures within three sequences (clones 5, 67, and 77). Within sequence family 3, clones 13, 28, and 84 exhibited a similar hairpin-like structure, whereas clones 9 and 91 had totally different folding patterns. Due to the identical sequences identified within family 4, the folding calculations were also identical. Within sequence family 5, clones 46 and 68 also exhibited the same folding pattern, although the sequences were not completely identical. Clones 80 and 88 had no degree of relationship to other families and differed in their folding patterns, as expected.
FIG. 4.

DNA folding patterns of the aptamer sequence families predicted by RNAfold software. Note that the folding calculations for aptamers 34, 54, and 74 within family 4 were identical because these aptamers had the exact same sequences. Color images available online at www.liebertpub.com/nat
Binding affinity analysis of generated aptamers by flow cytometry
Of the sequenced aptamers, 23 were synthesized and FITC labeled. For the analysis of the binding affinities, undifferentiated human JPCs were incubated with the FITC-labeled aptamers, and flow cytometry analyses were performed in which we subtracted the binding percentage of the starting library (Table 4). We determined the maximal binding affinities of the aptamer sequences derived from family 4 to the donor cells with which the SELEX procedure was performed and also to other varying donor cells. The 3 identical clones (34, 54, and 74) exhibited a binding affinity up to 29%–32%. Due to the clearly high binding affinities of these aptamers, aptamer 74 was chosen for all further analyses. The aptamer sequences from sequence family 5 also exhibited higher binding affinities to JPCs (clone 46: 15.14%; clone 68: 8.57%) in comparison to the other aptamer sequences. However, clone 74 showed by far the highest binding affinity to JPCs (32.2%).
Table 4.
List of the Binding Affinities of the Different Aptamers (in Percentage) Related to the Identified Sequence Families
| Clone number | % Positive counts | Sequence family |
|---|---|---|
| 24 | 0.69 | 1 |
| 92 | 0 | |
| 57 | 1.37 | |
| 87 | 0 | |
| 4 | 2.68 | |
| 7 | 3.15 | |
| 67 | 0.93 | 2 |
| 77 | 6.64 | |
| 5 | 2.7 | |
| 45 | 0.99 | |
| 60 | 2.71 | |
| 9 | 0 | 3 |
| 91 | 0.23 | |
| 13 | 0.27 | |
| 28 | 0 | |
| 24 | 0.69 | |
| 34 | 29.31 | 4 |
| 54 | 30.17 | |
| 74 | 32.2 | |
| 46 | 15.14 | 5 |
| 68 | 8.57 | |
| 80 | 0 | Independent |
| 88 | 0 | Independent |
To analyze whether aptamer 74 bound to osteogenic-induced cells, we performed binding assays with JPCs that were stimulated with osteoblast differentiating media for 5 days (Fig. 5). We could detect a lower or no binding affinity to cells treated with control medium (0%–10% positive counts). In contrast, aptamer 74 revealed donor-dependent affinities (between 8% and 35%) to osteogenic differentiated JPCs. Cells treated with chondrogenic or adipogenic differentiating media had a very low binding capacity with donor-dependent variations between 0%–3%.
FIG. 5.
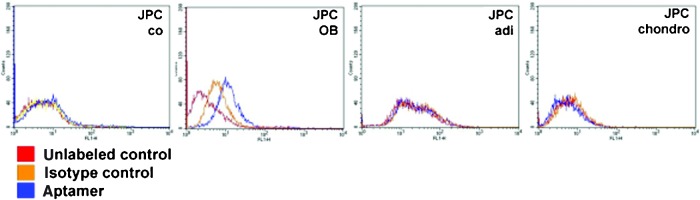
Representative FACS histograms illustrating the binding affinities of aptamer 74 to untreated human jaw periosteal cells (JPCs) (co) or to JPCs after 5 days of osteogenic (OB, n=12), adipogenic (adi, n=4), and chondrogenic (chondro, n=4) induction. The FACS analysis was performed with JPCs in the fifth passage. Red: unlabeled control cells; orange: isotype control-labeled cells (starting DNA library); blue: aptamer-labeled cells. Color images available online at www.liebertpub.com/nat
Binding affinity analysis of aptamer 74 to JPCs from different donors
The entire SELEX procedure was carried out using JPCs from only 1 patient. Because we intended to identify a donor-independent aptamer that could bind to an unknown jaw periosteal target, the binding affinities of aptamer 74 to JPCs from different patients were tested (Fig. 6). We did not detect higher binding affinities to cells that were able to mineralize in vitro (Fig. 6B; mineralizing JPCs: cells that were able to differentiate into the osteogenic lineage in vitro) compared to cells that did not show this ability (Fig. 6A; non-mineralizing JPCs: cells that were not able to differentiate into the osteogenic lineage in vitro). In conclusion, we detected no tendency of aptamer 74 to preferentially bind to mineralizing or to non-mineralizing JPCs.
FIG. 6.

