

Abstract
Posttranslational modifications such as phosphorylation are universally acknowledged regulators of protein function. Recently we characterised a striated muscle-specific isoform of the formin FHOD3 that displays distinct subcellular targeting and protein half-life compared to its non-muscle counterpart and which is dependent on phosphorylation by CK2 (formerly casein kinase 2). We now show that the two isoforms of FHOD3 are already expressed in the vertebrate embryonic heart. Analysis of CK2 alpha knockout mice showed that phosphorylation by CK2 is also required for proper targeting of muscle FHOD3 to the myofibrils in embryonic cardiomyocytes in situ. The localisation of muscle FHOD3 in the sarcomere varies depending on the maturation state, being either broader or restricted to the Z-disc proper in the adult heart. Following myofibril disassembly, such as that in dedifferentiating adult rat cardiomyocytes in culture, the expression of non-muscle FHOD3 is up-regulated, which is reversed once the myofibrils are reassembled. The shift in expression levels of different isoforms is accompanied by an increased co-localisation with p62, which is involved in autophagy, and affects the half-life of FHOD3. Phosphorylation of three amino acids in the C-terminus of FHOD3 by ROCK1 is sufficient for activation, which results in increased actin filament synthesis in cardiomyocytes and also a broader localisation pattern of FHOD3 in the myofibrils. ROCK1 can directly phosphorylate FHOD3, and FHOD3 seems to be the downstream mediator of the exaggerated actin filament formation phenotype that is induced in cardiomyocytes upon the overexpression of constitutively active ROCK1. We conclude that the expression of the muscle FHOD3 isoform is characteristic of the healthy mature heart and that two distinct phosphorylation events are crucial to regulate the activity of this isoform in thin filament assembly and maintenance.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1154-7) contains supplementary material, which is available to authorized users.
Keywords: Myofibril, Formin, Cardiac cytoarchitecture, Heart development
Introduction
The pumping action of the heart is carried out by highly specialised, terminally differentiated cells, the cardiomyocytes. Throughout life the heart has to adapt to changes in demand by changing the size of the individual cardiomyocyte, which results in physiological hypertrophy (induced by developmental growth, exercise or pregnancy), pathological hypertrophy (e.g. induced by hypertension) or atrophy [1]. At the level of the cardiomyocyte, these changes lead to increased assembly or disassembly of the contractile structures, the myofibrils, as well as to changes in their isoform composition, especially of myosin and actin [2, 3]. However, in addition to physiological adaptation, there is also a continuous replacement of proteins due to maintenance, which has been estimated to result in a half-life of between 3 and 4 days for some myofibrillar proteins [4]. Very little is currently known about how the maintenance and the additional synthesis of actin filaments are achieved in the developing and mature heart [5, 6]. Several laboratories have recently taken inspiration from the well-characterised events that occur during actin filament formation in migrating cells [7] and shown, for example, a role for well-known players, such as N-WASP, during myofibrillogenesis in skeletal myocytes [8]. In an earlier study, we characterised a striated muscle-specific splice variant of FHOD3, a member of the formin family of proteins, that appears to be required for myofibril maintenance in cultured cardiomyocytes and shown it to be down-regulated in heart disease [9]. This isoform is characterised by the presence of eight additional amino acids at the C-terminal end of the FH2 (formin homology 2) domain, which results in a target site for phosphorylation by CK2 (formerly known as casein kinase 2). CK2 is a constitutively active, ubiquitously expressed serine/threonine kinase with a plethora of substrates [10, 11]. In addition to its well-established role in cancer [12] and embryonic development [13, 14] and its recently described role in proliferation [15, 16], CK2 is starting to be recognised also as a regulator of the cytoskeleton and ultimately cell morphology [17]. Phosphorylation by CK2 affects the subcellular targeting as well as the half-life of muscle FHOD3 by preventing its interaction with p62, a protein involved in autophagy, but it does not appear to affect the ability of FHOD3 to support actin filament synthesis [9].
FHOD3 is a member of the diaphanous-related formins (DRF), which in addition to the formin domains FH1 and FH2 contain an N-terminal GTPase binding domain (GBD), a diaphanous autoinhibitory domain (DID) and a C-terminal diaphanous autoregulation domain (DAD) (for reviews on formins see [18–21]). Members of this formin family exist in an autoinhibited state, brought about by the intramolecular interaction between DID and DAD, which has to be relieved by the binding of a small GTPase, such as Rac1 or Rho, to lead to an active protein that can dimerise and stimulate the synthesis of actin filaments [22]. The possibility of a more direct activation was recently suggested for FHOD3’s closest relative, FHOD1, where the phosphorylation of three amino acids in the DAD domain by ROCK1 was sufficient for release of the autoinhibited state [23, 24].
The primary aim of our study was to establish whether the muscle FHOD3 isoform is also expressed during heart development. We also wanted to determine whether its subcellular localisation is indeed regulated by CK2 and how the activity of FHOD3 is controlled in cardiomyocytes. We were able to show that there is a tight correlation between the fully differentiated state of a myofibril and high levels of muscle FHOD3 expression and that the presence of CK2 is needed to target FHOD3 to the myofibrils. Non-muscle FHOD3 is expressed at higher levels in the embryonic atrium and it is also up-regulated in dedifferentiating adult rat cardiomyocytes in culture, similar to the situation already described for the diseased heart [9]. In addition, we were able to demonstrate that the activation of FHOD3 is regulated in a similar way as that of FHOD1 and that its constitutive activation leads to excessive actin filament synthesis in cultured cardiomyocytes. We propose a link between the subsarcomeric localisation of FHOD3 and the extent of actin filament synthesis in cardiomyocytes, which is governed by two separate phosphorylation events.
Materials and methods
Preparation of transfection constructs
Cloning of FHOD3-GFP (green fluorescent protein) and FHOD3-HA (hemagglutinin), the DAD deletion constructs, FHOD3-CT-GFP and FHOD3-CT-GST (glutathione-S-transferase), as well as the CK2 consensus site [T(D/E)5XE exon] splice variants and mutants has been described previously [9]. FHOD3-3D-GFP and FHOD3-CT-GFP (ROCK3D) were created by standard site-directed mutagenesis of S1590, S1596 and T1600 to aspartic acid using 5′-GAAACGAGACCGGGCCAACCGGAAAGACTTGCGAAGAGACCTGAAG-3′. Since the mutation of S1590 failed, 5′-GCCGAGGGAGAGGAAACGAGACCGGGCCAACCGGAAAGA-3′ primers were used in a second step. FHOD3-NT-GST was created using 5′-GGAATTCCGCCACCATGGCCACGCTGGCTTGCC-3′ forward and 5′-CGGGATCCCGCCCGGCACCCACTGGG-3′ reverse primers and FHOD3-GFP as a template by cloning into the EcoRI and BamHI sites of a GST-CI vector. p160-ROCK1-myc and p130-ROCK1-myc were kindly provided by S. Narumiya (Department of Pharmacology, Kyoto University, Japan).
Knockdown by shRNA was achieved by using plasmids described previously that encode for the shRNA under the control of an H1 expression cassette together with GFP under the control of a cytomegalovirus (CMV) promoter [9]. The vector just containing the H1 expression cassette and GFP was used as a control.
