

Abstract
The noradrenergic system of the brain is thought to facilitate neuronal processes that promote behavioral activation, alertness, and attention. It is known that norepinephrine (NE) can be significantly elevated in the prefrontal cortex under normal conditions such as arousal and attention, and following administration of psychostimulants and various other drugs prescribed for psychiatric disorders. However, how NE modulates neuronal activity and synapses in the local prefrontal circuitry remains elusive. In this study, we characterized the actions of NE on individual monosynaptic connections among layer V pyramidal neurons (P) and fast-spiking (FS) GABAergic interneurons in the juvenile (postnatal days 20–23) rat prefrontal local circuitry. We found that NE selectively depresses excitatory synaptic transmission in P-FS connections but has no detectable effect on the excitatory synapses in P-P connections and the inhibitory synapses in FS-P connections. NE apparently exerts distinctly different modulatory actions on identified synapses that target GABAergic interneurons but has no effect on those in the pyramidal neurons in this specific developmental period. These results indicate that, depending on the postsynaptic targets, the effects of NE in prefrontal cortex are synapse-specific, at least in the juvenile animals.
Keywords: norepinephrine, catecholamine, synaptic transmission, local circuit, neocortex
Introduction
The brain noradrenergic system is thought to facilitate neuronal processes that promote alertness and attention (Morilak et al., 2005, Miner et al., 2006, Milstein et al., 2007, Arnsten, 2009). One region in which norepinephrine (NE) may exert such effects is the medial prefrontal cortex (PFC), which receives a substantial input from the noradrenergic nucleus locus coeruleus and has been implicated in many cognitive functions including arousal, attention, working memory, and other executive functions (Berridge and Waterhouse, 2003, Torres et al., 2003, Arnsten and Li, 2005, Elliott and Beveridge, 2005, Berridge et al., 2006, Newman et al., 2008). Noradrenergic dysfunction in the PFC is linked to numerous psychiatric disorders including attention-deficit/hyperactivity disorder, depression, and schizophrenia (Arnsten, 2004, Friedman et al., 2004, Viggiano et al., 2004). Furthermore, it is clear that many drugs prescribed for psychiatric conditions, including psychostimulants, significantly elevate NE levels in the PFC (Torres et al., 2003, Elliott and Beveridge, 2005, Berridge et al., 2006, Arnsten, 2009). Although the effects of NE, NE agonists, and activation of the LC-NE pathway on neuronal and neural network response properties have been examined in many previous studies, the impact of NE on signal transmission at the level of individual synapses in the PFC has not been investigated in great detail. A full accounting of the actions of NE on identified cell types and different synapses in the PFC is critical for understanding noradrenergic regulation of prefrontal circuit operations and prefrontal functions such as sustained attention and working memory under normal circumstances and in psychiatric disorders which are associated with disrupted balance of excitation and inhibition in PFC circuits and where executive functions are severely compromised.
A large body of evidence has shown that activation of NE receptors alters both glutamatergic (Law-Tho et al., 1993, Marek and Aghajanian, 1999, Waterhouse et al., 2000, Delaney et al., 2007, Kobayashi, 2007, Ji et al., 2008a, Ji et al., 2008b, Dinh et al., 2009, Kobayashi et al., 2009) and gamma-aminobutyric acid (GABA)ergic synaptic transmission (Sessler et al., 1995, Bennett et al., 1998, Braga et al., 2004, Chen et al., 2006, Lei et al., 2007, Koyanagi et al., 2010, Salgado et al., 2011) in the cortex and other brain regions. NE also produces differential effects on neuronal excitability and synaptic plasticity in both pyramidal neurons and GABAergic interneurons in a cell-type specific manner (Bergles et al., 1996, Kawaguchi and Shindou, 1998, Devilbiss and Waterhouse, 2000, Carr et al., 2007, Tully et al., 2007, Dembrow et al., 2010, Wojtowicz et al., 2010). In general, these results demonstrate cell- and synapse-specific adrenergic modulation of synaptic transmission and excitability involving central neurons, and these effects appear to be mediated by different subtypes of adrenergic receptors, including alpha-1, alpha-2 or beta-receptors.
