

Abstract
Elevated resting heart rate is associated with greater risk of cardiovascular disease and mortality. In a 2-stage meta-analysis of genome-wide association studies in up to 181,171 individuals, we identified 14 new loci associated with heart rate and confirmed associations with all 7 previously established loci. Experimental downregulation of gene expression in Drosophila melanogaster and Danio rerio identified 20 genes at 11 loci that are relevant for heart rate regulation and highlight a role for genes involved in signal transmission, embryonic cardiac development and the pathophysiology of dilated cardiomyopathy, congenital heart failure and/or sudden cardiac death. In addition, genetic susceptibility to increased heart rate is associated with altered cardiac conduction and reduced risk of sick sinus syndrome, and both heart rate–increasing and heart rate–decreasing variants associate with risk of atrial fibrillation. Our findings provide fresh insights into the mechanisms regulating heart rate and identify new therapeutic targets.
A high resting heart rate has been associated with increased incidence of cardiovascular disease, as well as with cardiovascular and all-cause mortality, independent of traditional risk factors1–3. There are several potential mechanisms by which higher heart rate may contribute to greater cardiovascular risk. For example, higher heart rate entails elevated myocardial oxygen requirement and a shift in cardiac control from parasympathetic to sympathetic dominance, which may increase the likelihood of myocardial ischemia and electrical instability4. In addition, experimental alteration of heart rate by sinoatrial node ablation has been shown to influence the progression of atherosclerosis induced by an atherogenic high-cholesterol diet in cynomolgus monkeys5,6. In humans, selective reduction of heart rate using ivabradine was shown to reduce clinical events in individuals with heart failure, suggesting that elevated heart rate is a clinically relevant and modifiable risk factor7. However, whether the association of higher heart rate with cardiovascular risk is causal remains to be clarified.
Large twin studies with electrocardiogram (ECG) data have shown that genetic factors contribute to interindividual variation in heart rate, with heritability estimates ranging from 55 to 77% (refs. 8–10). So far, 3 genome-wide association studies (GWAS)11–13, incorporating data from up to 38,991 individuals each, have identified variants in 7 loci that show evidence of association with heart rate. These variants are common in the general population (minor allele frequency (MAF) ≥ 10%) and together explain ~0.7% of the variance in heart rate12. To gain more comprehensive insight into the genetic regulation of heart rate, we performed a 2-stage meta-analysis of GWAS in data from up to 181,171 individuals. Loci convincingly associated with heart rate were subsequently tested for association with cardiac conduction, rhythm disorders and cardiovascular disease to elucidate potential mechanisms underlying the association between heart rate and cardiovascular disease and mortality. Furthermore, we undertook experimental studies in D. melanogaster and D. rerio models as a first step toward identifying the causal genes within the associated loci.
RESULTS
Stage 1 GWAS identifies five new loci associated with heart rate
We performed a meta-analysis of the associations between 2,516,789 SNPs and heart rate in data from up to 85,787 individuals of European ancestry from 36 GWAS, including data from up to 11,207 individuals described previously13 and 6,568 individuals of Indian Asian ancestry (Online Methods, Supplementary Figs. 1–3 and Supplementary Tables 1–4). All studies included have been approved by local ethics committees, and all participants have provided their consent in writing. Our stage 1 meta-analysis showed associations with heart rate at genome-wide significance (P < 5 × 10−8) for variants in 12 loci (Table 1). These 12 loci included all 7 previously identified loci (in MYH6, CD46 and FADS1 and near GJA1, ACHE, SLC35F1 and LINC00477 (also known as C12orf67))11–13 and 5 additional loci (in KIAA1755, CCDC141, SYT10 and FLRT2 and near HCN4).
Table 1.
Stage 1 and stage 2 results for the loci that showed association with heart rate at genome-wide significance (P < 5 × 10−8)
| Alleles
|
Per-allele change in heart rateb
|
Stage 1 | Stage 2 | Stages 1 + 2
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Chr. | Nearest gene | Annotation | Heart rate SNP | Position (bp)a | Effect | Other | EAF | β | SE | P | P | N | P |
| Previously identified heart rate loci | ||||||||||||||
| 1 | 14 | MYH6 | N,P,S | rs365990 | 22931651 | G | A | 0.353 | 0.564 | 0.047 | 5.66 × 10−26 | 2.19 × 10−21 | 148,972 | 5.39 × 10−45 |
| 2 | 6 | GJA1 | S | rsl015451 | 122173184 | C | T | 0.102 | 0.713 | 0.072 | 7.06 × 10−20 | 2.14 × 10−17 | 155,303 | 1.14 × 10−32 |
| 3 | 7 | ACHE | N,Q,S,B | rsl3245899 | 100335067 | G | A | 0.195 | 0.447 | 0.055 | 1.54 × 10−15 | 3.27 × 10−14 | 176,643 | 7.67 × 10−27 |
| 4 | 1 | CD46 | S,B | rs11118555 | 206007476 | A | T | 0.124 | 0.612 | 0.069 | 4.35 × 10−14 | 7.49 × 10−15 | 166,654 | 3.88 × 10−26 |
| 5 | 11 | FADS1 | Q,S,B | rsl74549 | 61327958 | A | G | 0.310 | 0.358 | 0.047 | 1.13 × 10−12 | 1.57 × 10−12 | 172,847 | 1.38 × 10−22 |
| 6 | 6 | SLC35F1 | T,Q,P,S | rslll53730 | 118774215 | T | C | 0.509 | 0.381 | 0.044 | 7.01 × 10−15 | 8.79 × 10−9 | 156,783 | 7.55 × 10−21 |
| 7 | 12 | LINC00477 (C12orf67) | rsl7287293 | 24662145 | A | G | 0.850 | 0.444 | 0.062 | 6.90 × 10−12 | 3.98 × 10−11 | 151,085 | 3.07 × 10−20 | |
| Newly identified heart rate loci | ||||||||||||||
| 8 | 20 | KIAA1755 | N,S | rs6127471 | 36277452 | C | T | 0.540 | 0.429 | 0.045 | 5.96 × 10−23 | 2.98 × 10−10 | 162,593 | 5.22 × 10−29 |
| 9 | 2 | CCDC141 | N,P,S | rs17362588 | 179429291 | A | G | 0.114 | 0.736 | 0.077 | 3.25 × 10−18 | 4.22 × 10−11 | 136,061 | 3.57 × 10−26 |
| 10 | 12 | SYT10 | rs7980799 | 33468257 | A | C | 0.401 | 0.377 | 0.046 | 7.91 × 10−14 | 7.87 × 10−13 | 166,043 | 6.22 × 10−24 | |
| 11 | 15 | HCN4 | S | rs4489968 | 71452559 | T | G | 0.843 | 0.513 | 0.060 | 8.89 × 10−15 | 3.34 × 10−8 | 160,858 | 3.82 × 10−20 |
| 12 | 3 | GNB4 | S | rs7612445 | 180655673 | G | T | 0.816 | 0.358 | 0.060 | 2.41 × 10−7 | 7.78 × 10−10 | 140,395 | 1.86 × 10−14 |
| 13 | 14 | FLRT2 | S | rsl7796783 | 84879664 | T | C | 0.716 | 0.334 | 0.049 | 1.65 × 10−9 | 5.76 × 10−6 | 145,835 | 2.69 × 10−13 |
| 14 | 7 | CHRM2 | S | rs2350782 | 136293174 | C | T | 0.116 | 0.505 | 0.078 | 1.57 × 10−7 | 2.63 × 10−7 | 131,781 | 1.26 × 10−12 |
| 15 | 5 | NKX2-5 | Q,S | rs6882776 | 172596769 | G | A | 0.680 | 0.301 | 0.051 | 9.67 × 10−6 | 7.43 × 10−9 | 158,807 | 2.29 × 10−12 |
| 16 | 7 | GNG11 | C,Q,S,B | rsl80242 | 93387532 | T | A | 0.333 | 0.316 | 0.053 | 7.52 × 10−7 | 5.59 × 10−7 | 148,111 | 6.78 × 10−12 |
| 17 | 2 | B3GNT7 | N,Q,S,P,B | rsl3030174 | 231979528 | A | C | 0.733 | 0.300 | 0.051 | 1.13 × 10−6 | 3.66 × 10−4 | 144,810 | 1.04 × 10−10 |
| 18 | 3 | FNDC3B | S | rs9647379 | 173267862 | C | G | 0.400 | 0.206 | 0.047 | 2.59 × 10−7 | 3.07 × 10−4 | 137,314 | 1.17 × 10−9 |
| 19 | 12 | RFX4 | S | rs2067615 | 105673552 | A | T | 0.490 | 0.278 | 0.044 | 1.05 × 10−7 | 8.49 × 10−4 | 151,197 | 1.58 × 10−9 |
| 20 | 12 | CPNE8 | rs826838 | 37392998 | C | T | 0.443 | 0.234 | 0.045 | 3.32 × 10−7 | 6.20 × 10−4 | 166,632 | 3.73 × 10−9 | |
| 21 | 2 | TFPI | N,S | rs4140885 | 188041309 | A | G | 0.317 | 0.217 | 0.049 | 1.85 × 10−6 | 1.40 × 10−3 | 170,395 | 4.72 × 10−8 |
Chr., chromosome. EAF, effect allele frequency based on meta-analysis of stages 1 and 2 combined. Annotation shows whether the heart rate-associated SNP is (i) in strong LD (r2 > 0.8) with a copy number variant (C), nonsynonymous variant (N) or variant in a transcription factor binding site (T); (ii) associated with an eQTL (Q); (iii) expressed at the protein level in mouse heart and phosphorylated upon stimulation of the β1AR (P); (iv) located in or near a gene that was identified as being of potential relevance for heart rate using the automated literature search program SNIPPER (S); or (v) located in or near a biological candidate gene for heart rate (B).
Positions are according to HapMap Build 36, and allele coding is based on the positive strand.