In vitro phosphorylation assays
FHOD3-CT was subcloned into a pGEX-C1 vector using BglII and SacII restriction sites. The Rho kinase substrate motif was mutated using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technology, Santa Clara, CA) following the manufacturer’s instructions in three consecutive steps, with sense primers 5′-CGAGGGAGAGGAAACGAGCCCGGGCCA-3′, 5′-CGGGCCAACCGGAAAGCTTTGCGAAGAACCC-3′ and 5′-AACCGGAAAGCTTTGCGAAGAGCCCTGAAGAGC-3′ and the respective anti-sense primers. FHOD3-CT-GST and CT-3A-GST were expressed in Escherichia coli BL21 Star (Invitrogen, Carlsbad, CA). The peptides were purified using glutathion Sepharose (Amersham, Arlington Heights, IL). Equal amounts of CT-GST and CT-3A-GST were incubated with 20 mU recombinant active ROCK-I (Millipore, Billerica, MA) in 8 mM MOPS, 10 mM MgAc, 0.1 mM ATP and 1 mM EDTA at pH 7.0 for 5 min at 30 °C, then boiled in sodium dodecyl sulphate (SDS) buffer, and finally blotted with rabbit anti-phosphoserine/threonine (ECM Biosciences, Versailles, KY), mouse anti-GST (Cell Signaling Technology, Danvers, MA) and anti-His (Qiagen, Venlo, The Netherlands) antibodies.
Chick embryos
Fertilised hens eggs (mixed flock Henry Stewart, Peterborough, UK) were incubated in a humidified atmosphere at 37 °C. Embryos were staged according to Hamburger and Hamilton [25], and RNA was extracted either from whole embryos or from dissected atria and ventricles and used for reverse transcriptase (RT)-PCR assays. Alternatively, embryos were stored in 4 % paraformaldehyde (PFA) in phosphate buffered saline (PBS) at 4 °C until use for in situ hybridisation experiments.
Reverse transcriptase-PCR
RNA was extracted with Trizol (Invitrogen) following the manufacturer’s instructions. cDNA pools were prepared using M-MLV reverse transcriptase (Promega, Madison, WI).
RT-PCR assays for murine samples were performed as described previously [9]. For the amplification of the T(D/E)5XE exon containing and excluding, respectively, isoforms of Gallus gallus FHOD3, we used the 5′-AATGATCACAGATACTGATGAAGAAG-3′ and 5′-TGATCACAGATTCCGGAAAG-3′ forward and 5′-TCCTTCTCAATGACTTCCTATTTG-3′ reverse primers; for the amplification of FHOD3 independent of the T(D/E)5XE exon (“all isoforms”), we used an 5′-AGGTTTCACTCATTTTTGTTATTTATG-3′ forward and the same reverse primer; for the amplification of G. gallus glyceraldehyde 3-phosphate dehydrogenase (GAPDH), we used the 5′-CTGAAAATTGTCAGCAATGCATCG-3′ forward and 5′-CCAGTGGACGCTGGGATGATGTTC-3′ reverse primers.
In situ hybridisation
Fragments of FHOD3 were amplified with the 5′-GGGGTACCACAGATACTGATGAAGAAG-3′ forward and 5′-GGAATTCTCTCAATGACTTCCTATTTG-3′ reverse primers for the T(D/E)5XE-specific probes and with the 5′-GGGGTACCATCACAGATTCCGGAAAG-3′ forward and 5′-GGAATTCTCTCAATGACTTCCTATTTG-3′ reverse primers for the T(D/E)5XE-excluding probes [DT(D/E)5XE] and subsequently cloned into the KpnI and EcoRI sites of a pGEM-3Z vector (Promega). Digoxigenin (Roche Diagnostics, Indianapolis, IN) labelled sense and antisense RNA probes were created by digestion with EcoRI and KpnI, respectively, and subsequently transcription with SP6 and T7 (both Promega), respectively.
Whole-mount in situ hybridisation was carried out as described previously using a probe concentration of 1 μg/ml [26]. For detection purposes, embryos were incubated first with digoxigenin antibodies conjugated to alkaline phosphatase and then with NBT/BCIP (Roche Diagnostics) for visualisation. Embryos were embedded in a mix of gelatin type A, egg albumin and sucrose, crosslinked by glutaraldehyde [26], and cut into 50-μm sections on a VT1000S vibratome (Leica, Wetzlar, Germany); the sections were then placed on glass microscope slides and mounted in glycerol.
Pictures of embryos and sections were taken on a Leica MZ16 stereomicroscope.
Interaction assays
Glutathione-S-transferase pulldown assays and co-immunoprecipitations were performed as described previously [9]. COS cell lysates for the GST pulldown assays to assess the FHOD3 autointeraction were prepared in the presence of phosphatase inhibitor (0.5 μM calyculin A, 0.2 mM NaVO4 and 30 mM sodium pyrophosphate).
Immunofluorescence
Freshly dissected embryos were collected on embryonic day (E) 9.5 and E13.5, washed in PBS and fixed overnight at 4 °C with 4 % PFA in PBS. After fixation, embryos were washed in PBS and stored in 70 % ethanol until used. For embedding, embryos were first dehydrated by passage through a graded series of EtOH of increasing concentration [80, 90, 96, 100 % (2×) EtOH; 5 min at each concentration] and cleared by washing in 1:1 EtOH/xylene and xylene for 1 h. The embryos were then incubated in 1:1 xylene/paraplast (Oxford, St. Louis, MO) for 1 h at 60 °C and incubated in 100 % paraplast three times at 60 °C for 30 min to overnight. Embedded embryos were serially sectioned into 5-μm sections with an 820 microtome (American Optical Company, San Diego, CA), mounted on Superfrost microscope slides and dried overnight. Sections were deparaffinised by passage through a xylene–ethanol–water series, and epitopes were retrieved by boiling in 10 mM citric acid (pH 6.0) for 3 × 5 min. MAXblock blocking reagent was used before staining with the FHOD3 antiserum according to the manufacturer’s instructions (Active Motif, Carlsbad, CA). The sections were incubated with the primary antibody diluted in antibody dilution buffer [1 % bovine serum albumin (BSA), 20 mM Tris-base, 155 mM NaCl, 2 mM EGTA, 2 mM MgCl2, pH 7.5) for 1 h at room temperature (RT) and then washed three times in PBS, followed by incubation with the secondary antibody solution for 1 h at RT. Sections were mounted in 0.1 M Tris–HCl/glycerol (3:7) and 50 mg/ml n-propyl-gallate at pH 9.5.
Cells and culture
COS cells, HeLa cells and neonatal rat cardiomyocytes (NRCs) were prepared and cultured as described previously [9]. Adult rat cardiomyocytes were isolated by Langendorff perfusion of hearts and cultured in DMEM with 10 % foetal calf serum on collagen-coated plastic dishes [27]. Cells were fixed with 4 % PFA in PBS, permeabilised with 0.2 % Triton X-100 and stained as described above for the embryonic mouse sections.
Quantification of actin polymerisation activity was performed as reported previously [9].
Confocal microscopy
Confocal microscopy was performed on a LSM510 laser scanning confocal microscope (Carl Zeiss Micro Imaging GmbH, Jena, Germany) or an SP5 confocal microscope (Leica, Mannheim, Germany) with solid state (diode) laser excitation at 405 nm and emission at 420–480 nm, Ar-laser excitation at 488 nm and emission at 505–530 nm or He–Ne-laser excitation at 563 and 633 nm and emission at 560–615 and >650 nm, respectively, using LCI Plan-Neofluar 25×/0.8, Plan-Apochromat 63×/1.4 or Plan-Neofluar 100×/1.3 oil objectives.