Despite these findings, there has been no systematic and detailed assessment of how NE regulates synaptic communication between individual pyramidal neurons and interneurons in the local prefrontal circuitry. By contrast dopaminergic regulation of synaptic transmission in the PFC has been extensively studied. (Gao et al., 2001, Gao and Goldman-Rakic, 2003, Gao et al., 2003, Seamans and Yang, 2004). In the present study we examined the effects of NE on monosynaptically connected and individually identified pyramidal neurons and GABAergic interneurons in the PFC using multiple whole-cell patch clamp recordings. We found that NE selectively depresses excitatory synaptic transmission between pyramidal neurons and GABAergic cells without affecting the excitatory communication between pyramidal neurons or the inhibitory synapses between fast-spiking (FS) interneurons and pyramidal cells. These results demonstrate a differential modulatory effect of NE on individual synapses between pyramidal neurons and GABAergic FS interneurons in the PFC circuitry.
Materials and Methods
Animal treatment
We used 68 Sprague-Dawley rats aged PD20 to PD23 (Charles River Laboratories, Wilmington, MA). The rats were maintained on a 12-hour light/dark cycle and were fed ad libitum. The animals were treated under National Institutes of Health animal use guidelines, and the experimental protocol was approved by the Institutional Animal Care and Use Committee at Drexel University College of Medicine.
Slice preparation
The detailed procedure is described in our previous publications (Gao et al., 2001, Wang and Gao, 2009, 2010). The rats were deeply anesthetized with Euthasol (0.2 ml/kg, i.p.), rapidly perfused through the heart with ice-cold (< 4°C) sucrose solution bubbled with 95% O2 and 5% CO2 and then quickly decapitated with guillotine. The sucrose solution contained (mM) NaCl 87, KCl 2.5, NaH2PO4 1.25, NaHCO3 25, CaCl2 0.5, MgSO4 7.0, sucrose 75, and glucose 25. The brains were carefully removed and immersed in ice-cold sucrose solution. The brain tissue containing prelimbic region of the medial PFC was cut into 300-μm slices with a Leica Vibratome (VT 1000S; Leica). The PFC slices were harvested and incubated in oxygenated sucrose solution at 35°C for 1 hour. The cortical slices were kept at room temperature until being transferred into a submerged recording chamber that was controlled at 35 to 36°C and was perfused with Ringer’s solution bubbled with 95% O2 and 5% CO2 at a flow rate of 2 to 3 ml per minute. The Ringer’s solution contained (in mM) NaCl 128, KCl 2.5, NaH2PO4 1.25, CaCl2 2, MgSO4 1.0, NaHCO3 26, and dextrose 10.
Electrophysiological recordings
Only the slices in which apical dendrites of pyramidal neurons could be traced toward superficial layer 1 of the cortex under infrared differential interference contrast optics were selected for pair or multiple recordings. Multiple somatic whole-cell recordings (2–4 cells were recorded simultaneously) were performed with two Multiclamp 700B amplifiers (Molecular Devices) in both current and voltage clamp modes. The resistances of the glass pipettes were 4 to 7 MΩ and the electrodes were filled with intracellular solution containing (mM): 114 K-gluconate, 6 KCl, 0.5 CaCl2, 0.2 EGTA, 4 ATP-Mg, 10 HEPES, pH7.25, and 0.3% biocytin (Molecular Probes). The monosynaptic connections between and among layer V pyramidal neurons and GABAergic interneurons were identified by stimulating an individual neuron with a 10 + 1 pulse train (10 pulses at 20 Hz or 50-ms interpulse intervals and the 11th pulse at 500 ms after the 10th pulse) to evoke action potentials in the presynaptic neurons in current clamp mode. The pulse intensity was adjusted from 0.1 to 0.3 nA and synaptic responses (excitatory postsynaptic currents [EPSCs] or inhibitory postsynaptic currents [IPSCs]) in postsynaptic neurons were recorded. The stimulus pulses were delivered every 7.5 seconds, and at least 40 sweeps were recorded to obtain an average of the responses. The signals were amplified and filtered at 2 kHz, and the series resistances of the recorded neurons were constantly monitored with a negative 5 mV pulse (200 ms duration) and compensated during recordings. To avoid confounding drug effects, each brain slice was only used for one experiment. The data were acquired through Clampex 9.2 software and digitized through a DigiData 1322A (Molecular Devices). All drugs, including NE, the alpha-1 agonist phenylephrine, and alpha-1 antagonist prazosin, were purchased from Tocris Bioscience USA (Minneapolis, MN).