Representative FACS histograms illustrating the binding affinities towards different in vitro mineralizing (n=5) and non-mineralizing JPCs (n=7) following 5 days of osteogenic induction in comparison to the uninduced controls. Red: unlabeled control cells; orange: isotype control-labeled cells (starting library); blue: aptamer-labeled cells. Color images available online at www.liebertpub.com/nat
Binding affinity analysis of aptamer 74 to different MSC types
We then tested the suitability of aptamer 74 for use as a general mesenchymal stromal cell marker. Therefore, the binding affinities to placental and bone marrow stromal cells in addition to periosteal cells was determined (Fig. 7). Prostate fibroblasts served as a control for fully differentiated cells that were not able to differentiate into other lineages. For the flow cytometry analysis, we again used untreated cells (Fig. 7, upper panel) in comparison to osteogenic-stimulated cells (Fig. 7, lower panel). The histograms illustrating the binding affinities to bone marrow MSCs showed a similar pattern to those from jaw periosteal cells. The untreated cells again exhibited a low binding affinity (0%–2%) to aptamer 74. In contrast, the binding affinities to osteogenic-treated cells varied from 10%–25% depending on the donor. Human placental mesenchymal cells (maternal, not fetal fraction) exhibited binding affinities between 8% and 14%. Again, we detected much lower binding affinities of the untreated cells to aptamer 74 compared to those detected in osteogenic-induced placental mesenchymal stromal cells. The prostate fibroblasts did not show any capacity to bind to the aptamer, either in the unstimulated or in the osteogenic-stimulated cell state.
FIG. 7.

Representative FACS histograms illustrating the binding affinities towards placenta- and bone marrow-derived mesenchymal stromal cells (MSCs) (n=5), JPCs and prostate fibroblasts after five days of osteogenic induction in comparison to the uninduced controls. Red: unlabeled control cells; orange: isotype control-labeled cells (starting library); blue: aptamer-labeled cells. Color images available online at www.liebertpub.com/nat
Binding affinity analysis of aptamer 74 to different cell types
To verify whether aptamer 74 bound selectively to MSCs and not to any other cell types, the binding affinities of this aptamer to different cell lines and primary cells were determined (Fig. 8). As described in the previous section, human prostate fibroblasts showed only a very low binding affinity to aptamer 74, between 0% and 1.5%. Additionally, the following different cell types and cell lines were tested:
EAHy926 (human endothelial cell line)
SK-MES (human lung carcinoma cell line)
A594 (human lung carcinoma cell line)
MG63 (human osteosarcoma cell line)
Cal72 (human osteosarcoma cell line)
BICR56 (human oral squamous carcinoma cell line)
Human primary endothelial cells derived from vessels
Human primary endothelial cells derived from umbilical cord
PBMCs (peripheral blood mononuclear cells)
FIG. 8.

Representative FACS histograms illustrating the binding affinities towards different cell lines and primary cells. Red: unlabeled control cells; orange: isotype control-labeled cells (starting library); blue: aptamer-labeled cells. Cell lines: EaHy (human endothelial cell line), SK-MES (human lung carcinoma cell line), A594 (human lung carcinoma cell line), MG63 (human osteosarcoma cell line), Cal72 (human osteosarcoma cell line), BICR56 (human oral squamous carcinoma cell line). Primary cells: human primary endothelial cells derived from umbilical cord blood and from vessels, PBMCs (peripheral blood mononuclear cells). Color images available online at www.liebertpub.com/nat
We did not detect any binding specificity of aptamer 74 to the tested cell types compared to the starting library control. The binding affinities varied between 0% and 0.8%. The negative counterselection for the SELEX procedure was performed using the oral squamous carcinoma cell line BICR56 and peripheral blood mononuclear cells. As expected, we found no binding affinity of aptamer 74 to these two cell types (Fig. 8).
Live monitoring of JPC proliferation on oligonucleotide-coated as compared to non-coated E-plates
Using the xCELLigence system, JPC proliferation was analyzed for 42 hours on E-plates that were uncoated or had been coated with the starting library or aptamer 74. After 3 hours, we could already detect differences in the adhesion behaviors on the differently coated wells. In all 3 tested patients, the proliferation curve reflecting the JPC proliferation in uncoated wells was clearly below that reflecting the proliferation in the oligonucleotide-coated wells (Fig. 9A–C). However, no significant differences in terms of proliferation were detected between the aptamer- and the library-coated wells. This tendency was maintained over a time span of 13 days (data not shown).
FIG. 9.

Live monitoring of JPC proliferation on coated and non-coated wells. E-plates were coated with the DNA library or aptamer 74. The proliferation potentials of the JPCs growing in coated wells compared to uncoated wells were detected. (A, B, C) Adhesion behavior of JPCs derived from 3 different donors in the first 48 hours of adhesion and proliferation. Red: uncoated control; orange: DNA library; blue: aptamer 74. Color images available online at www.liebertpub.com/nat
Live monitoring of cell proliferation by aptamer 74-sorted cell fractions
To learn more about the differences between the aptamer 74-sorted cell fractions, JPCs from three different donors were separated using flow cytometry. Because cell viability is impaired after the sorting process due to the applied shear forces, the live monitoring of the aptamer 74-positive and -negative cell fractions was performed over a time period of 4 weeks (Fig. 10). The negative fraction clearly showed higher proliferation rates than the aptamer 74-positive fraction. The apparent and measurable proliferation capacity of the positive fraction was not detected until 10 days following the start of the experiment (Fig. 10A–C). The proliferation behaviors of the two fractions did not approach each other before 30 days of in vitro culture.