Transmission electron microscopy
Embryos were fixed at E10.5 in 4.3 % glutaraldehyde (Polysciences, Warrington, PA)/0.03 mol/L sodium barbital–sodium acetate buffer (pH 7.4)/0.07 mol/L potassium chloride for 24 h at RT. The samples were then rinsed with 1× PBS (3 × 15 min), and post-fixed in a solution of 1 % osmium tetroxide (Ted Pella, Redding, CA)/0.03 mol/L sodium barbital–sodium acetate buffer (pH 7.4)/0.07 mol/L potassium chloride, for 1–2 h at RT. Embryos were then dehydrated through a graded series of EtOH of increasing concentration (70, 80, 95, 100 %), 15–20 min for each step, and allowed to remain in 100 % EtOH for 1 h with two changes. A mixture of Araldite 502 and dodecenyl succinic anhydride (DDSA) was prepared by combining equal volumes of Araldite 502 and DDSA. The embryos were then placed in a 1:1 ratio of propylene oxide:Araldite 502–DDSA mixture for 30 min and treated with a 1:3 ratio of propylene oxide:Araldite 502–DDSA mixture for 60 min. The samples were then immersed in a freshly prepared 1:1 mixture of Araldite 502:DDSA with accelerator (2 % benzyl dimethyl amine) and incubated at 60 °C for 18 h for polymerisation. Sections were cut on an LKB Ultratome V (LKB, Bromma, Sweden). Ribbons of sections showing grey, silver or slightly gold interference colours were picked up on uncoated 200-mesh Athene Thin Bar copper grids (Ted Pella). Electron micrographs were obtained from sections stained with uranyl acetate followed by lead citrate using either a JEM-1011 or a Philips 300 transmission electron microscope.
Antibodies
The polyclonal rabbit anti-FHOD3 antibody was prepared and characterised as described previously [9]. The monoclonal mouse (mM) anti-sarcomeric alpha-actinin (clone EA-53) and anti-Troponin T (TnT, clone JLT-12) antibodies were obtained from Sigma (St. Louis, MO). The mM anti-GFP (mixture of clones 7.1 and 13.1), anti-c-myc (clone 9E10) and the monoclonal rat anti-HA antibody (clone 3F10) were purchased from Roche Applied Science (Mannheim, Germany). The mM anti-p62 was from Abcam (Cambridge, UK). The polyclonal goat anti-GST antibody was obtained from GE Healthcare (Amersham Place, UK). The mM anti-myomesin clone B4 was a gift from Prof. Jean-Claude Perriard’s Lab (Institute of Cell Biology, ETH-Zürich Hönggerberg, Zürich, Switzerland). Horseradish peroxidase (HRP)-conjugated anti-mouse immunoglobulins (Igs) were purchased from DAKO (Glostrup, Denmark). HRP-conjugated anti-rabbit Igs and anti-goat Igs were purchased from Calbiochem (San Diego, CA). Cy2-conjugated anti-rabbit Igs, Cy3-conjugated anti-mouse Igs and Cy5-conjugated anti-rabbit and anti-mouse Igs were all purchased from Jackson Immuno Research (West Grove, PA). Phalloidin (Alexa633- and Alexa546-conjugated, respectively) was bought from Invitrogen.
Results
Expression of FHOD3 isoforms is linked to the establishment of a dense myofibril arrangement
Our initial characterisation of muscle and non-muscle FHOD3 isoforms as distinguished by the presence or absence of the T(D/E)5XE exon suggested a striated muscle-specific expression pattern of T(D/E)5XE exon-containing isoforms (hence termed muscle FHOD3) [9]. Since this could be a hallmark only of fully differentiated muscle, we wanted to investigate the expression of different FHOD3 isoforms during embryonic heart development. RT-PCR analysis showed that FHOD3 lacking the T(D/E)5XE exon (chromosome 2: 85704901:85705524; see [9] for sequence alignment) is already expressed faintly in whole chick embryos of Hamburger–Hamilton (HH; [25]) stage 4 and that the first apparent expression of the T(D/E)5XE exon containing FHOD3 can be detected at HH11 [Electronic Supplementary Material (ESM) Fig. 1A]. Using isoform-specific probes in situ hybridisation experiments, we found a first difference in localisation of both probes at stage HH28 (Fig. 1), when expression of the non-muscle isoform is restricted to the atrium, while the muscle isoform is highly enriched in the ventricle. There it is localised in the compact and trabeculated myocardium, as confirmed on cross-sections through the embryos. Interestingly, even in the atrium a difference in the expression of both isoforms is clearly visible, with the muscle isoform being expressed in almost all atrial regions while the non-muscle isoform is mainly restricted to the atrial wall. Expression of both isoforms is also seen in the limb buds at this stage. These observations indicate that the muscle isoform, which contains the T(D/E)5XE exon, is expressed at higher levels in the ventricle, while the non-muscle isoform is expressed higher in the atria. RT-PCR with isoform-specific primer pairs on dissected embryonic atria and ventricles support this interpretation (ESM Fig. 1B).
Fig. 1.

Distinct expression of FHOD3 isoforms during early development in the chick embryo. In situ hybridisation experiments were carried out using probes specific for the muscle isoform [T(D/E)5XE exon] and probes excluding the T(D/E)5XE exon [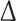 T(D/E)5XE exon] and thus specific for the non-muscle isoform, on chick embryos at Hamburger–Hamilton (HH) stage 28. Left, middle columns Overviews, right column cross sections taken at the level of the heart. H Heart, v ventricle, hlb hind limb bud, flb fore limb bud, lv left ventricle, la left atrium
T(D/E)5XE exon] and thus specific for the non-muscle isoform, on chick embryos at Hamburger–Hamilton (HH) stage 28. Left, middle columns Overviews, right column cross sections taken at the level of the heart. H Heart, v ventricle, hlb hind limb bud, flb fore limb bud, lv left ventricle, la left atrium
We turned to the mouse as an experimental system to determine whether this developmental shift in FHOD3 isoform expression levels is a general occurrence during vertebrate cardiac development. Analysis of the expression patterns of FHOD3 isoforms in developing mouse hearts by RT-PCR detected the expression of both isoforms in whole hearts of E14 mouse embryos, although the non-muscle isoform seems to be expressed at higher levels (Fig. 2a). This expression ratio changed in favour of muscle FHOD3 during the early postnatal phase of hypertrophic cardiac growth in mouse heart. These observations confirm our hypothesis that the expression of muscle FHOD3 is a hallmark of fully differentiated cardiomyocytes. To analyse the localisation of FHOD3 at a subcellular level we performed immunostainings of mouse heart paraffin sections. Since our FHOD3 antibodies recognise both isoforms and all attempts to produce a muscle isoform-specific FHOD3 antibody had failed, we were unable to study isoform-specific subcellular localisation differences. FHOD3 can be detected in cardiomyocytes as early as E9.5 (Fig. 2b) in a diffuse staining pattern with a more intense signal along the myofibrils. As myofibrillogenesis proceeds and the thin filaments become more organised (indicated by the anti-TnT staining) at E13.5, the FHOD3 staining pattern becomes restricted and is mostly found around the Z-disc. In the healthy adult heart, FHOD3 is exclusively found at the Z-disc, as indicated by its clear alternating pattern with myomesin, a marker of the M-band (Fig. 2b). Thus, FHOD3 subcellular localisation seems to be dependent on the maturity of the myofibrils, since while its localisation is restricted to the Z-disc in adult heart, it is found in a much broader distribution around the Z-disc in the embryonic heart. This localisation pattern nicely mirrors the results obtained by immunostaining or transfection of epitope-tagged FHOD3 in cultured NRCs [9] and may indicate a higher rate of turnover or myofibrillogenesis activity.