Histological and Morphological Analyses
All slices with recorded connections were preserved for biocytin immunostaining as previously reported in our laboratory (Gao et al., 2003, Gao, 2007, Wang and Gao, 2009). Briefly, slices were fixed in 4% paraformaldehyde until immunocytochemical processing occurred. The slices were placed in 3% H2O2 for 30 minutes to block the endogenous horseradish peroxidase. After thorough rinsing, ABC (Vector Laboratories, CA) reactions were conducted overnight, followed by the Ni-3,3-diaminobenzidine reaction. The slices were directly mounted from 0.2 mM phosphate buffer and covered with water-soluble mounting media. All labeled neurons were photographed and matched with their action potential firing patterns.
Data analysis
The pairs with significant rundown or input resistance changes (larger than 20%) were excluded from the data set for further analysis. The mean peak amplitudes of EPSCs or IPSCs were measured from the average of 40 sweeps in the baseline, the second 5 min in NE, or phenylephrine, or prazosin + NE (following a 5-min pre-application of prazosin), and the second 5 min in washout, respectively, with Clampfit 9.2 (Axon Instruments). Traces containing spike failures in the presynaptic neurons were manually removed and then averaged in several cases. The latency of the E/IPSC was determined from peak of action potential of presynaptic neuron and onset of the corresponding E/IPSC in the postsynaptic neurons. The rise time was measured as 20–80% of the rise phase of E/IPSC whereas decay time was fitted as 63% of the recovery phase of E/IPSC (see Table 1 and Figure 1 for the kinetics of individual EPSCs and IPSCs). A paired-pulse ratio (PPR) was obtained by measuring the ratio of the first two successive responses (2nd PSC/1st PSC) of the10 PSCs; the recovery ratio was calculated as the ratio of the 11th PSC/1st PSC. Synaptic failure was defined as an event in which the amplitude of the PSC was below the limit of 1.6-fold noise. The mean and standard deviation of the 1st PSC amplitudes were calculated, and the coefficients of variation (CVs) in the control, NE-, or phenylephrine-, or prazosin+NE-treated PSC amplitudes were computed as standard deviation/mean of the PSCs. The data were analyzed by ANOVA, the Kolmogorov-Smirnov test (K-S test, for cumulative probability), or Student’s t test and were presented as mean ± standard error. The histogram of PSC events per 5pA bin was analyzed with Gaussian fit to compare the changes that occurred with the different drugs.
Table 1.