FIG. 10.

Live monitoring of aptamer 74-positive and aptamer 74-negative cell proliferation sorted by flow cytometry. Purple: aptamer 74-negative fraction; magenta: aptamer 74-positive fraction; black: medium control. The cells were screened over a time period of 30 days with a sweep measurement every 15 minutes. Color images available online at www.liebertpub.com/nat
Analysis of cell mineralization by aptamer 74-sorted cell fractions
Stainings
To analyze the mineralization capacity of aptamer 74-sorted cells, only highly aptamer 74-positive and -negative cells were gated for the cell separation process. The aptamer 74-positive cell fraction had a slight tendency to show increased osteogenic capacity compared to the negative one, as detected by van Kossa stainings. However, no significant differences concerning the mineralization potential were detected by Alizarin Red stainings and following quantification of the stained calcium precipitates formed by aptamer 74-positive and -negative cells. Figure 11 illustrates Alizarin stainings (panel A), photometrical quantification of the precipitates stained by the Alizarin red dye (panel B), van Kossa stainings (panel C), and gene expression analysis (panel D).
FIG. 11.

Quantification of the calcium phosphate precipitates in the aptamer 74-positive and aptamer 74-negative fractions and gene expression analysis of osteogenic marker genes directly after the fluorescent cell sorting of JPCs that were osteogenic induced for 5 days. (A) Representative Alizarin red staining of aptamer 74-positive versus -negative cells after day 30 of osteogenic differentiation as compared to undifferentiated JPCs. Abbreviations: co,untreated; OB, osteogenic differentiation. (B) Photometric quantification of the secreted calcium phosphate precipitates in aptamer 74-sorted cells. (C) Representative van Kossa stainings of aptamer 74-positive versus -negative cells after day 30 of osteogenic induction compared to undifferentiated JPCs. (D) Gene expression analyses of the aptamer 74-positive versus -negative cell fractions. Induction factors of gene expression levels in aptamer 74-positive versus -negative cells are indicated in the diagram. Color images available online at www.liebertpub.com/nat
Gene expression
Additionally, we performed gene expression analysis of osteogenesis-associated genes, including alkaline phosphatase (AP), collagen 1 alpha 1 (Coll1a1), Runt-related transcription factor-2 (Runx-2), insulin-like growth factor-2 (IGF-2), laminin alpha 2 (Lama2), bone morphogenic protein 4 and 6 (BMP-4, BMP-6), cartilage oligomeric protein (COMP) and twist homolog 1 (Twist1) in the aptamer 74-positive and -negative cell fractions directly following the cell sorting process performed on day 5 of osteogenic induction (Fig. 11D). The gene expression levels in the cells from the aptamer 74-positive fraction were expressed as relative induction factors compared to those detected in cells from the aptamer 74-negative fraction. Therefore, the mRNA levels from the negative fraction were defined as 1.
The gene expression levels of Coll1a1 (2.05±1.0) and Runx-2 (2.34±1.69) were slightly higher in the aptamer 74-positive cells than those detected in the negative fraction. However, the differences were not significant. We could not detect any differential expression patterns of AP, IGF-2, Lama2, BMP-4, BMP-6, COMP, and Twist1 in the cell subsets analyzed. The aptamer 74-positive as well as the aptamer 74-negative fraction revealed similar gene expression patterns.
Nano-computed tomography analyses of 3D cultured aptamer 74-sorted cells
To get an overview over the degree of mineralization inside the constructs, nano-computed tomography (CT) analyses were performed. Within the constructs, a homogenous distribution of precipitates was detected. The untreated cells showed less mineralization than osteogenic induced cells within the scaffolds. Additionally, the aptamer 74-positive fraction mineralized stronger than the negative one, as shown in Fig. 12.
FIG. 12.
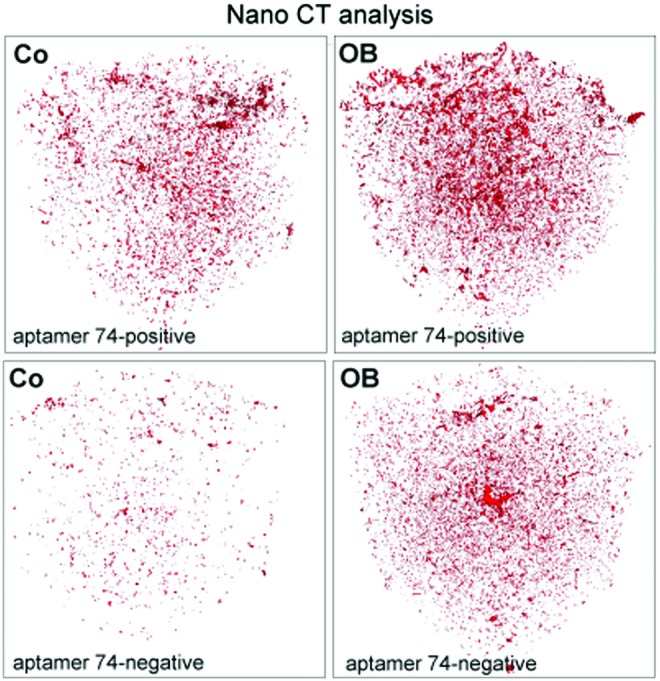
High resolution computer tomography of 3D constructs using the phoenix nanotom S (GE Healthcare). Aptamer 74-positive and negative cells were seeded into polylactic acid scaffolds and cultured for 40 days under untreated (co) and osteogenic induced (OB) conditions. After cell fixation and lyophilization constructs were used for nano-CT analyses. Color images available online at www.liebertpub.com/nat
Scanning electron microscopic analyses of 3D cultured aptamer 74-sorted cells
Scanning electron micrographs showed stronger mineralization on scaffold surfaces seeded with aptamer 74-positive cells in comparison to aptamer 74-negative cells seeded onto polylactic acid scaffolds (Fig. 13A). To confirm the components of formed nodules, we performed element analysis by EDX spectrometry (Fig. 13B). The results indicate the presence of calcium, phosphate, and oxygen in the precipitates. On scaffolds seeded with aptamer 74-positive cells we detected higher numbers of precipitates and an enhanced amount of the three elements within the precipitates. Three-dimensional cultured untreated cells did not show any precipitates containing the above-mentioned elements.