Effect sizes in bpm per effect allele obtained from stage 1 and stage 2 cohorts with heart rate as only outcome (N > 96,790; excludes data from the RRgen Consortium and ERF)
To validate associations of the loci that were significantly associated with heart rate in stage 1 and to identify additional loci (Supplementary Fig. 1), lead SNPs at 42 loci (associated at P < 3 × 10−5) were selected for follow-up (Online Methods and Supplementary Table 5). Conditional analyses based on summary statistics of stage 1 meta-analysis results14 identified two loci with secondary associations that remained significant (P < 5 × 10−8) after adjusting for the association of the lead SNP. These secondary associations were also selected for follow-up (Online Methods and Supplementary Table 6).
Stage 2 follow-up identifies nine additional new loci
In stage 2, we examined associations between the 42 loci identified in stage 1 and heart rate in data from up to 88,823 additional individuals of European descent from 27 GWAS, including data from up to 38,991 individuals from 15 GWAS described previously12, as well as 11 studies with Metabochip and 1 study with Cardiochip data (Online Methods, Supplementary Fig. 1 and Supplementary Tables 7–10).
In a joint analysis of results from stage 1 and stage 2, variants in 21 loci had associations that reached P < 5 × 10−8 in data from up to 181,171 individuals (Fig. 1, Table 1 and Supplementary Table 5). Among the 21 loci were all 12 loci with association P < 5 × 10−8 after stage 1, as well as 9 additional loci (in CHRM2, RFX4, CPNE8 and TFPI and near GNB4, NKX2-5, GNG11, B3GNT7 and FNDC3B). Hence, our study confirms the 7 previously identified loci11–13 and identifies 14 new loci robustly associated with heart rate.
Figure 1.
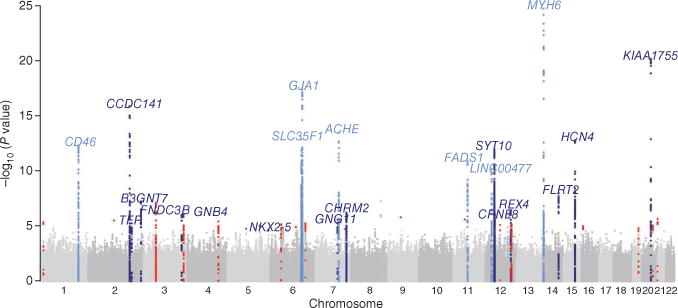
Manhattan plot of SNPs after meta-analysis of stage 1. The plot shows the significance of associations between all SNPs and heart rate in stage 1. The 7 loci that were previously identified are highlighted in light blue; the 14 newly associated loci are highlighted in dark blue. Loci that reached P < 3 × 10−5 after stage 1 but did not reach P < 5 × 10−8 after meta-analysis of stages 1 and 2 combined are highlighted in red.
Impact of the 21 confirmed loci on heart rate
The frequency of the heart rate–increasing alleles ranged from 10 to 85% for the 21 confirmed associations. Effect sizes of associations ranged from 0.21 to 0.74 beats per minute (bpm) per effect allele (mean ± s.d., 0.41 ± 0.15 bpm per effect allele) (Fig. 2 and Table 1).
Figure 2.
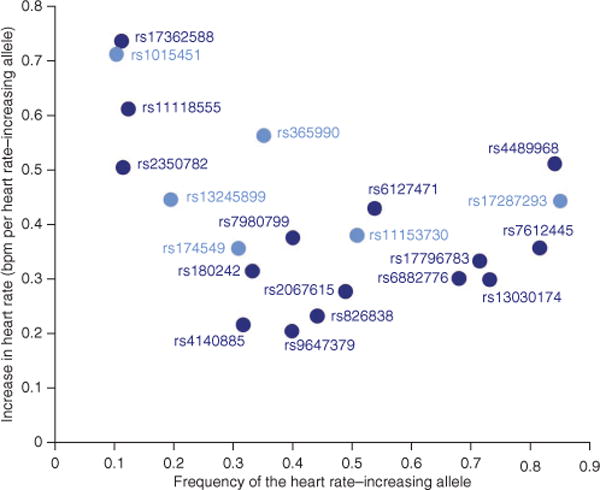
Effect size as a function of effect allele frequency. The plot shows the effect sizes of the 21 heart rate–associated SNPs after joint meta-analysis of stage 1 and stage 2 results as a function of their effect allele frequencies. Light-blue circles represent the 7 previously identified heart rate loci; dark blue circles represent the 14 newly identified heart rate loci.
To estimate the combined effect of the 21 loci on heart rate, we constructed a genetic predisposition score (GPS) by summing the number of heart rate-increasing alleles of the 21 associations. We examined associations between the GPS and heart rate in data from 5,053 adults from LifeLines2 (data for 19 loci available) and 4,000 12-year-old children from ALSPAC (data for 21 loci available) (Online Methods). The difference in average heart rate between individuals in the lowest and highest 5% of the GPS distribution was 4.1 bpm in adults (66.1 versus 70.2 bpm) and 4.9 bpm in children (73.7 versus 78.6 bpm) (Fig. 3a,b), differences that were previously shown to be clinically relevant15. The GPS explained 0.9% of the variance in heart rate in adults from LifeLines2 and 0.8% of the variance in children from ALSPAC.
Figure 3.
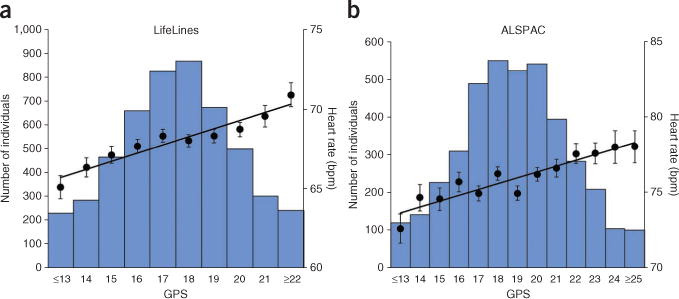
Combined effect of heart rate–increasing alleles on heart rate. (a) Combined effect of the 19 available heart rate loci in adults of European descent (LifeLines2, n = 5,053). (b) Combined effect of the 21 heart rate loci in 12-year-old children of European descent (ALSPAC, n = 4,000). In each plot, the number of heart rate–increasing alleles was summed across the heart rate–associated SNPs. The number of heart rate–increasing alleles is shown (x axis), grouped at the extremes, and mean heart rate ± s.e.m. is plotted (right y axis). The lines represent the regression of the mean heart rate values across the GPS distribution. The histogram shows the number of individuals in each GPS window (left y axis).
Conduction, rhythm disorders and cardiovascular disease
An altered heart rate reflects sinoatrial function and may reflect disturbed electrophysiological properties that are also present in other compartments of the heart. Such properties include atrial and atrioventricular nodal conduction (PR duration), ventricular depolarization (QRS duration) and myocardial repolarization (QT duration), which can be quantified on a 12-lead ECG. We examined whether the heart rate–associated loci showed evidence of association with cardiac conduction in data from previously reported GWAS for PR16, QRS17 and QT duration (QT-IGC Consortium (C.N.-C.), personal communication). Furthermore, we examined the association of the 21 loci with the risk of several conduction-related disorders, including atrial fibrillation, advanced (second- and third-degree) atrioventricular block and sick sinus syndrome (SSS, also known as sinus node dysfunction), as well as pacemaker implantation and sudden cardiac death13,18. Finally, elevated resting heart rate is a well-recognized precursor of increased blood pressure and hypertension, independent of initial blood pressure levels19,20, and predicts the incidence of coronary heart disease during up to 10 years of follow-up, independent of other major risk factors1,2. We therefore also examined associations of the heart rate loci with systolic blood pressure, diastolic blood pressure and the prevalence of hypertension, coronary artery disease (CAD) and myocardial infarction in data from the Global BPgen21 and CARDIoGRAM consortia22 (Online Methods).
For each of the ECG traits, we found a significant association with individual heart rate loci (P < 0.002). Heart rate–increasing alleles of these loci were associated with prolonged PR duration (near LINC00477 and NKX2-5) and reduced QT duration (near GJA1, FADS1, SLC35F1 and NKX2-5), independent of heart rate, as well as with both reduced (near GJA1, FADS1, SLC35F1 and NKX2-5) and prolonged (in CCDC141) QRS duration (Table 2). Common variants of the loci in or near GJA1, FADS1, CCDC141 and NKX2-5 were not previously identified as being associated with these cardiac conduction traits (Supplementary Table 11). In addition, stronger genetic susceptibility for increased heart rate as conferred by the GPS of 21 loci was associated with prolonged PR duration (P = 1.3 × 10−4) and reduced QT duration (P = 1.1 × 10−17), independent of heart rate, as well as with reduced QRS duration (P = 1.8 × 10−5) (Table 2 and Supplementary Fig. 4). These results suggest that, to some extent, similar cellular processes control heart rate and cardiac conduction through the atria and ventricles.
Table 2.