Fig. 2.

FHOD3 isoforms show a dynamic expression pattern and a developmental stage-specific subcellular localisation in the developing mouse heart. a Reverse transcription (RT)-PCR was carried out on RNA isolated from mouse hearts at embryonic day (E) 14, postnatal day (P) 0, P7, P16 and adult (Ad; 3 months) stages using forward primers in front of or within the mouse T(D/E)5XE exon (Exon 25) or specific for exon 25 (E25) lacking transcripts. Sarcomeric alpha-actinin (Actn2) was amplified as a loading control. Reactions were run longer than described previously [9] to allow expression levels to be compared between prenatal and postnatal hearts. b Confocal micrographs of paraffin sections of embryonic (E9.5, E13.5) and adult heart after staining with antibodies against FHOD3 (all isoforms, shown in green in the merged image) and against troponin T (TnT; embryonic hearts) or myomesin (adult heart) (shown in red in the merged image)
To investigate this further we utilised the model system of adult rat cardiomyocytes (ARC) in high serum culture [27, 28]. After isolation by Langendorff perfusion of the heart with collagenase and plating on collagen-coated plastic dishes in medium containing 10 % fetal calf serum (FCS), ARCs undergo a programme of de-differentiation and re-differentiation of their myofibrils and change from a rod-shaped morphology to a more flattened, fried egg-like shape that is similar to most cells in culture. After 2 weeks in culture and the re-assembly of their myofibrils, they also start again to beat, indicating full functionality of their contractile systems [27, 28]. This well-characterised disassembly and re-assembly process of the myofibrils allowed us to investigate potential changes in the FHOD3 isoform expression pattern in a purified population of cardiomyocytes and alterations in subcellular targeting pattern.
Analysis of the FHOD3 isoform expression pattern by RT-PCR showed a simultaneous up-regulation of the non-muscle and down-regulation of the muscle isoform of FHOD3 by day (d) 3 (Fig. 3a) and a reversion by d12, when muscle FHOD3 appeared to be the major isoform again. The shift in FHOD3 isoform expression appears to be correlated also with a differential localisation pattern: while displaying nice Z-disc association in freshly isolated ARC, FHOD3 localisation started to become more diffuse 3 days after plating (Fig. 3b). Interestingly, around this time FHOD3 started to appear in autophagosome-like vesicles, as judged by co-localisation with p62 (Fig. 3c). After day 8 post-plating, although FHOD3 was still strongly localising to autophagosomal structures in the cell body, we additionally detected targeting of FHOD3 to the newly forming stress-fibre like structures in the regions close to the cell attachments. During the following days more and more FHOD3 was recruited to the actin filaments, coinciding with increased expression levels of the muscle isoform. Two weeks after plating, FHOD3 is strongly localised to the actin structures in regions with a continuous alpha-actin staining pattern, indicative of areas of myofibrillogenesis [29]. Additionally, in line with our hypothesis that the changes in targeting depend on myofibril dynamics, FHOD3 was found in a broadly striated pattern similar to that observed in embryonic hearts and NRCs in regions with further advanced sarcomere formation and properly striated alpha-actinin staining (magnified image in Fig. 3b, lower panel). Therefore, the differential targeting in the sarcomere itself cannot be explained by attributing it to different FHOD3 isoforms, since at this stage muscle FHOD3 seems to be the prevalent isoform. The localisation of FHOD3 to autophagosome-like structures does seem to correlate with isoform identity, however, and thus with the ability/inability to interact with p62 [9]. These experiments support our hypothesis that muscle FHOD3 isoform expression and FHOD3 localisation at the Z-disc is a hallmark of mature myofibrils [30] and that challenges to this strictly organised cytoarchitecture lead to an up-regulation of the non-muscle isoform and hence increased FHOD3 protein turnover.
Fig. 3.
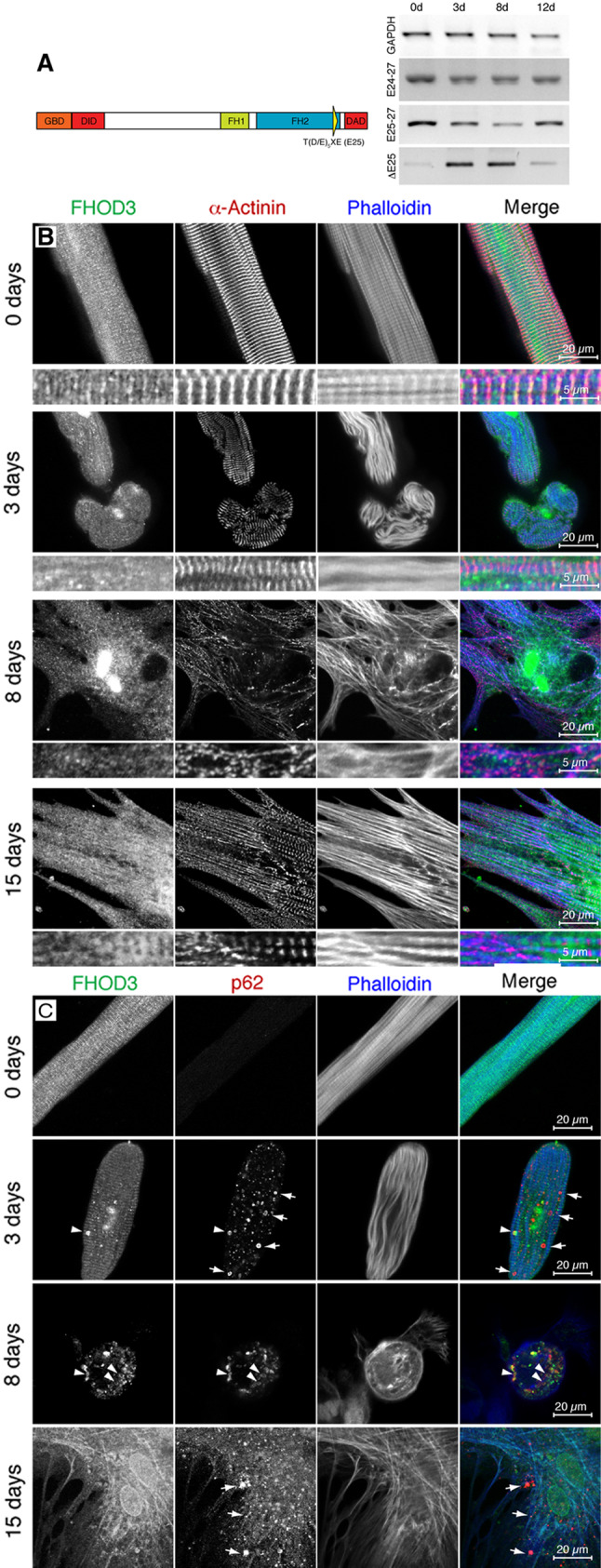
Adult rat cardiomyocytes (ARC) display a shift in FHOD3 isoform expression and altered subcellular targeting of FHOD3 during their myofibril degeneration–regeneration phase induced by high serum culture conditions. a Schematic drawing of FHOD3, indicating the muscle exon at the C-terminus of the formin homology 2 (FH2) domain as a yellow triangle. RT-PCR was carried out on RNA isolated from ARC immediately after collagenase digestion on 0 days or after 3, 8 and 12 days of culture in DMEM with 10 % fetal calf serum, with primer sets amplifying all FHOD3 isoforms (E24–27), specifically the muscle isoform (E25–27) or specifically the non-muscle isoform (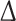 E25). A transient up-regulation of the non-muscle FHOD3 is seen at 3 and 8 days, with re-expression of muscle FHOD3 by 12 days. b Confocal micrographs of ARC at 0, 3, 8 and 15 days in culture, showing the degeneration and regeneration of myofibrils as visualised by staining for sarcomeric alpha-actinin (red signal in the merged image); confocal sections were taken close to the substrate where maximal cell spreading was seen. At day 0 FHOD3 (green signal) is co-localised at the Z-disc, becomes more diffuse during de-differentiation and is re-localised to the myofibrils only after 15 days in culture in the most mature sarcomeres (indicated in the blow-up). c Confocal micrographs of ARC at 0, 3, 8 and 15 days in culture showing a partial co-localisation of FHOD3 with p62 containing speckles at 3 and 8 days (arrowheads), but a total absence of co-localisation at 15 days (arrows, also at 3 days), when the myofibrils are regenerated and muscle FHOD3 expression levels have returned to normal (data not shown). The confocal section for the 8-day cell was taken from the top of the cell, where most of the degradation seems to take place, and not close to the substrate