FIG. 13.

(A) Representative scanning electron micrographs (SEMs) of 3D constructs and (B) element analysis by energy dispersive x-ray (EDX) spectrometry of formed precipitates within the analyzed scaffolds. Aptamer 74-positive and negative cells were seeded into polylactic acid scaffolds and cultured for 40 days under untreated (co) and osteogenic induced (OB) conditions. After cell fixation and lyophilization the degree of mineralization was visualized and analyzed. SEMs are illustrated in a 1,000-fold magnification. Gold (Au) and palladium (Pd) peaks are caused by sputter coatings of 3D constructs. Color images available online at www.liebertpub.com/nat
Discussion
The aim of this study was to establish a suitable approach for generating an aptamer with high specificity to human jaw periosteal cells. We intended to identify a DNA aptamer that was able to specifically bind the osteogenic progenitors within the entire heterogeneous JPC cell population. For tissue engineering purposes, aptamer-coated scaffolding materials should recruit the osteogenic progenitor cells in vitro and in vivo. Additionally, it was of interest to determine whether the aptamer could be employed as a general mesenchymal stromal cell marker.
Aptamers display a high potential for the treatment of acute and chronic diseases (Guo et al., 2007a) and have been successfully used in different disciplines [i.e., diagnostics (Chen et al., 2008), therapy (Chapman and Beckey, 2006) and target validation (Blank and Blind, 2005)]. The only FDA-approved aptamer for clinical application to date is pegaptanib, an inhibitor of ocular neovascularization that is used for the treatment of age-related macular degeneration (Vavvas and D'Amico, 2006; Apte et al., 2007).
Until now, little has been learned and published about the use of aptamers in the growing field of tissue engineering (Guo et al., 2007a). However, the group of H.P. Wendel generated a capture molecule to isolate porcine MSCs from bone marrow (Guo et al., 2006). In the same group, an aptamer against osteosarcoma cells was identified. Polystyrene plates and titan implants were then coated with this aptamer, resulting in a significant enhancement in cell adhesion (Guo et al., 2005; Guo et al., 2007b). In addition, an aptamer that specifically bound to porcine endothelial progenitor cells was generated and used to coat polymers (Hoffmann et al., 2008). Other published studies have dealt with different combinations of biomaterials and aptamers (Zhou and Rossi, 2011).
In our study, an aptamer with an over-30% binding affinity to JPCs was generated. The other identified aptamers that were classified into sequence families other than family 4 exhibited only weak affinities to JPCs, as described in the results section. JPCs from a single donor patient were used for the entire SELEX procedure. Thus, we tested the binding affinity of aptamer 74 to JPCs derived from different donors and detected donor-dependent binding affinities up to 32%.
Live measurements using the xCELLigence system provided an insight into aptamer 74-dependent physiology. JPCs incubated in aptamer 74-coated wells exhibited noticeably higher proliferation rates (Fig. 9). JPCs cultured in DNA library-coated plates also showed enhanced proliferation. This finding could be explained by unspecific interactions of the JPCs with the immobilized DNA. Alternatively, it could be possible that the DNA coating had a disadvantageous effect on the accuracy of the live measurements.
In other analyses, JPCs were induced into osteo-, chondro-, and adipogenic lineages, and the binding affinities of the aptamer to these cells were determined. Flow cytometry provided evidence that aptamer 74 clearly had higher specificity for osteogenic-induced JPCs than for undifferentiated, chondrogenic, and adipogenic cells. However, analysis of in vitro mineralization revealed a slight tendency of an enhanced osteogenic potential of aptamer 74-positive cells as shown by van Kossa stainings. This tendency could not be confirmed by Alizarin red stainings. However, the specificity of the Alizarin red staining is discussed controversially.
In order to confirm the results from van Kossa stainings, aptamer 74-sorted cell fractions were cultured on 3D polylactic acid scaffolds. Nano-CT analyses indicate higher mineralization capacities of aptamer 74-positive cells compared to the respective negative fraction, as shown in Fig. 12. Unfortunately, during nano-CT imaging the structure of the polylactic acid scaffolding material was slightly destroyed resulting in SEM images of lesser quality. Due to limited cell numbers after the cell sorting procedure, the same constructs have been used for both analyses approaches. However, considering all results obtained by nano-CT, SEM, and EDX spetromery analyses it can be assumed that aptamer 74-positive cells show a higher mineralization capacity compared to that of aptamer 74-negative cells.