Association of the heart rate loci with cardiac conduction and rhythm disorders
| Alleles
|
PR duration
|
QRS duration
|
QT duration
|
Atrial fibrillation
|
SSS
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Nearest gene | Heart rate SNP | Effect | Other | N | β (ms) | SE (ms) | P | N | β (ms) | SE (ms) | P | N | β (ms) | SE (ms) | P | OR (95% CI) | P | OR (95% CI) | P |
| Previously identified heart rate loci | ||||||||||||||||||||
| 1 | MYH6 | rs365990 | G | A | 22,711 | −0.53 | 0.23 | 0.02 | 36,680 | −0.00 | 0.07 | 0.98 | 67,414 | 0.05 | 0.10 | 0.65 | 1.027 (0.99–1.06) |
0.12 | 0.931 (0.84–1.03) |
0.18 |
| 2 | GJA1 | rsl015451 | C | T | 26,528 | 0.66 | 0.34 | 0.05 | 40,090 | −0.38 | 0.11 | 6.51 × 10−4 | 72,806 | −0.50 | 0.16 | 1.67 × 10−3 | 0.903 (0.86–0.95) |
1.58 × 10−4 | 0.961 (0.82–1.13) |
0.63 |
| 3 | ACHE | rsl3245899 | G | A | 25,317 | 0.35 | 0.27 | 0.19 | 37,902 | −0.07 | 0.09 | 0.41 | 72,980 | 0.11 | 0.12 | 0.39 | 0.985 (0.95–1.03) |
0.46 | 0.951 (0.83–1.09) |
0.46 |
| 4 | CD46 | rs11118555 | A | T | 27,701 | −0.03 | 0.32 | 0.93 | 39,530 | −0.00 | 0.11 | 0.97 | 73,601 | −0.25 | 0.15 | 0.09 | 0.964 (0.92–1.01) |
0.15 | 0.895 (0.77–1.04) |
0.16 |
| 5 | FADS1 | rsl74549 | A | G | 28,482 | −0.53 | 0.22 | 0.02 | 40,085 | −0.27 | 0.07 | 1.85 × 10−4 | 74,604 | −0.57 | 0.10 | 4.33 × 10−6 | 1.003 (0.97–1.04) |
0.85 | 0.971 (0.88–1.07) |
0.57 |
| 6 | SLC35F1 | rslll53730 | T | C | 27,585 | 0.56 | 0.20 | 6.20 × 10−3 | 39,200 | −0.59 | 0.07 | 1.26 × 10−16 | 74,932 | −1.65 | 0.10 | 2.23 × 10−67 | 1.061 (1.03–1.10) |
5.19 × 10−4 | 0.973 (0.88–1.08) |
0.59 |
| 7 | LINC00477 (C12orf67) | rsl7287293 | A | G | 27,912 | 2.06 | 0.29 | 4.91 × 10−13 | 39,548 | −0.10 | 0.09 | 0.31 | 74,180 | −0.01 | 0.14 | 0.93 | 1.132 (1.08–1.19) |
2.38 × 10−7 | 0.811 (0.71–0.93) |
2.83 × 10−3 |
| Newly identified heart rate loci | ||||||||||||||||||||
| 8 | KIAA1755 | rs6127471 | C | T | 27,459 | 0.47 | 0.21 | 0.02 | 39,775 | −0.06 | 0.07 | 0.40 | 72,570 | −0.28 | 0.10 | 4.17 × 10−2 | 0.978 (0.95−1.01) |
0.18 | 0.973 (0.88−1.08) |
0.61 |
| 9 | CCDC141 | rs17362588 | A | G | 24,111 | −0.83 | 0.33 | 0.01 | 35,425 | 0.55 | 0.11 | 4.75 × 10−7 | 64,780 | 0.27 | 0.16 | 0.09 | 1.016 (0.96–1.07) |
0.55 | 0.873 (0.75–1.02) |
0.09 |
| 10 | SYT10 | rs7980799 | A | C | 24,965 | 0.52 | 0.22 | 0.02 | 36,338 | 0.03 | 0.07 | 0.64 | 69,431 | −0.18 | 0.10 | 0.08 | 0.990 (0.96–1.02) |
0.57 | 1.027 (0.92–1.13) |
0.60 |
| 11 | HCN4 | rs4489968 | T | G | 25,991 | 0.67 | 0.29 | 0.02 | 38,579 | 0.09 | 0.09 | 0.33 | 72,387 | 0.05 | 0.13 | 0.69 | 0.856 (0.82–0.89) |
1.85 × 10−12 | 0.924 (0.81–1.06) |
0.26 |
| 12 | GNB4 | rs7612445 | G | T | 23,153 | −0.37 | 0.28 | 0.18 | 38,398 | −0.14 | 0.09 | 0.11 | 67,376 | −0.11 | 0.13 | 0.40 | 0.959 (0.92–1.00) |
0.04 | 0.912 (0.81–1.03) |
0.14 |
| 13 | FLRT2 | rsl7796783 | T | C | 27,993 | −0.12 | 0.22 | 0.61 | 38,467 | −0.05 | 0.07 | 0.54 | 73,380 | −0.24 | 0.11 | 0.02 | 0.959 (0.93–0.99) |
0.02 | 0.898 (0.81–1.00) |
0.05 |
| 14 | CHRM2 | rs2350782 | C | T | 23,414 | −0.26 | 0.37 | 0.48 | 34,417 | −0.07 | 0.12 | 0.57 | 58,549 | 0.25 | 0.19 | 0.17 | 0.941 (0.89–1.00) |
0.05 | 0.911 (0.76–1.09) |
0.32 |
| 15 | NKX2-5 | rs6882776 | G | A | 18,484 | 1.29 | 0.27 | 1.46 × 10−6 | 30,877 | −0.27 | 0.09 | 1.87 × 10−2 | 60,768 | −0.49 | 0.12 | 2.27 × 10−5 | 1.110 (1.07–1.15) |
4.41 × 10−6 | 1.056 (0.94–1.18) |
0.33 |
| 16 | GNG11 | rsl80242 | T | A | 21,101 | 0.33 | 0.24 | 0.17 | 34,141 | 0.00 | 0.08 | 0.96 | 63,609 | 0.02 | 0.11 | 0.84 | 0.995 (0.96–1.03) |
0.80 | 1.025 (0.93–1.14) |
0.63 |
| 17 | B3GNT7 | rsl3030174 | A | C | 27,169 | 0.18 | 0.24 | 0.45 | 38,446 | −0.23 | 0.08 | 4.14 × 10−3 | 72,111 | −0.14 | 0.11 | 0.19 | 0.997 (0.96−1.03) |
0.88 | 1.025 (0.91−1.15) |
0.67 |
| 18 | FNDC3B | rs9647379 | C | G | 21,503 | 0.65 | 0.23 | 4.23 × 10−3 | 35,642 | −0.02 | 0.07 | 0.74 | 63,885 | 0.03 | 0.10 | 0.74 | 1.002 (0.97–1.04) |
0.89 | 0.969 (0.87–1.08) |
0.55 |
| 19 | RFX4 | rs2067615 | A | T | 27,782 | −0.17 | 0.20 | 0.40 | 39,494 | -0.05 | 0.07 | 0.42 | 74,116 | −0.02 | 0.10 | 0.83 | 1.029 (1.00–1.06) |
0.08 | 1.054 (0.96–1.16) |
0.29 |
| 20 | CPNE8 | rs826838 | C | T | 28,059 | 0.62 | 0.21 | 2.78 × 10−3 | 39,313 | 0.02 | 0.07 | 0.80 | 74,402 | −0.04 | 0.10 | 0.71 | 0.999 (0.97–1.03) |
0.93 | 0.926 (0.84–1.02) |
0.12 |
| 21 | TFPI | rs4140885 | A | G | 26,384 | −0.15 | 0.23 | 0.50 | 37,447 | 0.05 | 0.08 | 0.55 | 68,919 | −0.18 | 0.11 | 0.10 | 1.011 (0.98–1.05) |
0.55 | 1.126 (1.02–1.25) |
0.02 |
| Multi-:SNP predispostion score | 2.55 | 0.67 | 1.33 × 10−4 | −0.95 | 0.22 | 1.83 × 10−5 | −2.66 | 0.31 | 1.12 × 10−17 | 0.921 (0.83–1.02) |
0.11 | 0.589 (0.44–0.78) |
2.34 × 10−4 | |||||||
Association analyses were performed in data from the PR GWAS Consortium (PR duration), the QRS GWAS Consortium (QRS duration), the QT-IGC Consortium (QT duration), deCODE Genetics and the CHARGE-AF Consortium (atrial fibrillation), and deCODE Genetics (SSS), Analyses were performed in data from 9,183 cases and 91,625 controls for atrial fibrillation and in data from 903 cases and 40,722 controls for SSS. CI, confidence interval; effect allele, the allele that is associated with higher heart rate. P values for association with PR duration are not adjusted for population stratification, as are the ones for the remaining traits. The multi-SNP predisposition score shows the combined effect of the 21 heart rate–associated SNPs per genetically predicted 5-bpm increase in heart rate, based on single-SNP summary statistics and weighting by effect sizes for heart rate. P values smaller than 2.0 × 10−3 are considered statistically significant.
Five of the 21 heart rate loci are associated with atrial fibrillation (P < 0.002). Heart rate–increasing alleles of these loci were associated with both increased (near SLC35F1, LINC00477 and NKX2-5; odds ratio (OR) = 1.06–1.13) and decreased (near GJA1 and HCN4; OR = 0.86–0.90) risk of atrial fibrillation (Table 2). Common variants of the loci in or near GJA1, SLC35F1 and NKX2-5 were not previously identified as being associated with atrial fibrillation (Supplementary Table 11). Stronger genetic susceptibility for increased heart rate in the 21 loci combined was not associated with atrial fibrillation, which reflects the bidirectionality of the associations in the individual loci (Table 2).
None of the heart rate loci showed evidence of association with the risk of atrioventricular block, SSS, pacemaker implantation or sudden cardiac death individually (Supplementary Table 12). However, a higher GPS was associated with reduced risk of SSS (P = 2.3 × 10−4) and pacemaker implantation (P = 3.6 × 10−4) (Table 2 and Supplementary Fig. 4). SSS encompasses a group of sinus rhythm disorders, including pathological sinus bradycardia (slow heart rate), sinus arrest, sinoatrial block and paroxysmal tachycardias (bradycardia-tachycardia syndrome). SSS is the most common indicator for permanent pacemaker implantation23, and ~80% of individuals with SSS in our data set had undergone pacemaker implantation18. Hence, the association between the heart rate loci and pacemaker implantation is likely secondary to the association with SSS in this study population.