E25). A transient up-regulation of the non-muscle FHOD3 is seen at 3 and 8 days, with re-expression of muscle FHOD3 by 12 days. b Confocal micrographs of ARC at 0, 3, 8 and 15 days in culture, showing the degeneration and regeneration of myofibrils as visualised by staining for sarcomeric alpha-actinin (red signal in the merged image); confocal sections were taken close to the substrate where maximal cell spreading was seen. At day 0 FHOD3 (green signal) is co-localised at the Z-disc, becomes more diffuse during de-differentiation and is re-localised to the myofibrils only after 15 days in culture in the most mature sarcomeres (indicated in the blow-up). c Confocal micrographs of ARC at 0, 3, 8 and 15 days in culture showing a partial co-localisation of FHOD3 with p62 containing speckles at 3 and 8 days (arrowheads), but a total absence of co-localisation at 15 days (arrows, also at 3 days), when the myofibrils are regenerated and muscle FHOD3 expression levels have returned to normal (data not shown). The confocal section for the 8-day cell was taken from the top of the cell, where most of the degradation seems to take place, and not close to the substrate
Muscle FHOD3 targeting as regulated by CK2 seems required for myofibril maturation in the ventricle
Our previously reported results in cultured NRCs indicate that muscle FHOD3 has to be phosphorylated by CK2 to target to the myofibrils [9]. To investigate this experimentally in an in situ setting, we made use of the CK2alpha knockout mice [14]. The myofibrils from hearts of CK2alpha knockout mice at E9.5 are less developed than those of their wild-type littermates (Fig. 4). Nevertheless, sarcomeric myosin could be detected in a cross-striated pattern, albeit with slightly reduced sarcomere length. However, a clear myofibrillar association was never observed for FHOD3 in CK2alpha knockout hearts, since it localised in a more diffuse fashion (Fig. 4b). There is a higher occurrence of FHOD3 speckles in the absence of CK2alpha (arrows in Fig. 4b), which is similar to the localisation that we described earlier for non-muscle FHOD3 [9]. This isoform lacks the CK2 consensus site and is targeted to autophagosomes due to its interaction with p62, both in transient transfection experiments as well as in cardiomyopathy [9]. Unfortunately, we were unable to demonstrate the co-localisation of FHOD3 and p62 in the CK2alpha knockout hearts due to the lack of reactivity of the monoclonal mouse anti-p62 on paraffin-embedded tissue. Electron micrographs of the CK2alpha knockout hearts also showed a more sparse distribution of myofibrils with slightly reduced sarcomere length (Fig. 5). Interestingly, this phenotype is only apparent in the ventricles, while atrial tissue appears to be spared (Fig. 5, bottom row). The finding that CK2alpha in wildtype embryos is expressed equally in atria and ventricles (Degano et al., submitted) suggests that CK2alpha activity—and possibly its action on muscle FHOD3—is only required for the assembly and maintenance of myofibrils in the embryonic ventricle.
Fig. 4.

FHOD3 needs CK2 activity for targeting to the myofibrils. Confocal micrographs of paraffin sections of E9.5 wildtype (a) and CK2alpha knockout mice (b), stained for FHOD3 (all isoforms, green signal in the merged image) and sarcomeric myosin (red signal in the merged image). FHOD3 shows clear myofibril association in the wildtype hearts (arrows); this targeting seems lost in the CK2alpha knockout hearts and an increased number of speckles is observed (arrowheads)
Fig. 5.

Lack of CK2alpha expression (CK2α−/−) affects myofibrils in the ventricle more dramatically than in the atria. Electron micrographs of E10.5 wildtype (left column) and CK2alpha knockout embryos (right column) taken from the right ventricle (RV), the left ventricle (LV) and the atrium (A). Sparse, misaligned myofibrils with reduced sarcomere length are apparent in both CK2alpha knockout ventricles, while atrial myofibrils appear to be indistinguishable between wildtype and knockout mice
C-terminal phosphorylation by ROCK1 is sufficient for FHOD3 activation
Members of the subfamily of diaphanous-related formins, such as FHOD3, are normally found in an autoinhibited form in cells [31]. Active dimers, which can aid the assembly of actin filaments, can only be formed once the autointeraction between the N-terminal DID and the C-terminal DAD domain is prevented. FHOD1, FHOD3’s closest paralog, can be activated by ROCK1 [24], which phosphorylates a consensus site in the DAD domain. A similar potential consensus site is conserved in the FHOD3 DAD domain between human and mouse (ESM Fig. 2a). We therefore wanted to determine whether ROCK1 is also sufficient for the activation of FHOD3. Indeed, we found an interaction of ROCK1 with FHOD3, which was increased when using a constitutively active form of ROCK1 (Fig. 6a). Constructs containing or lacking the T(D/E)5XE exon bound equally well to constitutively active ROCK1 (ESM Fig. 2b). To test for the ability of ROCK1 to activate FHOD3 by preventing its autointeraction, pulldown assays with GST-tagged N-terminal (DID-) and GFP-tagged C-terminal (DAD-) constructs (Fig. 6b) were performed. The constructs interacted with each other, but the interaction was strongly reduced in the presence of ROCK1 (Fig. 6c) or after mutation of the ROCK1 consensus site to mimic the phosphorylated state (Fig. 6d). These results confirm that the autointeraction can be inhibited via phosphorylation of the DAD domain by ROCK1. To prove that this activation also has functional consequences, we carried out actin polymerisation assays in HeLa cells transfected with different FHOD3 constructs. Increased actin polymerisation activity, as indicated by increased phalloidin staining, was detected in HeLa cells transfected with the phosphomimetic mutant compared to control or wildtype FHOD3 transfected cells (Fig. 6e). In addition, the activity of the phosphomimetic mutant (FHOD3-3D) seemed to be comparable to a constitutively active C-terminal truncation (FHOD3-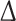 DAD; Fig. 6f). Therefore, FHOD3 is regulated in an analogous fashion to FHOD1 and does not appear to require the binding of a small GTPase of the Rho family to its GBD to relieve the autoinhibited state, as most other members of the DRF family do [22].
DAD; Fig. 6f). Therefore, FHOD3 is regulated in an analogous fashion to FHOD1 and does not appear to require the binding of a small GTPase of the Rho family to its GBD to relieve the autoinhibited state, as most other members of the DRF family do [22].