The fact that aptamer 74 bound to osteogenic differentiated JPCs with a higher affinity seemed to contradict the fact that the aptamer bound to mineralizing JPCs as well to non-mineralizing JPCs in a similar manner. A possible explanation for this observation might be that both mineralizing and non-mineralizing cells display very similar expression patterns during the first 5 days of osteogenesis. The cell target bound by aptamer 74 is most likely a factor in the early stages of osteogenesis of JPCs.
However, as shown in Fig. 10, the proliferation potential of the aptamer 74-positive cell fraction seemed to be clearly delayed. In our recent studies, we observed that after the magnetically activated cell sorting of JPCs, the positive fraction always proliferated more slowly (unpublished data). Osteogenesis follows a defined sequential arrangement and includes three phases, beginning with the strong proliferation of the differentiating progenitor cells. Following this stage, there is a phase of extensive synthesis of the extracellular matrix, finally followed by the late phase of differentiation and mineral deposits. However, if the proliferation phase is elongated, it can be assumed that the mineralization phase of the aptamer-positive cell fraction is also delayed. Therefore, a certain inaccuracy in the quantification of the calcium phosphate precipitates must be considered. Similarly, using the gene expression analysis, we did not detect differential expression patterns of osteogenic factors, such as AP, Runx-2, TWIST, and Coll1a1, after the cell sorting between the positive and negative fractions. Only a slight inductive tendency for osteogenic factors was observed in the aptamer 74-positive cell subpopulation.
Another interesting question was the identity of the target structure on the osteogenic differentiated cells bound by aptamer 74. Therefore, we performed different analysis to identify this target. The aptamer 74 was coupled to streptavidin-labeled Dynabeads and incubated with whole-cell lysates of osteogenic differentiated cells to allow target protein binding. After dissociation of Dynabeads from the bound protein/structure, proteins were loaded and separated on a sodium dodecyl sulfate gel and visualization of the detected proteins was performed by silver staining. Bands which were not detected in control samples but in JPC extracts were excised and analyzed by mass spectrometrical analyses (not shown). The putative targets (vimentin and annexin 5 were identified as potential targets) were then analyzed by western blot, fluorescence microscopy and flow cytometry using an antibody towards the protein hypothesized to be the target (data not shown). Unfortunately, the putative target could not be confirmed by the used approaches and we could not detect any co-localization of the potential target protein and the bound aptamer 74. Additionally, we did not detect any enhanced gene expression of the presumed target in aptamer 74-positive cells in comparison to the negative ones. Therefore, the results were not convincing enough to address a target molecule binding to aptamer 74.
To date, there is no reliable stem cell marker that exactly characterizes MSCs. The minimal criteria for MSCs were defined previously (Dominici et al., 2006), but these markers remain controversial. For example, 2 subpopulations isolated from bone marrow, namely the MSCA1+CD56 + and MSCA1+CD56- (MSCA1: mesenchymal stem cell antigen 1, CD56: neural cell adhesion molecule [NCAM]) subsets, both fulfill the minimal criteria for MSCs but exhibit disparate differentiation potentials (Buhring et al., 2009a; Buhring et al., 2009b). The MSCA1+CD56 +subpopulation has been described to elicit a higher chondrogenic potential. In contrast, the MSCA1+CD56-cell fraction displays an enhanced adipogenic differentiation potential. Furthermore, there is a general disagreement concerning the precise definition of stem cells. It has even been postulated that pericytes (cell-stabilizing vessels, which were described to be multipotent) are the MSCs, at least when studied ex vivo (Meirelles Lda and Nardi 2009; Augello et al., 2010). Therefore, it was desirable to develop an MSC marker aptamer with an unknown target. We examined whether aptamer 74 was able to bind to other MSC types by determining the binding affinities of the molecule to bone marrow- and placenta-derived MSCs in comparison to periosteum-derived MSCs. The undifferentiated controls showed only weak binding affinities, whereas similar amounts of positive counts were detected for osteogenic differentiated cells as found for JPCs. This result leads to the assumption that upon osteogenic differentiation, the MSCs from different sources investigated in this study all expressed the aptamer 74 target.
To test the specificity of aptamer 74 and to determine that the target was not expressed by other cell types, the binding affinities to other cell types, such as primary endothelial cells, osteoblasts, oral squamous carcinoma cells, lung carcinoma cells and endothelial cells, were analyzed. None of the tested cell types showed a binding affinity to aptamer 74.
The cell population isolated from jaw periosteum is quite heterogeneous and contains a large proportion of tissue fibroblasts. Furthermore, other cells, including endothelial, muscle, and adipogenic cells, are present. To ensure that the aptamer did not bind to these contaminating cells, human prostate fibroblasts and primary endothelial cells were analyzed using flow cytometry, and the binding affinities of aptamer 74 to these cells were determined. We obtained no positive counts in the undifferentiated or in the osteogenic-stimulated fibroblasts. The negative counterselection was performed using the human oral squamous carcinoma cell line BICR56 and primary PBMCs. Aptamer 74 did not show any binding affinity to PBMCs, which was expected because all of the aptamers that bound to these cells were eliminated during the negative counterselection in the SELEX procedure.