NE effects on the properties of both pre- and post-synaptic neurons
| P-FS (n=7) | FS-P (n=7) | P-P (n=12) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-NE | Post-NE | P value | Pre-NE | Post-NE | P value | Pre-NE | Post-NE | P value | |
| Presynaptic Neuron | |||||||||
| Resting membrane potential (mV) | −67.9±2.04 | −66.0±2.10 | 0.131 | −70.0±1.74 | −66.0±1.80 | 0.222 | −65.7±5.10 | −66.1±6.92 | 0.878 |
| input resistance (MΩ) | 112±16.3 | 118±22.3 | 0.572 | 200±31.6 | 211±38.8 | 0.50 | 135±48.9 | 137±38.8 | 0.820 |
| Membrane time constant (ms) | 29.5±3.15 | 28.0±3.26 | 0.640 | 7.55±1.36 | 8.70±1.99 | 0.221 | 26.5±1.33 | 25.0±1.55 | 0.303 |
| AP threshold (mV) | −44.2±1.88 | −3.7±1.32 | 0.811 | −5.2±1.12 | −3.4±1.33 | 0.255 | −2.6±1.23 | −1.2±0.85 | 0.374 |
| AP half-width (ms) | 1.79±0.17 | 2.04±0.21 | 0.122 | 0.46±0.12 | 0.47±0.12 | 0.136 | 2.24±0.25 | 2.35±0.23 | 0.697 |
| Postsynaptic neuron | |||||||||
| inject current (pA) | −17.5±16.2 | −35.4±14.6 | 0.147 | 20.6±14.9 | −2.8± 15.6 | 0.084 | −6.17±12.1 | −2.99±11.4 | 0.605 |
| Latency (ms) | 0.52±0.10 | 0.58±0.06 | 0.331 | 0.50±0.06 | 0.48±0.06 | 0.278 | 0.99±0.10 | 0.99±0.09 | 0.918 |
| Amplitude (pA) | 36.9±15.6 | 10.4±4.76 | 0.058 | 30.5±15.5 | 25.0±13.4 | 0.140 | 11.5±2.06 | 11.0±3.02 | 0.861 |
| 20–80% rise time (ms) | 0.71±0.09 | 0.93±0.19 | 0.272 | 0.68±0.07 | 0.96±0.16 | 0.123 | 1.41±0.29 | 1.32±0.17 | 0.577 |
| 63% decay time (ms) | 5.50±0.94 | 5.90±1.19 | 0.7196 | 8.23±0.85 | 10.2±1.55 | 0.0967 | 11.2±1.79 | 9.50±1.55 | 0.2649 |
| Half-duration (ms) | 5.81±1.10 | 5.88±0.87 | 0.9304 | 12.7±1.18 | 12.8±1.33 | 0.9411 | 10.8±1.80 | 10.7±1.72 | 0.8806 |
Figure 1.

Identification of prefrontal cortical neurons. A, Sample firing patterns of a layer V pyramidal neuron (P) and a fast-spiking (FS) interneuron. The P cell was easily identified by its regular firing pattern with adaptive firing, wider action potentials, and smaller fAHP; the FS cell was identified by its high-frequency firing of action potentials without adaptation, short half-width, and large fAHP. B, The 1st unitary EPSC in a train of 10 pulses in P–P and P–FS and IPSC in FS–P connections. The uEPSC and uIPSC in these pairs exhibit distinct different kinetics as shown in the Table 1. C, Scatter plot shows a sample recording of EPSCs in a P-FS synaptic connection (EPSC amplitude, upper panel, and synaptic failure rate per min, lower panel). The EPSCs recordings were typically stable and reliable for more than 20 min without rundown. This fact ensures the reliability of the depressive NE effects observed in the Figure 2.
Results
To distinguish the effects of NE on monosynaptically connected and individually identified connections between pyramidal neurons and GABAergic interneurons, we used multiple whole-cell patch clamp recordings in PFC slices to record 3–4 cells simultaneously to increase the probability of obtaining monosynaptic connections among individual excitatory (P-P and P-FS) and inhibitory (FS-P) synapses.