None of the heart rate loci showed evidence of association with blood pressure or prevalent hypertension, CAD or myocardial infarction, either individually or when combined in the GPS of 21 loci (Supplementary Fig. 4 and Supplementary Tables 13 and 14). In addition, we showed, at most, limited evidence of association with heart rate for loci previously identified as being associated with blood pressure or prevalent hypertension, CAD or myocardial infarction (Supplementary Tables 15 and 16).
Pathway analyses
The 21 confirmed loci contain 234 genes that are located within 500 kb of the associations with heart rate (Supplementary Fig. 5 and Supplementary Table 17). To systematically identify biological connections between these genes and to identify new pathways associated with heart rate, we tested whether biological processes or molecular functions that were predefined in five databases were enriched for multiple modest heart rate associations using MAGENTA24 (Online Methods). We found evidence of enrichment of associations in pathways involved in dilated, hypertrophic and arrhythmogenic right ventricular cardiomyopathy, (cardiac) muscle contraction, regulation of heart contraction, integration of energy metabolism, positive regulation of cell adhesion and Alzheimer’s disease (P < 2 × 10−3, false discovery rate (FDR) < 0.1) (Supplementary Tables 18 and 19).
Follow-up in D. melanogaster and D. rerio
Variants identified by GWAS typically implicate genomic regions rather than individual genes. We used a range of approaches to identify promising candidate genes for heart rate regulation within the 21 loci, including proteomics experiments aimed at identifying genes expressed at the protein level in mouse heart that are phosphorylated upon stimulation of the β1 adrenergic receptor (β1AR), gene expression quantitative trait locus (eQTL) analysis in blood, in silico search for potentially functional variants in high linkage disequilibrium (LD) with lead variants (r2 > 0.8), an automated literature search using the program SNIPPER and biological candidacy (Online Methods and Supplementary Tables 20–23). These approaches labeled 49 of the 234 genes located within the 21 loci as candidate genes for heart rate regulation (Supplementary Table 24).
To examine whether some of the 49 candidate genes are likely to underlie the associations identified by GWAS, we performed 2 series of experiments using animal models. First, we compared heart rate and risk of arrhythmia in D. melanogaster control pupae and pupae in which orthologs of the candidate genes were downregulated using RNA-mediated gene interference (RNAi), both at rest and after 20 min of tachypacing (Online Methods and Supplementary Tables 25 and 26). Second, we compared heart rate and fractional shortening of the ventricular chamber in control embryos of the zebrafish D. rerio and embryos in which orthologs of the candidate genes were downregulated using morpholino oligonucleotides (Online Methods and Supplementary Table 27).
Results were available for the orthologs of 25 candidate genes from 13 loci in D. melanogaster pupae and for orthologs of 12 genes from 7 loci in D. rerio embryos; results from orthologs of 6 genes were available in both species (Supplementary Fig. 6 and Supplementary Table 24). Results from these experiments support a role in heart rate regulation for 20 of the 31 candidate genes tested across the 2 models: ACHE, UFSP1, TRIP6, EPHB4 and PCOLCE (locus 3), PLXNA2 (locus 4), FADS1, FADS2, FADS3 and BEST1 (locus 5), TTN (locus 9), MFN1 (locus 12), CHRM2 (locus 14), GNG11 (locus 15), NCL and HTR2B (locus 17), PLD1 and GHSR (locus 18), CRY1 (locus 19) and CALCRL (locus 21) (P < 2 × 10−3 in D. melanogaster pupae and P < 4 × 10−3 in D. rerio embryos) (Fig. 4 and Supplementary Tables 28 and 29).
Figure 4.
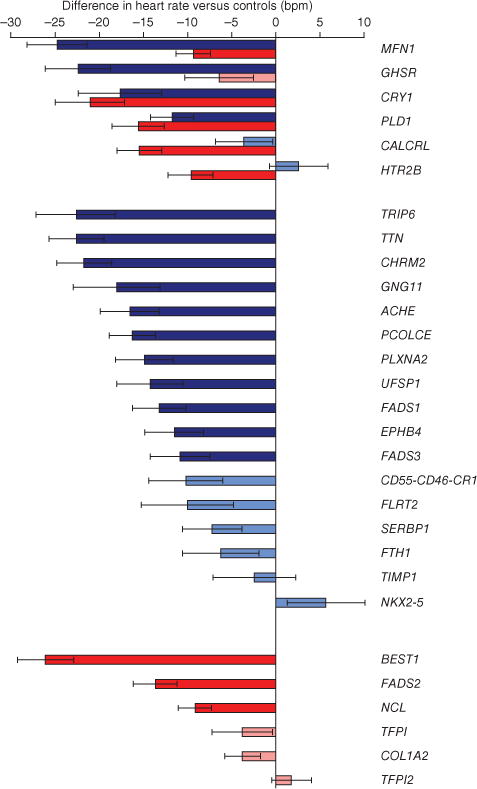
Effects on heart rate of reduced or ablated expression of orthologs of positional candidate genes from GWAS in D. melanogaster and D. rerio. Bars show the heart rate (± s.e.m.) of (i) D. melanogaster pupae with orthologs of positional candidate genes located within 500 kb of GWAS associations downregulated using RNAi compared with control pupae (blue bars) and (ii) D. rerio (zebrafish) embryos with expression of orthologs of positional candidate genes reduced by injecting morpholino oligonucleotides versus PBS (red bars). Darker coloring indicates that heart rate is significantly different in targeted animals compared with controls after Bonferroni correction for 23 tests in D. melanogaster (P < 2 × 10−3) and 12 tests in D. rerio (P < 4 × 10−3); lighter coloring indicates that differences do not reach significance. Results are ordered by availability (D. melanogaster and D. rerio, D. melanogaster only, D. rerio only) and by effect size.
The most convincing results were observed for orthologs of MFN1 and PLD1 (Supplementary Note), for which downregulated gene expression reduced resting heart rate in both models (P < 1 × 10−5). Moreover, D. melanogaster pupae with downregulated expression of the MFN1 ortholog were characterized by reduced heart rate after 20 min of tachypacing (P = 9.5 × 10−5) and by increased risk of arrhythmia, both at rest (P = 4.3 × 10−4) and after tachypacing (P = 2.0 × 10−6). In D. rerio embryos, in addition to reduced heart rate, downregulation of mfn1 (the ortholog of MFN1) was accompanied by edema in 73% of embryos, whereas 51% of embryos with reduced expression of pld1a (the ortholog of PLD1) had an unlooped heart (Supplementary Table 29).
In addition to reduced heart rate, downregulated gene expression of orthologs of ACHE, PCOLCE and FADS3 was associated with increased risk of arrhythmia after 20 min of tachypacing in D. melanogaster pupae. Furthermore, D. rerio embryos with reduced gene expression of D. rerio orthlogs that was accompanied by reduced heart rate were also characterized by edema (BEST1 and MFN1), blood pooling (FADS2), an unlooped heart (HTR2B, NCL, PLD1 and CALCRL) and atrioventricular canal malformation (CALCRL). Downregulated expression of the CRY1 ortholog was accompanied by a highly penetrant developmental malformation that likely mediates the heart rate effect (Supplementary Fig. 7 and Supplementary Table 29). Reduced expression of the COL1A2 ortholog did not affect heart rate in D. rerio embryos (P = 0.07) but resulted in reduced fractional shortening of the ventricular chamber (P = 1.8 × 10−3).
In summary, results from experiments in D. melanogaster and D. rerio models support a role in heart rate regulation for 20 genes found within 11 loci associated with heart rate (Supplementary Table 30). Notably, results from animal models confirmed the eQTL associations observed in humans for all available orthologs, with a consistent direction of effect across species for all genes except TRIP6 (Supplementary Tables 21 and 30).
DISCUSSION
Using a 2-stage meta-analysis of GWAS in up to 181,171 individuals, we identified 14 loci previously unknown to be robustly associated with heart rate and confirmed the 7 previously established loci, increasing the total number of heart rate loci to 21. Results from experiments in D. melanogaster and D. rerio models support a role in heart rate regulation for 20 candidate genes from 11 loci. These experiments highlight a role for genes that are essential for embryonic cardiovascular development and signal transmission, as well as for genes with a role in the pathophysiology of dilated cardiomyopathy, congestive heart failure and/or sudden cardiac death (Supplementary Note). In addition, stronger genetic susceptibility to higher heart rate is associated with prolonged PR duration and reduced QT duration, both independent of heart rate, as well as with reduced QRS duration and lower risk of SSS, a group of sinus rhythm disorders that result from sinus node dysfunction and are characterized by bradycardia.
Prevalent SSS is unlikely to explain the association between common variants and heart rate shown by GWAS, as the associations were essentially unchanged with a priori exclusion of individuals with prevalent cardiovascular disease, heart rate outside the range of 50–100 bpm and/or using heart rate–altering medication. This suggests that the confirmed loci have subtle effects on sinus node function in the general population, which manifest themselves in higher heart rate and reduced risk of SSS, showing the clinical relevance of our findings. Future studies should address whether such effects on sinus node function also affect the risk of mortality. The associations with higher heart rate do not translate into significantly higher risk of CAD or myocardial infarction, either individually or in combination, which may reflect low statistical power given the effect sizes for heart rate and the number of available CAD and myocardial infarction cases.
Heart rate–increasing alleles of the loci near GJA1, SLC35F1 (PLN) and NKX2-5 show unidirectional associations with reduced QRS and QT duration but bidirectional associations with atrial fibrillation. These findings suggest that both tails of the ventricular depolarization and myocardial repolarization distributions are associated with increased risk of atrial fibrillation and that altered heart rate associated with genetic predisposition in these loci may reflect adaptations to disturbed electrophysiological properties (compartments) of the heart. Results from experiments in a D. melanogaster model highlight genes in additional loci that show suggestive evidence of a role in both heart rate regulation and arrhythmia susceptibility (ACHE, PCOLCE, FADS3 and MFN1). Taken together, these results may enable the discovery of new druggable targets for the prevention and treatment of cardiovascular endpoints by selective reduction of heart rate and arrhythmia susceptibility, similar to the way ivabradine likely exerts its effects through targeting of HCN4 (refs. 7,25).