Due to the preferential binding affinity of aptamer 74 to osteogenic differentiated cells, we also tested the binding of the aptamer towards the human osteosarcoma cell lines Cal72 and MG63. However, we could not detect any positive counts, which at first seemed inconsistent. Taking into consideration that the expression pattern of sarcoma cell lines differs severely from primary cells, it is possible that—in contrast to the primary cells—these tumor cells simply do not express the target of aptamer 74 (Rochet et al., 2003). As mentioned earlier, the target is most likely a protein expressed in the early phase of osteogenesis, and it is likely that the dedifferentiated tumor cells no longer express this target. Due to the high autofluorescence of the primary osteoblastic cell cultures isolated by enzymatic digestion, it was not possible to perform flow cytometry analysis with these cells, even with an attempt to eliminate the autofluorescence through quenching.
In sum, we can state that aptamers, in general, are a promising tool in the growing field of tissue engineering.
In the present study, we succeeded in the generation of an aptamer that preferentially bound to osteogenic-stimulated human jaw periosteal cells. We showed that aptamer 74 binds to MSCs derived from different cell sources and that aptamer 74-positive 3D cultured cells reveal a higher osteogenic potential compared to cells from the negative fraction. However, the target and the phenotype of the aptamer 74 binding population remain unclear.
Chemical modifications to enhance the binding properties of aptamer 74 should be undertaken to increase its specificity to osteogenic-induced JPCs. In this regard, subsequent studies are required to develop a suitable approach for the immobilization of aptamer 74 onto scaffolding materials and to analyze the influence of the aptamer on JPC adhesion, proliferation, and differentiation in 3D culture.
Acknowledgments
We thank Dr. H.J. Bühring for giving us the opportunity to perform cell sorting experiments within the FACS core facility and Prof. Gerd Klein and Tatjana Kaiser for providing us with the Cal72 and MG 63 cell lines. Additionally, we would like to thank Dr. Andrea Nolte for providing us with the A594, SK-MES, and EAHy926 cell lines, the human umbilical vein endothelial cells, and the prostate fibroblasts. N.A. was financed by DF6 Graduate College 794 (German Research Foundation).
Author Disclosure Statement
No competing financial interests exist.
References
- AGATA H. ASAHINA I. YAMAZAKI Y. UCHIDA M. SHINOHARA Y. HONDA M.J. KAGAMI H. UEDA M. Effective bone engineering with periosteum-derived cells. J. Dent. Res. 2007;86:79–83. doi: 10.1177/154405910708600113. [DOI] [PubMed] [Google Scholar]
- ALEXANDER D. ARDJOMANDI N. MUNZ A. FRIEDRICH B. REINERT S. ECM remodeling components regulated during jaw periosteal cell osteogenesis. Cell. Biol. Int. 2011;35:973–80. doi: 10.1042/CBI20100836. [DOI] [PubMed] [Google Scholar]
- ALEXANDER D. HOFFMANN J. MUNZ A. FRIEDRICH B. GEIS-GERSTORFER J. REINERT S. Analysis of OPLA scaffolds for bone engineering constructs using human jaw periosteal cells. J. Mater. Sci. Mater. Med. 2008;19:965–974. doi: 10.1007/s10856-007-3351-8. [DOI] [PubMed] [Google Scholar]
- ALEXANDER D. SCHAFER F. MUNZ A. FRIEDRICH B. KLEIN C. HOFFMANN J. BUHRING H.J. REINERT S. LNGFR induction during osteogenesis of human jaw periosteum-derived cells. Cell Physiol. Biochem. 2009;24:283–290. doi: 10.1159/000233253. [DOI] [PubMed] [Google Scholar]
- ALEXANDER D. SCHAFER F. OLBRICH M. FRIEDRICH B. BUHRING H.J. HOFFMANN J. REINERT S. MSCA-1/TNAP selection of human jaw periosteal cells improves their mineralization capacity. Cell Physiol. Biochem. 2010;26:1073–1080. doi: 10.1159/000323985. [DOI] [PubMed] [Google Scholar]
- APTE R.S. MODI M. MASONSON H. PATEL M. WHITFIELD L. ADAMIS A.P. Pegaptanib 1-year systemic safety results from a safety-pharmacokinetic trial in patients with neovascular age-related macular degeneration. Ophthalmology. 2007;114:1702–1712. doi: 10.1016/j.ophtha.2007.02.021. [DOI] [PubMed] [Google Scholar]
- AUGELLO A. KURTH T.B. DE BARI C. Mesenchymal stem cells: a perspective from in vitro cultures to in vivo migration and niches. Eur. Cell Mater. 2010;20:121–133. doi: 10.22203/ecm.v020a11. [DOI] [PubMed] [Google Scholar]