Fig. 6.

Interaction of active ROCK1 with the C-terminus of FHOD3 prevents the autoinhibitory interaction between the diaphanous autoinhibitory domain (DID) and diaphanous autoregulation domain (DAD) and leads to an activation of FHOD3, as evidenced by an increase in F-actin in HeLa cells. a Co-immunoprecipitation of myc-tagged full-length (p160-ROCK1) and constitutively active (p130-ROCK1) ROCK1 with the C-terminus of FHOD3 [green fluorescent protein (GFP) tagged] shows a stronger interaction with p130-ROCK1. b Schematic drawing of the autoinhibitory state and the proposed interaction of C- and N-terminal FHOD3 constructs in the absence or the presence of activation by phosphorylation (indicated by P in the DAD domain). c Glutathione-S-transferase (GST) pulldown assays of GST-tagged N-terminal FHOD3 constructs with GFP-tagged C-terminal FHOD3 constructs on their own show a strong interaction, which is weakened by the presence of constitutively active myc-tagged ROCK1. d GST pulldown assays of GST-tagged N-terminal FHOD3 constructs with GFP-tagged C-terminal FHOD3 constructs show an interaction (lane 2) which is abolished in pulldown assays with GFP-tagged C-terminal FHOD3 constructs that mimic phosphorylation (ROCK3D; lane 5) despite equal inputs, as shown with the anti-GST western blot (5 % of input loaded). e HeLa cells were transfected with GFP, or with different GFP-tagged FHOD3 constructs (full-length FHOD3; FHOD3- DAD, which lacks the DAD domain; FHOD3-3D, which phosphomimics the three ROCK1 phosphorylation sites), stained for F-actin with phalloidin and analysed by confocal microscopy. More filamentous actin is obvious in all cases where activated FHOD3 is present (FHOD3-
DAD, which lacks the DAD domain; FHOD3-3D, which phosphomimics the three ROCK1 phosphorylation sites), stained for F-actin with phalloidin and analysed by confocal microscopy. More filamentous actin is obvious in all cases where activated FHOD3 is present (FHOD3- DAD, FHOD3-3D), which is quantified in f. Values are presented as the means (± standard deviation) between three separate experiments, with nine quantified images per construct per experiment
DAD, FHOD3-3D), which is quantified in f. Values are presented as the means (± standard deviation) between three separate experiments, with nine quantified images per construct per experiment
While the effect of activated formins on the cytoskeleton of fibroblasts has been studied intensively, very little is known about any effect of constitutively active formins in muscle cells. To investigate the effect of activated FHOD3 on the cytoskeleton of cardiomyocytes, FHOD3 and ROCK1 were co-transfected into NRCs. In a cellular environment a dramatic effect on the actin cytoskeleton was already observed for full-length ROCK1 and was even more pronounced for constitutively active ROCK1 (Fig. 7). While control cells that just expressed HA-tagged muscle FHOD3 displayed nicely cross-striated thin filaments, as visualised by phalloidin (Fig. 7a, top row), both double-transfections with ROCK1 resulted in much more pronounced actin cables with a lack of striations (Fig. 7a, middle and bottom row), possibly indicating an excessive amount of actin filament synthesis. Patches of strong FHOD3 expression coincided with higher F-actin staining (arrows in Fig. 7a). The same observations were made for endogenous FHOD3 in ROCK1 overexpressing cardiomyocytes (Fig. 7b), as well as for constitutively active FHOD3 constructs (both DAD deletion constructs as well as ROCK1 phosphomimetic mutants; Fig. 7c).
Fig. 7.

Activation of FHOD3 leads to increased F-actin staining in neonatal rat cardiomyocytes (NRCs). a Confocal micrographs of NRCs that were transiently transfected with hemagglutinin (HA)-tagged full-length FHOD3 either on its own or together with wildtype (p160) or constitutively active (p130) myc-tagged ROCK1 constructs. While FHOD3 on its own targets to the myofibrils, and distinct I-band striations are visualised by Alexa633-phalloidin staining, activation in a cellular environment by ROCK1 leads to increased formation of actin cables (arrows). b Endogenous FHOD3 shows a more pronounced co-localisation with actin cables in NRCs that were co-transfected with myc-tagged p130 ROCK1 compared to neighbouring control cells. c Activation of FHOD3 by truncating the DAD domain or by a phosphomimetic construct of the ROCK1 phosphorylation sites also leads to increased actin cable formation in transiently transfected NRCs
To provide evidence that FHOD3 is a direct target for ROCK1 phosphorylation, we performed in vitro kinase assays (Fig. 8a). His-tagged ROCK1 was either incubated with a wildtype construct of the FHOD3 C-terminus or with a construct, where the three putative ROCK1 phosphorylation sites had been mutated to alanines. A robust phosphorylation signal was only detected with the wildtype construct and not with the unphosphorylatable mutant, which indicates that ROCK1 can indeed phosphorylate FHOD3 directly and that the three amino acids, which we had identified by sequence comparison, are the target amino acids.
Fig. 8.

ROCK1 directly phosphorylates the C-terminus of FHOD3 in vitro, and the expression of FHOD3 is necessary to obtain the actin filament polymerisation phenotype that is induced by ROCK1 overexpression. a In vitro kinase assay using His-tagged ROCK1 on either the wildtype C-terminus of FHOD3 or a mutant construct, where the serines and the threonine were mutated to alanine. A signal with the anti-phospho-serine/threonine antibodies could only be detected for the wildtype construct. b NRCs were double transfected with myc-tagged constitutively active ROCK1 (p130-ROCK1-myc) and either the control (top row) or the FHOD3 knockdown shRNA plasmid also expressing GFP (bottom row). After 5 days, pronounced actin cables (visualised by Alexa633-phalloidin) induced by ROCK1 can only be seen in the control cardiomyocytes, but not in cardiomyocytes with reduced FHOD3 expression levels, which show only rudimentary myofibrils, as described previously [9]. The cell labelled with an asterisk is a contaminating fibroblast in the cardiomyocyte primary culture. Bar: 10 μm
As shown above, overexpression of constitutively active ROCK1 results in excessive actin filament polymerisation in NRCs (Fig. 7a, b). Since this could be due to other proteins than FHOD3, we investigated double-transfected NRCs, which expressed p130-ROCK1 but also had been subjected to FHOD3 knockdown for 5 days (Fig. 8b). The FHOD3 knockdown cells showed the same fragmented myofibril phenotype that we had observed in our previous study [9], which also included the actin filaments, despite expressing constitutively active ROCK1 (bottom row). Control cells transfected with p130-ROCK1 and the negative control knockdown plasmid displayed an exaggerated actin filament phenotype compared to untransfected neighbouring cells, suggesting that FHOD3 is quite likely to be the downstream effector of this phenotype.
These results indicate that ROCK1 on its own is sufficient to activate FHOD3 by relieving its intramolecular autoinhibition and that this subsequently leads to increased F-actin polymerisation in HeLa cells as well as in cultured cardiomyocytes. This increased F-actin formation is probably directly due to the activated FHOD3.
Taken together, our results are the first to show the developmental regulation of expression and differentiation state-specific subcellular localisation of a formin family member, FHOD3, in cardiomyocytes and its dependence on CK2 function. In addition, we were able to throw light on the mechanism of activation of FHOD3 by ROCK1, as well as demonstrate the effects of constitutively active FHOD3 on actin filament synthesis in cardiomyocytes. We conclude that muscle FHOD3 plays an important role in fine-tuning the extent of thin filament assembly in the sarcomere, in particular in ventricular cardiomyocytes.