Results from experiments in D. melanogaster and D. rerio models support a role in heart rate regulation for genes that are essential for embryonic cardiovascular development (EPHB4, PLXNA2, PLD1 and CALCRL), as well as for genes with a role in the pathophysiology of dilated cardiomyopathy, congestive heart failure and/or sudden cardiac death (TTN, MFN1, CHRM2 and PLD1). In congruence, we show that zebrafish embryos with downregulated expression of orthologs of these genes have edema (MFN1), an unlooped heart (PLD1 and CALCRL) and atrioventricular canal malformation (CALCRL). Such defects in cardiovascular development can be hypothesized to mediate the reduced heart rate that we observe in these embryos. Future studies are required to determine whether individuals with genetic susceptibility for reduced heart rate in these loci are enriched for mild forms of such cardiovascular phenotypes.
In conclusion, our results provide new insights into the mechanisms that regulate or modulate heart rate in health and disease and provide a new perspective on the well-recognized association of heart rate with cardiovascular disease and mortality.
ONLINE METHODS
Stage 1 genome-wide association meta-analysis
The discovery sample encompassed 36 studies with data on heart rate in up to 85,787 individuals of European ancestry. Heart rate was derived from ECG in 12 studies (32% of the total sample) and peripheral pulse rate in 22 studies (49%) and was self-reported in 2 studies with data from health professionals (19%) (Supplementary Table 1). All studies included have been approved by local ethics committees, and all participants have provided their consent in writing.
Samples were genotyped using Affymetrix and Illumina genome-wide genotyping arrays (Supplementary Tables 2 and 3) and were imputed for polymorphic HapMap Phase 2 European CEU SNPs using MACH26, IMPUTE27, BIMBAM28 or Beagle29 (Supplementary Table 2).
Each study performed SNP association analyses with heart rate using an additive genetic model implemented in MACH2QTL30, Merlin31, SNPTEST27, ProbAbel32, GenABEL33, LME in R, MMAP, Matlab or PLINK34 (Supplementary Table 2). Associations were adjusted for age, age2, body mass index (BMI) and study-specific covariates when appropriate (for example, principal components). Analyses were stratified by sex and case status for samples ascertained for diseases or conditions. To allow for relatedness in the deCODE, HAPI Heart, Heritage, Korcula, NBS, NSPHS and SPLIT studies, regression coefficients were estimated in the context of a variance component model that took into account relatedness in men and women, with sex as an additional covariate. Before meta-analysis of the results from the 36 GWAS, we excluded SNPs with poor imputation quality score (r2 hat ≤ 0.3 in MACH, proper_info ≤ 0.4 in IMPUTE and BIMBAM, info ≤ 0.8 in Beagle), low minor allele count (n × MAF ≤ 3) and/or extreme effect size (β > ±50 bpm per effect allele, that is, ~5 times the standard deviation of heart rate as typically observed in the contributing studies) in each sex- and case-specific stratum. Individual GWAS were corrected by genomic control before meta-analysis when appropriate (λGC > 1.000) to adjust for population stratification.
We performed the stage 1 fixed-effects meta-analysis using the inverse variance method in METAL35. Before SNPs were selected for follow-up, a final genomic control correction of the meta-analysis results was performed (λGC= 1.106), giving conservative association estimates.
Lead SNPs at 42 independent loci were selected for follow-up in stage 2 (P < 3 × 10−5) (Supplementary Table 5). Loci were considered to be independent if pairwise r2 for LD was less than 0.2 and if they were separated by at least 1 Mb. We subsequently performed conditional analyses using summary statistics of stage 1 results14 to examine whether any of the 42 loci contained secondary associations with heart rate that remained significant after adjusting for the association of the lead SNP (P < 5 × 10−8). Before embarking on the follow-up analysis using all available data, we made sure that the inclusion of subgroups of the population did not affect the results (Supplementary Table 4).
Stage 2 follow-up
We tested for association of the 42 lead SNPs and 3 secondary associations in data from up to 88,823 individuals of European descent from 37 in silico replication studies with heart rate in stage 2. Heart rate was derived from ECG in 22 studies (57% of the total sample) and from peripheral pulse rate in 15 studies (43%) (Supplementary Table 7). GWAS data were available for up to 60,396 individuals of European descent from 2 sources: a previously reported meta-analysis of 15 GWAS for RR interval (RRgen Consortium)12 and 12 GWAS that have not been described previously in this context (Supplementary Tables 7–9). Additional data were available for 24,334 individuals of European ancestry from 11 studies who were genotyped using the Metabochip36 and from 5,171 individuals of European descent from 1 study who were genotyped using the Cardiochip (Supplementary Tables 8 and 10).
Samples and SNPs that did not meet the quality control criteria described by each individual study and for stage 1 were excluded. Minimum genotyping quality control criteria were defined as Hardy-Weinberg equilibrium P > 1 × 10−6 and call rate > 95% in each of the follow-up studies.
We tested the association of the 42 lead SNPs and 3 secondary associations with heart rate in each stage 2 study separately as described for stage 1 studies. Missing SNPs in GWAS of stage 2 were replaced by one of up to three proxies selected a priori (r2 > 0.8) (if available). Prioritizing of proxies was based on (in order of importance) (i) availability on the Metabochip and/or Cardiochip with r2 > 0.8; (ii) r2 for LD; and (iii) proximity to the lead SNP. This resulted in the inclusion of three proxies in LifeLines2 and one proxy in ACTS, all of which had r2 > 0.9 with the lead SNP at the locus (Supplementary Table 10). None of the loci for which these proxies were included has association reaching P < 5 × 10−8 after meta-analysis of stages 1 and 2 together.
We performed meta-analysis on summary statistics from the stage 1 meta-analysis and stage 2 studies using the weighted z-score method in data from up to 181,171 individuals (Supplementary Table 5). Genomic control–adjusted P values were used throughout stages 1 and 2 for GWAS. For studies with data from Metabochip and Cardiochip, little evidence for population stratification was previously observed for associations with other cardiovascular and metabolic traits, and, hence, no correction of P values was applied.
For loci with secondary associations, the SNP with the lowest P value for association with heart rate after combined meta-analysis of stage 1 and 2 results was considered the most representative for the locus. For loci with confirmed associations after meta-analysis of stage 1 and 2 results, an estimate of the effect size was obtained by fixed-effects meta-analysis of summary statistics from the stage 1 meta-analysis and stage 2 studies with heart rate, using the inverse variance method.
Additional analyses and functional follow-up experiments
Cumulative effects of confirmed loci and interindividual variation in heart rate
To estimate the cumulative effect of the 21 heart rate–associated loci, we calculated the GPS in 5,053 adults from the LifeLines2 study and 4,000 12-year-old children from the ALSPAC study by summing the number of heart rate–increasing alleles carried by an individual at the lead SNP of each heart rate locus. The number of heart rate–increasing alleles ranged from 9 to 26 for the 19 available loci in LifeLines2 (data from rs6127471 and rs2340782 were not available) and from 10 to 29 for the 21 loci in ALSPAC.
We compared the explained variance (r2) between covariate-adjusted models with and without the GPS to assess the variance in heart rate that can be explained by these loci in the LifeLines2 and ALSPAC studies.
Association analyses with related traits
Associations between the 21 heart rate loci and related cardiovascular intermediates and endpoints were extracted from GWAS data of the CHARGE Consortium (PR duration)16, the QRS GWAS Consortium (QRS duration)17 and the QT-IGC Consortium (QT duration; QT-IGC Consortium (C.N.-C.), personal communication) (Table 2), as well as from deCODE Genetics (prevalent advanced (second-or third-degree) atrioventricular block, SSS, pacemaker implantation and sudden cardiac death)13,18 (Table 2 and Supplementary Table 12), deCODE Genetics13 and the CHARGE-AF Consortium25 (atrial fibrillation) (Table 2), the Global BPgen Consortium (systolic and diastolic blood pressure, as well as prevalent hypertension)21 (Supplementary Table 13) and the CARDIoGRAM Consortium (prevalent CAD and myocardial infarction)22 (Supplementary Table 14). All associations were adjusted for covariates as described previously13,16–18,21,22,25,37.
We calculated multi-SNP predisposition scores for each trait to examine the association of the 21 heart rate loci combined with each of the before-mentioned traits, on the basis of single-SNP summary statistics and weighting by effect sizes for association with heart rate after meta-analysis of stages 1 and 2 together.
Associations of the 21 heart rate loci, both individually and in combination in a multi-SNP predisposition score, with related traits were considered statistically significant at P < 0.002, that is, α = 0.05 with Bonferroni correction for 21 independent tests.
Enrichment analysis of heart rate associations in biological pathways
We used MAGENTA24 to test whether predefined biological processes or molecular functions were enriched for multiple modest heart rate associations, aiming to discover new pathways associated with heart rate and to test whether the 21 heart rate loci cluster near genes that constitute specific biological connections (Supplementary Tables 18 and 19). First, we calculated a corrected gene association P value for each gene in the genome and grouped genes into pathways using annotations from the Kyoto Encyclopedia of Genes and Genomes (KEGG), BIOCARTA, Protein Analysis THrough Evolutionary Relationships (PANTHER)38, Biological Processes (PANTHER, BP) and Molecular Functions (PANTHER, MF), REACTOME, Gene Ontology (GO) and Ingenuity databases. Finally, for each pathway, we evaluated potential enrichment of highly ranked gene scores by comparing the fraction of genes within each gene set whose corrected P value was more significant than the 95th percentile of all gene P values to that of 10,000 randomly sampled gene sets of identical size from the genome24. In significantly enriched gene sets, in addition to genes in validated association regions, the top ranked genes above the enrichment cutoff may suggest new modest associations for follow-up (Supplementary Table 19).