- BAGALKOT V. FAROKHZAD O.C. LANGER R. JON S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew. Chem. Int., Ed. 2006;45:8149–8152. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]
- BIEBACK K. KERN S. KOCAOMER A. FERLIK K. BUGERT P. Comparing mesenchymal stromal cells from different human tissues: bone marrow, adipose tissue and umbilical cord blood. Biomed. Mater. Eng. 2008;18:S71–S76. [PubMed] [Google Scholar]
- BLANK M. BLIND M. Aptamers as tools for target validation. Curr. Opin. Chem. Biol. 2005;9:336–342. doi: 10.1016/j.cbpa.2005.06.011. [DOI] [PubMed] [Google Scholar]
- BUHRING H.J. BATTULA V.L. TREML S. BAREISS P.M. GIESEKE F. ROELOFS H. DE ZWART P. MULLER I. SCHEWE B. SKUTELLA T., et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen-1. Haematologica. 2009a;94:173–184. doi: 10.3324/haematol.13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUHRING H.J. TREML S. CERABONA F. DE ZWART P. KANZ L. SOBIESIAK M. Phenotypic characterization of distinct human bone marrow-derived MSC subsets. Ann. N. Y. Acad. Sci. 2009b;1176:124–134. doi: 10.1111/j.1749-6632.2009.04564.x. [DOI] [PubMed] [Google Scholar]
- BUNKA D.H. STOCKLEY P.G. Aptamers come of age: at last. Nat. Rev. Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- CERCHIA L. DUCONGE F. PESTOURIE C. BOULAY J. AISSOUNI Y. GOMBERT K. TAVITIAN B. DE FRANCISCIS V. LIBRI D. Neutralizing aptamers from whole-cell SELEX inhibit the RET receptor tyrosine kinase. PLoS Biol. 2005;3:e123. doi: 10.1371/journal.pbio.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- CHAN J. KENNEA N.L. FISK N.M. Placental mesenchymal stem cells. Am. J. Obstet. Gynecol. 2007;196:e18. doi: 10.1016/j.ajog.2006.08.002. author reply e18–19. [DOI] [PubMed] [Google Scholar]
- CHAPMAN J.A. BECKEY C. Pegaptanib: a novel approach to ocular neovascularization. Ann. Pharmacother. 2006;40:1322–1326. doi: 10.1345/aph.1G604. [DOI] [PubMed] [Google Scholar]
- CHEN H.W. MEDLEY C.D. SEFAH K. SHANGGUAN D. TANG Z. MENG L. SMITH J.E. TAN W. Molecular recognition of small-cell lung cancer cells using aptamers. ChemMedChem. 2008;3:991–1001. doi: 10.1002/cmdc.200800030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE BARI C. DELL'ACCIO F. LUYTEN F.P. Human periosteum-derived cells maintain phenotypic stability and chondrogenic potential throughout expansion regardless of donor age. Arthritis Rheum. 2001;44:85–95. doi: 10.1002/1529-0131(200101)44:1<85::AID-ANR12>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- DE BARI C. DELL'ACCIO F. VANLAUWE J. EYCKMANS J. KHAN I.M. ARCHER C.W. JONES E.A. MCGONAGLE D. MITSIADIS T.A. PITZALIS C., et al. Mesenchymal multipotency of adult human periosteal cells demonstrated by single-cell lineage analysis. Arthritis Rheum. 2006;54:1209–1221. doi: 10.1002/art.21753. [DOI] [PubMed] [Google Scholar]
- DEMARCO F.F. CONDE M.C. CAVALCANTI B.N. CASAGRANDE L. SAKAI V.T. NOR J.E. Dental pulp tissue engineering. Braz. Dent. J. 2011;22:3–13. doi: 10.1590/s0103-64402011000100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHAR S. GU F.X. LANGER R. FAROKHZAD O.C. LIPPARD S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOMINICI M. LE BLANC K. MUELLER I. SLAPER-CORTENBACH I. MARINI F. KRAUSE D. DEANS R. KEATING A. PROCKOP D. HORWITZ E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- ELLINGTON A.D. SZOSTAK J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- ESPOSITO C.L. CATUOGNO S. DE FRANCISCIS V. CERCHIA L. New insight into clinical development of nucleic acid aptamers. Discov. Med. 2011;11:487–496. [PubMed] [Google Scholar]
- FAMULOK M. MAYER G. Aptamer Modules as Sensors and Detectors. Acc. Chem. Res. 2011;44:1349–1358. doi: 10.1021/ar2000293. [DOI] [PubMed] [Google Scholar]
- GUO K. WENDEL H.P. SCHEIDELER L. ZIEMER G. SCHEULE A.M. Aptamer-based capture molecules as a novel coating strategy to promote cell adhesion. J. Cell. Mol. Med. 2005;9:731–736. doi: 10.1111/j.1582-4934.2005.tb00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO K.T. SCHAFER R. PAUL A. GERBER A. ZIEMER G. WENDEL H.P. A new technique for the isolation and surface immobilization of mesenchymal stem cells from whole bone marrow using high-specific DNA aptamers. Stem Cells. 2006;24:2220–2231. doi: 10.1634/stemcells.2006-0015. [DOI] [PubMed] [Google Scholar]