Discussion
We previously reported the existence of a muscle-specific splice variant of FHOD3 that is important for myofibril maintenance [9]. In the current work we show that this muscle FHOD3 isoform is already expressed in the developing embryo heart and that its presence at Z-discs is tightly correlated with the existence of mature myofibrils. In the case of dedifferentiation events, such as in adult cardiomyocytes that are adapting to culture conditions or in cardiomyopathy, a shift to the increased expression of the non-muscle FHOD3 is observed, with the consequences of a lack of sarcomeric targeting, increased interaction with p62 and shorter half-life of FHOD3 [9]. The expression of FHOD3 in mouse embryos from E8 to E10.5 has already been studied by in situ hybridisation by Xu and colleagues [32], who detected a cardiac-specific expression pattern at these early stages. It is interesting to note that the expression of non-muscle FHOD3 is maintained in the atrium at a time when it has already disappeared from the ventricle. A potential explanation could be that atrial cardiomyocytes are known to be smaller and less differentiated and have a higher proliferative activity also later on during development. Consequently, they are the only cardiomyocyte population in the heart from which it has been possible to establish a cell line, the HL-1 cells [33]. We could also show here that the atrial myofibrils are not affected in the CK2alpha knockout mice. This observation suggests that non-muscle FHOD3 is sufficient for the assembly and maintenance of the more loose myofibrillar network in atrial cardiomyocytes, but that the presence of muscle FHOD3 and its proper targeting to sarcomeres is required for the more dense myofibrillar arrangement in ventricular cardiomyocytes. Interestingly, an actin-bundling activity in addition to a nucleation activity has been shown for other formin family members, such as FRL 1 and mDia2, but not for mDia1 [34]. We also found previously that mDia1 is much less efficient than FHOD3 in the restoration of actin filaments in NRCs following latrunculin B treatment [9] and that there is a possibility that muscle FHOD3 may also be involved in the lateral integration of thin filaments in the ventricle.
Analysis of the phenotype of the CK2alpha knockout embryos revealed predominantly a defect in proliferation, while the extent of apoptosis was comparable to that of the controls [16]. It has previously been shown that CK2alpha is the principal isoform in the postnatal heart and that its expression is developmentally regulated, with lower levels in the adult heart compared to the perinatal one [35]. Forced expression of CK2alpha in transgenic mice leads to cardiac hypertrophy [36]. Our results suggest that muscle FHOD3 may be an effector molecule in these events, since elevated levels of CK2 activity would lead to lower FHOD3 protein turnover and therefore improved myofibril maintenance and, potentially, even more myofibrillogenesis.
The timing of the observed change in expression patterns between muscle and non-muscle FHOD3 isoforms fits extremely well with the switch from hyperplastic to hypertrophic heart growth in the early postnatal phase of mouse heart development [37] and suggests that while muscle FHOD3 can be regarded as a marker for mature myocardium, non-muscle FHOD3 can be added to the list of foetal isoforms that are re-expressed in pathological hypertrophy [27, 38–40].
DRFs are inhibited by the interaction of the C-terminal DAD with the N-terminal DID. This can be either due to autointeraction [22] or, at least in vitro, to homo- and heterodimerisation [41]. FHOD1, unlike other DRFs, is activated upon phosphorylation of three residues in the DAD by ROCK1, which releases the inhibitory interaction between DAD and DID [23, 24]. In our study, we were able to show that the same activation mechanism applies for FHOD3. Our GST pulldown assay and co-immunoprecipitation results confirm the binding of ROCK1 to FHOD3, and we also demonstrated direct phosphorylation by ROCK1 in an in vitro kinase assay. Furthermore, we were able to show that ROCK1 activity inhibits the autointeraction and induces assembly of (linear) actin filaments in HeLa cells, as well as the formation of thick actin cables in NRCs. The mutation of the suspected phospho-acceptor resides S1590, S1596 and T1600 (based on the homology to the FHOD1 residues S1131, S1137, T1141 [24]) to aspartic acids mimicked the effect and confirmed the activation of FHOD3 via ROCK1 phosphorylation. ROCK1 is highly expressed in the embryonic heart and has been reported to be involved in several steps of heart development [42, 43]. Consistent with a predominant early cardiogenic role, its expression is down-regulated after birth, but ROCK1 gets re-expressed again during pathological cardiac hypertrophy [40]. Inhibition of ROCK1 disrupts the formation of striated myofibrils in cultured chicken blastoderm [44, 45]. Considering the expression pattern of ROCK1 as well as FHOD3’s ability to polymerise actin filaments in cardiac myocytes [9], activation of FHOD3 by ROCK1 could be a crucial event during myofibrillogenesis.
In conclusion, we have described the relevance of two different phosphorylation events that determine the localisation and the activity of muscle FHOD3 (for schematic representation see Fig. 9). Phosphorylation of the C-terminal end of the FH2 domain of FHOD3 by CK2 is crucial for its targeting to myofibrils, where FHOD3 is involved in their maintenance and potentially also in novel assembly and lateral integration. For full activation of FHOD3, it seems to be sufficient that three amino acids in its C-terminus are phosphorylated by ROCK1. This regulation by posttranslational modification together with the alterations in the expression levels of different FHOD3 isoforms, CK2 and ROCK1 over time in the heart appear to make an important contribution to the hypertrophic growth seen during early postnatal development and in hypertrophic cardiomyopathy.
Fig. 9.

Scheme depicting the two distinct phosphorylation (P) events that govern the localisation and activity of muscle FHOD3. Only the muscle isoform of FHOD3 can be phosphorylated by CK2, which is required for its proper targeting to the myofibrils. Phosphorylation by ROCK1 at the C-terminus is alone sufficient for the activation of FHOD3. GBD GTPase binding domain
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are extremely grateful to Prof. S. Narumiya (Department of Pharmacology, Kyoto University Faculty of Medicine, Kyoto, Japan) for providing the ROCK1 constructs and to Dr. F. Cuello (Cardiovascular Division, King’s College London, London, UK) for supplying us with freshly isolated adult rat cardiomyocytes. Work in the laboratory of Elisabeth Ehler was supported by a Medical Research Council Career Establishment Grant. Thomas Iskratsch was the recipient of a King’s College London Strategic Investment PhD Fellowship that was awarded to the Cardiovascular Division and a British Heart Foundation Research Excellence Centre Pump Priming Fellowship. Work in the laboratory of Isabel Dominguez was supported by the American Heart Association and the National Institutes of General Medical Sciences (1R01GM098367) and Environmental Health Sciences (P01 ES11624). Irene R. Dégano was the recipient of a Beatriu de Pinos postdoctoral fellowship from the Catalonian Government.