Proteomics experiments in mouse heart and genetic enrichment analysis
We used results from proteomics experiments to identify genes located within the heart rate loci that are expressed at the protein level in mouse heart and that are phosphorylated upon stimulation of β1AR (A.L., M.N. Andersen, A.B. Steffensen, H. Horn, C.D. Kelstrup et al., unpublished data). Briefly, male C57BL/6 mice were either treated with β1AR- and β2AR-specific antagonists (control group, n = 3) or with a β2AR-specific antagonist followed by β1AR-specific agonist (test group, n = 3). Cardiac proteins were extracted and digested, enriched for phosphopeptides by TiO2 chromatography and analyzed by nanoflow liquid chromatography tandem mass spectrometry, as described previously39.
A total of 8,518 phosphorylation sites that could be mapped to a specific residue were identified. Mice in the test and control groups were compared using a two-sided t test with permutation-based FDR < 0.01. The number of genes encoding proteins identified in the experiments was 4,096. Forty-one of these were located within 500 kb of confirmed heart rate associations, four of which were regulated by β1AR stimulation (MYH6, PLN, TTN and NCL).
eQTL analyses
We examined associations between each of the heart rate loci and expression of genes in cis in 1,469 whole-blood samples (PAXgene), reflecting primary leukocyte gene expression40 (Supplementary Table 21). Transcriptional components were applied to reduce a substantial proportion of interindividual non-genetic expression variation. An eQTL meta-analysis was subsequently performed on the residual expression variation. We used FDR < 0.05 to correct for multiple testing. We removed 50 principal components by linear regression to remove non-genetic variation in gene expression. In addition, we performed conditional analyses to examine to what extent each heart rate–associated SNP explains the association between the gene transcript and the SNP most significantly associated with the gene transcript.
Significant cis associations were observed between five heart rate–associated SNPs and the levels of nine nearby transcripts in blood (Table 1 and Supplementary Table 21). The heart rate–associated SNPs explained a substantial proportion of the association with the most significant SNP for the gene transcript in conditional analyses (adjusted P > 0.05) for TRIP6, TMEM258(C11orf10), FADS1, BEST1-FTH1, FADS2, CEP85L(C6orf204) and NCL-SNORD20.
Potentially functional variants within the 21 loci
To identify SNPs in the confirmed loci that may be causal for the association with heart rate, we explored whether the heart rate–associated SNPs were in strong LD (r2 > 0.8) with variants in transcription factor binding sites, nonsynonymous SNPs or copy number variants (deletion variants and mobile element insertion polymorphisms) identified in the 1000 Genomes Project CEU Pilot 1 or HapMap CEU reference panels41. For nonsynonymous SNPs, PANTHER38 was used to assess whether the variant was likely to have a detrimental effect on protein function, based on alignment of evolutionarily related proteins (Supplementary Table 20).
One association tagged a variant in a transcription factor binding site near CEP85L(C6orf204; near SLC35F1). Nonsynonymous variants in strong LD with heart rate–associated SNPs were present in six genes (Supplementary Table 22), with the p.Arg1045Trp alteration encoded in KIAA1755 likely having a deleterious effect on protein function38. Of interest, the rs180242 allele that was associated with lower heart rate tagged a common 723-bp deletion variant located 8 kb upstream of GNG11 (ref. 42) (1000 Genomes Pilot ID P2_M_061510_7_474; r2 = 0.96) and was additionally associated with lower expression of GNG11 in blood (Supplementary Table 21).
Candidate genes based on the literature
To identify additional candidate genes in the heart rate loci, we identified all genes within 500 kb of the 21 heart rate–associated SNPs and performed an automated literature search using the search term ‘heart’ in the program SNIPPER (Supplementary Table 23). We identified many genes with established connections to embryonic cardiac development, cardiac conduction, cardiac contractile proteins, calcium regulation, angiogenesis and endothelial function (Supplementary Note). Many of the loci harbored genes in which mutations lead to dilated and hypertrophic cardiomyopathy (in MYH6 and MYH7, PLN (near SLC35F1), TTN (near CCDC141), MFN1 (near GNB4) and CHRM2).
Experimental follow-up of positional candidate genes in D. melanogaster and D. rerio
We used D. melanogaster and the zebrafish D. rerio as models to examine whether candidate genes within the heart rate loci were likely to underlie the associations identified by GWAS. Forty-nine positional candidate genes were identified on the basis of results from proteomics experiments and genetic enrichment analysis in mouse heart and eQTL analyses, as well as by the presence of functional variants and results from the automated literature search. In addition, we searched the genes located within 500 kb of associations for biological candidates (Supplementary Table 24).
Experiments in D. melanogaster
BLAST searches were performed to identify obvious D. melanogaster orthologs of positional candidate genes (Supplementary Table 25). We subsequently used RNAi43,44 to downregulate orthologs of candidate genes (Vienna Drosophila RNAi Center). Expression of RNAi was induced by crossing with a D. melanogaster line expressing GAL4 driven by an actin promoter (stock 4414, Bloomington Drosophila Stock Center) (Supplementary Table 25).
D. melanogaster stocks were kept at 25°C on standard medium. Pre-pupae were selected for tachypacing, an established D. melanogaster model for atrial fibrillation, as previously described45. We recorded videos through a microscope at 10× magnification before and after tachypacing to visualize heart contractions in triplicate periods of 10 s. Heart rate was subsequently quantified using ImageJ software. An arrhythmia index was calculated as the ratio of arrhythmic periods and total measurement duration using the same software.
The number of positional candidate genes for which we analyzed results was reduced from 49 to 25 owing to the absence of orthologs (n = 13 genes), lack of RNAi lines (n = 3), non-viability of offspring (n = 1) and reduced viability, defined as the generation of fewer than 5 live offspring (n = 7) (Supplementary Fig. 6 and Supplementary Table 25). For the 25 remaining genes, represented by 23 orthologs, we compared heart rate and risk of arrhythmia in 11 ± 5 (mean ± s.d.) pupae with downregulated gene expression and 30 6000V controls. Ten additional pupae were available with downregulation of stwl (stonewall), a gene that is not anticipated to have a role in heart rate regulation and which can thus be interpreted as an extra control group. We targeted 13 orthologs using multiple independent RNAi lines.
We compared differences in heart rate between the offspring of RNAi-treated D. melanogaster and 6000V controls using a multilevel approach, adjusting for the dependence of repeated measures within pupae as a random effect. We compared RNAi lines with 6000V controls at baseline and after tachypacing. Differences in heart rate after tachypacing were examined with and without adjusting for average heart rate at baseline as a fixed effect. Results of analyses from multiple independent RNAi lines targeting the same ortholog were combined using the fixed-effects meta-analysis with inverse variance method (Supplementary Table 26).
We examined the risk of arrhythmia by comparing the number of arrhythmic cases and controls between RNAi-targeted orthologs and 6000V controls, both before and after tachypacing, using Fisher’s exact test. Differences were considered statistically significant at P < 0.002, that is, α = 0.05 with Bonferroni correction for 23 independent tests.
Experiments in D. rerio
In the zebrafish D. rerio, we excluded genes in eight loci that mapped in or near positional candidate genes with extensive a priori evidence of a role in cardiovascular processes (Supplementary Table 27). A maximum of two positional candidate genes per locus were selected in the remaining loci, which together with a lack of zebrafish orthologs in 12 genes resulted in the selection of 12 positional candidate genes for follow-up in zebrafish experiments (Supplementary Fig. 6 and Supplementary Tables 25 and 27).
Wild-type D. rerio stocks from Ekwill Fish Farm were maintained using standard procedures. Morpholino oligonucleotides (GeneTools) were designed against orthologs of the 12 positional candidate gene primary transcripts targeting the first exon-intron boundaries, except for tfpia (the ortholog of TFPI), which was designed to target the intron 1–exon 2 boundary (Supplementary Table 27). Embryos were injected at the single-cell stage and were scored and analyzed 48 h later. Downregulation of candidate genes was confirmed using quantitative PCR (Supplementary Table 29).
Heart rate analysis was performed as previously described46. For each ortholog, we performed the procedure twice on different days, comparing heart rate in embryos from the same embryonic aliquot that were injected with either morpholino oligonucleotides or PBS. Heart rate was measured in 26 ± 4 embryos injected with morpholino oligonucleotides and in 27 ± 5 embryos injected with PBS.
Measures of heart rate alone do not provide information on cardiac contractility. We therefore additionally measured ventricular fractional shortening in a subsample of embryos (6 ± 1 embryos injected with morpholino oligonucleotides and 6 ± 2 embryos injected with PBS) as previously described47.
Differences in heart rate and fractional shortening of the ventricular chamber were examined using linear regression and were adjusted for variation in the timing of the heart rate measurement.
For each group of embryos injected with morpholino oligonucleotides (42 ± 18 embryos), we assessed whether downregulation of positional candidate gene expression resulted in visible phenotypes that distinguished treated embryos from those injected with PBS.
Supplementary Material
Acknowledgments
A full list of acknowledgments appears in the Supplementary Note. Funding sources had no involvement in the collection, analysis and interpretation of the data.