- GUO K.T. SCHAFER R. PAUL A. ZIEMER G. WENDEL H.P. Aptamer-based strategies for stem cell research. Mini Rev. Med. Chem. 2007a;7:701–705. doi: 10.2174/138955707781024481. [DOI] [PubMed] [Google Scholar]
- GUO K.T. SCHARNWEBER D. SCHWENZER B. ZIEMER G. WENDEL H.P. The effect of electrochemical functionalization of Ti-alloy surfaces by aptamer-based capture molecules on cell adhesion. Biomaterials. 2007b;28:468–474. doi: 10.1016/j.biomaterials.2006.09.021. [DOI] [PubMed] [Google Scholar]
- HE X. MA J. JABBARI E. Effect of grafting RGD and BMP-2 protein-derived peptides to a hydrogel substrate on osteogenic differentiation of marrow stromal cells. Langmuir. 2008;24:12508–12516. doi: 10.1021/la802447v. [DOI] [PubMed] [Google Scholar]
- HERSEL U. DAHMEN C. KESSLER H. RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- HOFFMANN J. PAUL A. HARWARDT M. GROLL J. REESWINKEL T. KLEE D. MOELLER M. FISCHER H. WALKER T. GREINER T., et al. Immobilized DNA aptamers used as potent attractors for porcine endothelial precursor cells. J. Biomed. Mater. Res. A. 2008;84A:614–621. doi: 10.1002/jbm.a.31309. [DOI] [PubMed] [Google Scholar]
- HUTMACHER D.W. SITTINGER M. Periosteal cells in bone tissue engineering. Tissue Eng. 2003;9(Suppl 1):S45–S64. doi: 10.1089/10763270360696978. [DOI] [PubMed] [Google Scholar]
- MAYER G. The chemical biology of aptamers. Angew. Chem., Int. Ed. 2009;48:2672–2689. doi: 10.1002/anie.200804643. [DOI] [PubMed] [Google Scholar]
- MAYER G. AHMED M.S. DOLF A. ENDL E. KNOLLE P.A. FAMULOK M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 2010;5:1993–2004. doi: 10.1038/nprot.2010.163. [DOI] [PubMed] [Google Scholar]
- MEIRELLES LDA S. NARDI N.B. Methodology, biology, and clinical applications of mesenchymal stem cells. Front. Biosci. 2009;14:4281–4298. doi: 10.2741/3528. [DOI] [PubMed] [Google Scholar]
- PILZ G.A. ULRICH C. RUH M. ABELE H. SCHAFER R. KLUBA T. BUHRING H.J. ROLAUFFS B. AICHER W.K. Human term placenta-derived mesenchymal stromal cells are less prone to osteogenic differentiation than bone marrow-derived mesenchymal stromal cells. Stem Cells Dev. 2011;20:635–646. doi: 10.1089/scd.2010.0308. [DOI] [PubMed] [Google Scholar]
- RINGE J. LEINHASE I. STICH S. LOCH A. NEUMANN K. HAISCH A. HAUPL T. MANZ R. KAPS C. SITTINGER M. Human mastoid periosteum-derived stem cells: promising candidates for skeletal tissue engineering. J. Tissue Eng. Regen. Med. 2008;2:136–146. doi: 10.1002/term.75. [DOI] [PubMed] [Google Scholar]
- ROCHET N. LEROY P. FAR D.F. OLLIER L. LOUBAT A. ROSSI B. CAL72: a human osteosarcoma cell line with unique effects on hematopoietic cells. Eur. J. Haematol. 2003;70:43–52. doi: 10.1034/j.1600-0609.2003.02766.x. [DOI] [PubMed] [Google Scholar]
- SCHOFER M.D. FUCHS-WINKELMANN S. GRABEDUNKEL C. WACK C. DERSCH R. RUDISILE M. WENDORFF J.H. GREINER A. PALETTA J.R. BOUDRIOT U. Influence of poly(L-lactic acid) nanofibers and BMP-2-containing poly(L-lactic acid) nanofibers on growth and osteogenic differentiation of human mesenchymal stem cells. ScientificWorldJournal. 2008;8:1269–1279. doi: 10.1100/tsw.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUERK C. GOLD L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- VAVVAS D. D'AMICO D.J. Pegaptanib (Macugen): treating neovascular age-related macular degeneration and current role in clinical practice. Ophthalmol. Clin. North Am. 2006;19:353–360. doi: 10.1016/j.ohc.2006.05.008. [DOI] [PubMed] [Google Scholar]
- WENDEL H.P. AVCI-ADALI M. PAUL A. WILHELM N. ZIEMER G. Upgrading SELEX technology by using lambda exonuclease digestion for single-stranded DNA generation. Molecules. 2010;15:1–11. doi: 10.3390/molecules15010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU J. ROSSI J.J. Cell-specific aptamer-mediated targeted drug delivery. Oligonucleotides. 2011;21:1–10. doi: 10.1089/oli.2010.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU S.J. CHOI B.H. HUH J.Y. JUNG J.H. KIM B.Y. LEE S.H. A comparative qualitative histological analysis of tissue-engineered bone using bone marrow mesenchymal stem cells, alveolar bone cells, and periosteal cells. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2006;101:164–169. doi: 10.1016/j.tripleo.2005.04.006. [DOI] [PubMed] [Google Scholar]
- ZUK P.A. ZHU M. MIZUNO H. HUANG J. FUTRELL J.W. KATZ A.J. BENHAIM P. LORENZ H.P. HEDRICK M.H. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–228. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]