Abbreviations
- CK2
Casein kinase 2
- DAD
Diaphanous autoregulation domain
- DID
Diaphanous autoinhibitory domain
- DRF
Diaphanous related formins
- E
Embryonic day
- FH
Formin homology
- HH
Hamburger–Hamilton
- P
Postnatal day
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315–2324. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suurmeijer AJ, Clement S, Francesconi A, Bocchi L, Angelini A, Van Veldhuisen DJ, Spagnoli LG, Gabbiani G, Orlandi A. Alpha-actin isoform distribution in normal and failing human heart: a morphological, morphometric, and biochemical study. J Pathol. 2003;199:387–397. doi: 10.1002/path.1311. [DOI] [PubMed] [Google Scholar]
- 4.Martin AF. Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I. J Biol Chem. 1981;256:964–968. [PubMed] [Google Scholar]
- 5.Sparrow JC, Schöck F. The initial steps of myofibril assembly: integrins pave the way. Nat Rev Mol Cell Biol. 2009;10:293–298. doi: 10.1038/nrm2634. [DOI] [PubMed] [Google Scholar]
- 6.Dwyer J, Iskratsch T, Ehler E. Actin in striated muscle: recent insights into assembly and maintenance. Biophys Rev. 2012;4:17–25. doi: 10.1007/s12551-011-0062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr Opin Cell Biol. 2011;23:569–578. doi: 10.1016/j.ceb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Takano K, Watanabe-Takano H, Suetsugu S, Kurita S, Tsujita K, Kimura S, Karatsu T, Takenawa T, Endo T. Nebulin and N-WASP cooperate to cause IGF-1-induced sarcomeric actin filament formation. Science. 2010;330:1536–1540. doi: 10.1126/science.1197767. [DOI] [PubMed] [Google Scholar]
- 9.Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E. Formin follows function: a muscle specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol. 2010;191:1159–1172. doi: 10.1083/jcb.201005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 11.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB Signalling: linking development and cancer. Cell Mol Life Sci. 2009;66:1850–1857. doi: 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: cK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66:1858–1867. doi: 10.1007/s00018-009-9154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23:908–915. doi: 10.1128/MCB.23.3.908-915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28:131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huillard E, Ziercher L, Blond O, Wong M, Deloulme JC, Souchelnytskyi S, Baudier J, Cochet C, Buchou T. Disruption of CK2beta in embryonic neural stem cells compromises proliferation and oligodendrogenesis in the mouse telencephalon. Mol Cell Biol. 2010;30:2737–2749. doi: 10.1128/MCB.01566-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez I, Degano IR, Chea K, Cha J, Toselli P, Seldin DC. CK2alpha is essential for embryonic morphogenesis. Mol Cell Biochem. 2011;356:209–216. doi: 10.1007/s11010-011-0961-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canton DA, Litchfield DW. The shape of things to come: an emerging role for protein kinase CK2 in the regulation of cell morphology and the cytoskeleton. Cell Signal. 2006;18:267–275. doi: 10.1016/j.cellsig.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 20.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- 21.Schönichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803:152–163. doi: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 22.Li F, Higgs HN. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. doi: 10.1016/S0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- 23.Hannemann S, Madrid R, Stastna J, Kitzing T, Gasteier J, Schonichen A, Bouchet J, Jimenez A, Geyer M, Grosse R, Benichou S, Fackler OT. The Diaphanous-related Formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J Biol Chem. 2008;283:27891–27903. doi: 10.1074/jbc.M801800200. [DOI] [PubMed] [Google Scholar]
- 24.Takeya R, Taniguchi K, Narumiya S, Sumimoto H. The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. EMBO J. 2008;27:618–628. doi: 10.1038/emboj.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- 26.Reijntjes S, Zile MH, Maden M. The expression of Stra6 and Rdh10 in the avian embryo and their contribution to the generation of retinoid signatures. Int J Dev Biol. 2010;54:1267–1275. doi: 10.1387/ijdb.093009sr. [DOI] [PubMed] [Google Scholar]
- 27.Eppenberger ME, Hauser I, Baechi T, Schaub MC, Brunner UT, Dechesne CA, Eppenberger HM. Immunocytochemical analysis of the regeneration of myofibrils in long-term cultures of adult cardiomyocytes of the rat. Dev Biol. 1988;130:1–15. doi: 10.1016/0012-1606(88)90408-3. [DOI] [PubMed] [Google Scholar]
- 28.Eppenberger HM, Hertig C, Eppenberger-Eberhardt M. Adult rat cardiomyocytes in culture: a model system to study the plasticity of the differentiated cardiac phenotype at the molecular and cellular levels. Trends Cardiovasc Med. 1994;4:187–192. doi: 10.1016/1050-1738(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 29.LoRusso SM, Rhee D, Sanger JM, Sanger JW. Premyofibrils in spreading adult cardiomyocytes in tissue culture: evidence for reexpression of the embryonic program for myofibrillogenesis in adult cells. Cell Motil Cytoskeleton. 1997;37:183–198. doi: 10.1002/(SICI)1097-0169(1997)37:3<183::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Iskratsch T, Ehler E. Formin-g muscle cytoarchitecture. BioArchitecture. 2011;1:66–68. doi: 10.4161/bioa.1.2.15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu XQ, Soo SY, Sun W, Zweigerdt R. Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells. 2009;27:2163–2174. doi: 10.1002/stem.166. [DOI] [PubMed] [Google Scholar]
- 33.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esue O, Harris ES, Higgs HN, Wirtz D. The filamentous actin cross-linking/bundling activity of mammalian formins. J Mol Biol. 2008;384:324–334. doi: 10.1016/j.jmb.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 35.Kim SO, Hasham MI, Katz S, Pelech SL. Insulin-regulated protein kinases during postnatal development of rat heart. J Cell Biochem. 1998;71:328–339. doi: 10.1002/(SICI)1097-4644(19981201)71:3<328::AID-JCB2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.Eom GH, Cho YK, Ko JH, Shin S, Choe N, Kim Y, Joung H, Kim HS, Nam KI, Kee HJ, Kook H. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation. 2011;123:2392–2403. doi: 10.1161/CIRCULATIONAHA.110.003665. [DOI] [PubMed] [Google Scholar]
- 37.Leu M, Ehler E, Perriard JC. Characterisation of postnatal growth of the murine heart. Anat Embryol (Berl) 2001;204:217–224. doi: 10.1007/s004290100206. [DOI] [PubMed] [Google Scholar]
- 38.Schlüter KD, Piper HM. Regulation of growth in the adult cardiomyocytes. FASEB J. 1999;13(Suppl):17–22. doi: 10.1096/fasebj.13.9001.s17. [DOI] [PubMed] [Google Scholar]
- 39.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 40.Ahuja P, Perriard E, Pedrazzini T, Satoh S, Perriard JC, Ehler E. Re-expression of proteins involved in cytokinesis during cardiac hypertrophy. Exp Cell Res. 2007;313:1270–1283. doi: 10.1016/j.yexcr.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 41.Copeland SJ, Green BJ, Burchat S, Papalia GA, Banner D, Copeland JW. The diaphanous inhibitory domain/diaphanous autoregulatory domain interaction is able to mediate heterodimerization between mDia1 and mDia2. J Biol Chem. 2007;282:30120–30130. doi: 10.1074/jbc.M703834200. [DOI] [PubMed] [Google Scholar]
- 42.Wei L, Roberts W, Wang L, Yamada M, Zhang S, Zhao Z, Rivkees SA, Schwartz RJ, Imanaka-Yoshida K. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development. 2001;128:2953–2962. doi: 10.1242/dev.128.15.2953. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Z, Rivkees SA. Rho-associated kinases play an essential role in cardiac morphogenesis and cardiomyocyte proliferation. Dev Dyn. 2003;226:24–32. doi: 10.1002/dvdy.10212. [DOI] [PubMed] [Google Scholar]
- 44.Kaarbo M, Crane DI, Murrell WG. RhoA is highly up-regulated in the process of early heart development of the chick and important for normal embryogenesis. Dev Dyn. 2003;227:35–47. doi: 10.1002/dvdy.10283. [DOI] [PubMed] [Google Scholar]
- 45.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.