AUTHOR CONTRIBUTIONS
Steering committee (oversaw the project): M. den Hoed (lead) and R.J.F.L. (chair). Writing group (drafted the manuscript): M.E., M. den Hoed (chair), R.J.F.L. and N.J.S. Editing group (edited the manuscript): B.J.J.M.B., P.T.E., T.E., D.M.E., E.J.C.d.G., M. den Hoed (chair), E.I., D.J.M., R.J.F.L., A.L., D.J.M., I.M.N., A.V. Segrè, O.C.M.S., H.S., J.R.T. and N.J.T. Meta-analysis working group (performed stage 1 and stage 2 meta-analyses): T.E. and M. den Hoed (chair). Data preparation working group (prepared data from contributing cohorts for meta-analyses): M.E., T.E., M. den Hoed (lead) and R.J.F.L. (chair). Conditional analyses: M. den Hoed, R.J.F.L., P.M.V. (chair) and J.Y. (lead). Genetic predisposition score analyses: D.M.E., M. den Hoed (chair) and I.M.N. Association analyses with related traits: C.M.A., P.I.W.d.B., CARDIoGRAM Consortium, CHARGE-AF Consortium, Y.S.C., M.C., D.D., P.T.E., J. Erdmann, Global BPgen Consortium, M.J.G., M. den Hoed (chair), H.H., A.I., T.J., S. Kääb, Y.J.K., K.L.L., P.B.M., C.N.-C., A. Pfeufer, PR GWAS Consortium, QRS GWAS Consortium, QT-IGC Consortium, N.J.S., S. Sharp, N. Sotoodehnia and J.R.T. Copy number variant analyses: R.E.H. (lead), M. den Hoed (chair), S.A.M. and C. Stewart. Gene eQTL analyses: L. Franke (chair), M. den Hoed and H.-J.W. (lead). Proteomics experiments and genetic enrichment analyses: M. den Hoed, R.J.F.L., A.L. (lead), E.J.R. and J.V.O. (chair). SNIPPER analyses for selection of positional candidate genes: M. den Hoed (chair), R.J.F.L. and C. Willer (lead). Pathway analyses: M. den Hoed (chair), R.J.F.L. and A.V. Segrè (lead). D. melanogaster experiments: B.J.J.M.B. (lead), M. den Hoed, F.H.-B., B.K., R.J.F.L., O.C.M.S. (chair) and H.S. D. rerio experiments: M. den Hoed, R.J.F.L., S.N.L., D.J.M. (chair), D.S.P. (lead) and J.T.S.
Project design, management and coordination of contributing cohorts
Stage 1–GWAS: (ADVANCE) T.L.A., C.I. and T.Q.; (ALSPAC) G.D.S.; (ASCOT cases) N.R.P., P.S.S., D.C.S. and A.V. Stanton; (ATBC) D. Albanes and J. Virtamo; (B58C) W.L.M.C. and D.P.S.; (BLSA) S. Bandinelli and L. Ferrucci; (BRIGHT) M.C., T.J., P.B.M. and N.J.S.; (CoLaus) J.S.B., P.V. and G. Waeber; (COROGENE) M.-L.L.L., M.S.N., M.P. and J.S.; (deCODE) D.O.A., K.S. and U.T.; (DGI) L.G. and B.I.; (EGCUT) A.M.; (EPIC-Norfolk) N.J.W.; (Fenland) U.E., N.G.F., R.J.F.L. and N.J.W.; (Fingesture) H.V.H., J.D.R. and J.-C.T.; (Finrisk07) M.P. and V.S.; (FUSION) M. Boehnke and J.T.; (GOOD) C.O.; (HAPI) B.D.M. and A.R.S.; (HBCS) J. Eriksson, M.P. and E.W.; (Health 2000) A.J. and M.P.; (Health ABC) W.-C.H.; (HERITAGE) C. Bouchard, T.R. and D.C.R.; (HPFS) G.C., F.B.H., D.J.H., P.K., L.Q. and E.B.R.; (Hypergenes) D.C., N.G., L.I. and F.R.; (InCHIANTI) S. Bandinelli and L. Ferrucci; (Korcula) I.R.; (LifeLines) R.A.d.B., M.M.v.d.K., H.S. and R.P.S.; (Lolipop) J.C.C. and J.S.K.; (NBS) J.d.G. and L.A.K.; (NFBC1966) M.-R.J.; (NHS) G.C., F.B.H., D.J.H., P.K., L.Q. and E.B.R.; (NSPHS) U.G.; (PREVEND) W.H.v.G., G.N. and D.J.v.V.; (SPLIT) I.R.; and (YFS) M. Kähönen, T.L., M.P., O.T.R. and J. Viikari. Stage 2– in silico replication studies: (AGES, RRgen) V.G. and T.B.H.; (ACTS) N.G.M.; (ALSPAC) G.D.S.; (ARIC, RRgen) A.A.; (CHS, RRgen) B.M.P.; (DESIR) N.B.-N.; (EGCUT) A.M.; (Ely) N.J.W.; (EPIC-NL) J.M.A.B., Y.T.v.d.S. and W.M.M.V.; (EPIC-Norfolk) N.J.W.; (ERF) C.M.v.D. and B.A.O.; (FamHS) I.B.B.; (Fenland) U.E., N.G.F., R.J.F.L. and N.J.W.; (FHS, RRgen) C.J.O.; (Finrisk07) M.P. and V.S.; (KORA, RRgen) A. Peters and S. Kääb; (LifeLines2) R.A.d.B., M.M.v.d.K., H.S. and R.P.S.; (MESA) R.A.K. and J.I.R.; (MICROS, RRgen) P.P.P.; (NSHD) D.K.; (NTR) D.I.B. and E.J.C.d.G.; (ORCADES, RRgen) J.F.W.; (RISC) M.W.; (PIVUS) E.I. and L.L.; (RS1-3) A. Hofman, B.H.Ch.S. and J.C.M.W.; (SardiNIA, RRgen) E.G.L. and K.V.T.; (SHIP, RRgen) M.D. and S.B.F.; (Stanford IST) T.Q.; (STR) E.I. and N.L.P.; (Twins UK, RRgen) Y.J. and T.D.S.; (ULSAM) E.I.; and (Whitehall II) A. Hingorani and M. Kivimaki.
Genotyping of contributing cohorts
Stage 1–GWAS: (ADVANCE) D. Absher; (ALSPAC) S.M.R. and W.L.M.; (ATBC) S.J.C.; (BLSA) L. Ferrucci and A.B.S.; (BRIGHT) M.C. and P.B.M.; (COROGENE) P.S.; (EGCUT) T.E., L.M. and M.N.; (EPIC-Norfolk) R.J.F.L. and J.H.Z.; (Fenland) J.L.; (Fingesture) P.G. and J.D.R.; (Finrisk07) P.S.; (FUSION) P.S.C.; (GOOD) M. Lorentzon and C.O.; (HBCS) P.S.; (Health2000) P.L. and P.S.; (Health ABC) Y.L.; (HERITAGE) C. Bouchard and T.R.; (HPFS) M.C.C. and M.K.J.; (Hypergenes) C. Barlassina and P.B.; (InCHIANTI) L. Ferrucci and A.B.S.; (Korcula) C.H.; (LifeLines) L. Franke; (Lolipop) J.C.C. and J.S.K.; (NBS) L.A.K.; (NFBC1966) P.E., A.-L.H., M.-R.J. and P.Z.; (NHS) M.C.C. and M.K.J.; (NSPHS) Å.J.; (PREVEND) P.v.d.H.; (SPLIT) C.H. and V.V.; and (YFS) M. Kähönen, T.L., M.P., O.T.R., P.S. and J. Viikari. Stage 2– in silico replication studies: (ACTS) N.G.M., S.E.M. and G.W.M.; (ALSPAC) S.M.R. and W.L.M.; (ARIC, RRgen) D.E.A.; (DESIR) N.B.-N.; (EGCUT) T.E., L.M. and M.N.; (EPIC-NL) N.C.O.-M. and C. Wijmenga; (ERF) C.M.v.D., A.I. and B.A.O.; (Ely) R.J.F.L. and J.L.; (EPIC-Norfolk) R.J.F.L. and J.H.Z.; (FamHS) I.B.B. and M.F.F.; (Fenland) J.L.; (Finrisk07) P.S.; (LifeLines2) L. Franke; (MESA) J.I.R.; (NSHD) D.K., K.K.O. and A.W.; (NTR) D.I.B. and J.-J.H.; (PIVUS) E.I. and L.L.; (RS1-3) A.G.U.; (Stanford IST) T.L.A. and J.W.K.; (STR) E.I. and N.L.P.; (ULSAM) E.I.; and (Whitehall II) M. Kumari and C. Langenberg.
Phenotyping of contributing cohorts
Stage 1–GWAS: (ADVANCE) C.I.; (ASCOT cases) N.R.P., P.S.S. and A.V. Stanton; (ATBC) D. Albanes and J. Virtamo; (B58C) D.P.S.; (BLSA) S. Bandinelli and L. Ferrucci; (BRIGHT) M.C. and N.J.S.; (CoLaus) P.M.-V.; (COROGENE) M.P.; (deCODE) D.O.A. and H.H.; (DGI) B.I.; (EGCUT) K.F. and A.M.; (EPIC-Norfolk) K.-T.K.; (Fingesture) H.V.H. and J.J.; (Finrisk07) M.P.; (FUSION) H.M.S.; (GOOD) M. Lorentzon, C.O. and L.V.; (HBCS) J. Eriksson, M.P. and E.W.; (Health2000) A.J. and M.P.; (Health ABC) A.B.N.; (HERITAGE) C. Bouchard; (Hypergenes) D.C., N.G., L.I. and F.R.; (InCHIANTI) S. Bandinelli and L. Ferrucci; (Korcula) O.P.; (LifeLines) R.A.d.B., M.M.v.d.K. and R.P.S.; (Lolipop) J.C.C., A.S.K., J.S.K., K.A.M. and J.S.S.; (NBS) S.H.; (NFBC1966) A.-L.H., M.-R.J., A. Pouta and P.Z.; (PREVEND) R.A.d.B., W.H.v.G. and P.v.d.H.; (SPLIT) D.R.; and (YFS) M. Kähönen, T.L., M.P., O.T.R., P.S. and J. Viikari. Stage 2– in silico replication studies: (AGES, RRgen) V.G.; (ACTS) N.G.M. and J.B.W.; (CHS, RRgen) N. Sotoodehnia; (DESIR) B.B. and P.F.; (EGCUT) K.F. and A.M.; (Ely) S. Brage and U.E.; (EPIC-NL) J.M.A.B., Y.T.v.d.S. and W.M.M.V.; (EPIC-Norfolk) K.-T.K.; (ERF) C.M.v.D., A.I., J.A.K. and B.A.O.; (FamHS) I.B.B. and M.F.F.; (FHA, RRgen) C.N.-C.; (Finrisk07) M.P.; (LifeLines2) R.A.d.B., M.M.v.d.K. and R.P.S.; (MESA) S.R.H. and R.A.K.; (MICROS, RRgen) A.A.H.; (NSHD) D.K.; (NTR) D.I.B., E.J.C.d.G. and G. Willemsen; (ORCADES, RRgen) S.H.W.; (PIVUS) E.I. and L.L.; (RISC) M.W.; (RS1-3) B.H.Ch.S. and A.G.U.; (SHIP, RRgen) M.D. and M.R.P.M.; (Stanford IST) T.L.A. and J.W.K.; (STR) E.I. and N.L.P.; (ULSAM) E.I.; and (Whitehall II) M. Kumari.
Analyses of contributing cohorts
Stage 1–GWAS: (ADVANCE) T.L.A. and L.W.; (ALSPAC) D.M.E., J.P.K., B.S.P. and N.J.T.; (ASCOT cases) T.J.; (ATBC) W.W.; (B58C) D.H. and D.P.S.; (BLSA) T.T.; (BRIGHT) T.J. and S.P.; (CoLaus) M. Bochud and Z.K.; (COROGENE) P.S.; (deCODE) D.G. and H.H.; (DGI) P.A., C. Ladenvall and R.A.S.; (EGCUT) T.E. and E.M.; (EPIC-Norfolk) M. den Hoed, R.N.L. and J.H.Z.; (Fenland) M. den Hoed and J.L.; (Fingesture) G.B. and P.G.; (Finrisk07) A.S.H., K.K. and P.S.; (FUSION) A.U.J.; (GOOD) M. Lorentzon, C.O. and L.V.; (HAPI) M.E.M. and J.R.O.; (HBCS,) P.S.; (Health2000) P.S.; (Health ABC) W.-C.H. and O.T.N.; (HERITAGE) C. Bouchard, T.R. and D.C.R.; (HPFS) M.C.C. (InCHIANTI) T.T.; (Korcula) C.H.; (LifeLines) I.M.N. and H.S.; (Lolipop) J.C.C., J.S.K., J.S.S. and W.Z.; (NBS) M. den Heijer; (NFBC1966) P.F.O.; (NHS) M.C.C.; (Hypergenes) D.C.; (NSPHS) W.I.; (PREVEND) P.v.d.H. and I.M.L.; (SPLIT) C.H. and V.V.; and (YFS) P.S. Stage 2– in silico replication studies: (AGES, RRgen) A.V. Smith; (ACTS) P.A.L.; (ALSPAC) D.M.E., J.P.K., B.S.P. and N.J.T.; (ARIC, RRgen) A.C.M.; (CHS, RRgen) J.C.B. and N. Sotoodehnia; (DESIR) C.D., N.B.-N. and L.Y.; (EGCUT) T.E. and E.M.; (Ely) M. den Hoed and J.L.; (EPIC-NL) M. Leusink and N.C.O.-M.; (EPIC-Norfolk) M. den Hoed, R.N.L. and J.H.Z.; (ERF) A.I.; (FamHS) M.F.F. and S. Ketkar; (Fenland) M. den Hoed and J.L.; (FHS, RRgen) C.N.-C. and S.-J.H.; (Finrisk07) A.S.H. and K.K., P.S.; (KORA, RRgen) M.M.-N.; (LifeLines2) I.M.N. and H.S.; (MESA) K.F.K. and Q.W.; (MICROS, RRgen) C.F.; (NSHD) M. den Hoed, J.L. and A.W.; (NTR) H.H.M.D. and J.-J.H.; (ORCADES, RRgen) P.N.; (PIVUS) E.I. and C. Song; (RISC) M.N.W. and W.X.; (RRgen) P.I.W.d.B.; (RS1-3) P.I.W.d.B. and M.E.; (SardiNIA, RRgen) S. Sanna; (Stanford IST) W.X.; (STR) E.I. and C. Song; (Twins UK, RRgen) N. Soranzo; (ULSAM) E.I. and C. Song; and (Whitehall II) M. den Hoed and J.L.
The corresponding author (R.J.F.L.) had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
URLs. PANTHER, http://www.pantherdb.org/; Gene Ontology (GO), http://www.geneontology.org/; Molecular Signatures Database (MsigDB), http://www.broad.mit.edu/gsea/msigdb/collections.jsp; Mascot Search algorithm, http://www.matrixscience.com; Ensembl, http://www.ensembl.org/index.html; SNIPPER, http://csg.sph.umich.edu/boehnke/snipper/.
Note: Supplementary information is available in the online version of the paper.
References
- 1.Dyer AR, et al. Heart rate as a prognostic factor for coronary heart disease and mortality: findings in three Chicago epidemiologic studies. Am J Epidemiol. 1980;112:736–749. doi: 10.1093/oxfordjournals.aje.a113046. [DOI] [PubMed] [Google Scholar]
- 2.Gillum RF, Makuc DM, Feldman JJ. Pulse rate, coronary heart disease, and death: the NHANES I Epidemiologic Follow-up Study. Am Heart J. 1991;121:172–177. doi: 10.1016/0002-8703(91)90970-s. [DOI] [PubMed] [Google Scholar]
- 3.Nauman J, Janszky I, Vatten LJ, Wisloff U. Temporal changes in resting heart rate and deaths from ischemic heart disease. J Am Med Assoc. 2011;306:2579–2587. doi: 10.1001/jama.2011.1826. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Billman GE, Stone HL. Autonomic mechanisms in ventricular fibrillation induced by myocardial ischemia during exercise in dogs with healed myocardial infarction. An experimental preparation for sudden cardiac death. Circulation. 1984;69:790–800. doi: 10.1161/01.cir.69.4.790. [DOI] [PubMed] [Google Scholar]
- 5.Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226:180–182. doi: 10.1126/science.6484569. [DOI] [PubMed] [Google Scholar]
- 6.Beere PA, Glagov S, Zarins CK. Experimental atherosclerosis at the carotid bifurcation of the cynomolgus monkey. Localization, compensatory enlargement, and the sparing effect of lowered heart rate. Arterioscler Thromb. 1992;12:1245–1253. doi: 10.1161/01.atv.12.11.1245. [DOI] [PubMed] [Google Scholar]
- 7.Böhm M, et al. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886–894. doi: 10.1016/S0140-6736(10)61259-7. [DOI] [PubMed] [Google Scholar]
- 8.Dalageorgou C, et al. Heritability of QT interval: how much is explained by genes for resting heart rate? J Cardiovasc Electrophysiol. 2008;19:386–391. doi: 10.1111/j.1540-8167.2007.01030.x. [DOI] [PubMed] [Google Scholar]
- 9.De Geus EJ, Kupper N, Boomsma DI, Snieder H. Bivariate genetic modeling of cardiovascular stress reactivity: does stress uncover genetic variance? Psychosom Med. 2007;69:356–364. doi: 10.1097/PSY.0b013e318049cc2d. [DOI] [PubMed] [Google Scholar]
- 10.Russell MW, Law I, Sholinsky P, Fabsitz RR. Heritability of ECG measurements in adult male twins. J Electrocardiol. 1998;30(suppl):64–68. doi: 10.1016/s0022-0736(98)80034-4. [DOI] [PubMed] [Google Scholar]
- 11.Cho YS, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 12.Eijgelsheim M, et al. Genome-wide association analysis identifies multiple loci related to resting heart rate. Hum Mol Genet. 2010;19:3885–3894. doi: 10.1093/hmg/ddq303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm H, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 14.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue T, et al. Higher heart rate predicts the risk of developing hypertension in a normotensive screened cohort. Circ J. 2007;71:1755–1760. doi: 10.1253/circj.71.1755. [DOI] [PubMed] [Google Scholar]
- 16.Pfeufer A, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sotoodehnia N, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm H, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43:316–320. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrison RJ, Kannel WB, Stokes J, III, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med. 1987;16:235–251. doi: 10.1016/0091-7435(87)90087-9. [DOI] [PubMed] [Google Scholar]
- 20.Levy RL, et al. Transient tachycardia; prognostic significance alone and in association with transient hypertension. Med Press Egypt. 1946;38:207–212. [PubMed] [Google Scholar]
- 21.Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusumoto FM, Goldschlager N. Cardiac pacing. N Engl J Med. 1996;334:89–97. doi: 10.1056/NEJM199601113340206. [DOI] [PubMed] [Google Scholar]
- 24.Segrè AV, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6:pii: e1001058. doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinor PT, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 28.Guan Y, Stephens M. Practical issues in imputation-based association mapping. PLoS Genet. 2008;4:e1000279. doi: 10.1371/journal.pgen.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellinghaus D, Schreiber S, Franke A, Nothnagel M. Current software for genotype imputation. Hum Genomics. 2009;3:371–380. doi: 10.1186/1479-7364-3-4-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pei YF, Zhang L, Li J, Deng HW. Analyses and comparison of imputation-based association methods. PLoS ONE. 2010;5:e10827. doi: 10.1371/journal.pone.0010827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genomewide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voight BF, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newton-Cheh C, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas PD, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lundby A, et al. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dubois PC, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clemens JC, et al. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci USA. 2000;97:6499–6503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dietzl G, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 45.Zhang D, et al. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for atrial fibrillation. J Mol Cell Cardiol. 2011;51:381–389. doi: 10.1016/j.yjmcc.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 46.Burns CG, et al. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1:263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- 47.Shin JT, Pomerantsev EV, Mably JD, MacRae CA. High-resolution cardiovascular function confirms functional orthology of myocardial contractility pathways in zebrafish. Physiol Genomics. 2010;42:300–309. doi: 10.1152/physiolgenomics.00206.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.