We used a pipette solution containing 7 mM [Cl]i and recorded IPSCs at −50 mV in the voltage clamp mode. Because the expected reversal potential of IPSC is − 78.4 mV ([Cl−]out = 134.5 mM and [Cl−]in = 7 mM; Nernst potential = − 78.4 mV; junction potential = + 9 mV), the IPSC appears as an outward current in this solution when membrane potential is held at − 50 mV whereas EPSC appears as an inward current when recorded at −70 mV. The IPSCs/EPSCs were confirmed, in some cases, at the end of the experiment by bath application of picrotoxin (0.1 mM) or CNQX (20 μM), respectively (data not shown). The advantages of using this relatively low chloride intracellular solution are clear. First, the firing patterns of both pre- and postsynaptic neurons can be recorded in current clamp mode, which is critical for identifying the types of recorded neurons, as exhibited in Figure 1. In addition, the driving force for GABAA channels is suitable for detecting the IPSC. Unitary synaptic responses in the postsynaptic cell were unambiguously identified as IPSC or EPSC based on their pharmacological characteristics (see Table 1), their reversal potential (reversal potential for EPSCs is ~ 0 mV), and the firing patterns of presynaptic neurons which were recorded in current clamp mode with a 15-sweep step current, 50 pA increment at each step (Fig. 1).
All recorded neurons were categorized by their electrophysiological properties, and some were verified morphologically by biocytin labeling. In these latter cases, both neuronal morphological characteristics, such as dendritic and axonal branch patterns, and the action potential firing pattern were used to determine the cell types (Kawaguchi, 1995, Galarreta and Hestrin, 1999, Gibson et al., 1999, Gao et al., 2003, Wang and Gao, 2009). Cells with triangular soma and pial-oriented apical dendrites were deemed pyramidal cells whereas cells with characteristics of narrow action potentials, deep afterhyperpolarization (AHP), high firing rates (100–250 Hz) and no frequency adaptation were identified as FS interneurons according to the criteria established by Kawaguchi (Kawaguchi, 1995) and Gibson et al. (Gibson et al., 1999), as well as our recent studies (Gao et al., 2003, Wang and Gao, 2009).
NE significantly suppressed excitatory synaptic transmission in P-FS connections
We first tested the effects of bath-applied NE on the monosynaptic excitatory connections between layer V pyramidal neurons and FS interneurons (P-FS) with a 10+1 pulse train applied in the presynaptic neurons. As shown in the Figure 1C, the EPSC recordings in the P-FS connection were usually stable and reliable for more than 20 min without rundown under control conditions. We then tested the effects of bath-applied NE at 10 μM on both excitatory and inhibitory connections. The concentration of NE was based on concentrations used in similar studies that reported a significant effect of NE on both excitatory and inhibitory synaptic transmission in cortical and amygdala neurons with an EC50 = ~ 4–5 μM and near plateau effects at 10 μM (Kawaguchi and Shindou, 1998, Delaney et al., 2007, Lei et al., 2007). We first tested the effects of bath-applied NE on the monosynaptic excitatory connections between layer V pyramidal neurons and FS interneurons (P-FS, n = 7). As shown in the Figure 2A, the unitary EPSCs were induced by applying a 10+1 pulse train in the presynaptic neuron. The unitary EPSCs were recorded for 5 min to establish the baseline, and then 10 μM NE was bath-applied for 10 min to prevent potential instability of the recordings, followed by a 15-min washout period with normal Ringer’s solution. We found that the peak amplitude of the first EPSC was significantly decreased by bath-applied NE and this effect lasted during the wash period (Fig. 2A–D), consistent with a previous study reported for IPSCs (Lei et al., 2007). Cumulative probability analysis showed a clear shift to the left of the curve with administration of NE and a significant difference between baseline values and those after administration of NE (P < 0.001 K-S test, Fig. 2C). The peak amplitude of the first EPSC decreased significantly by average of 63.8 ± 8.08% in P-FS pairs after NE administration (from 36.9 ± 15.6 pA at baseline to 15.4 ± 9.22 pA with NE, and to 10.4 ± 4.76 pA in wash; n = 7, baseline vs. NE, P = 0.028; baseline vs. wash, P = 0.058; NE vs. wash, P = 0.335; F = 5.72, P = 0.018; Fig. 2D). Correspondingly, the CV of the first EPSCs increased significantly (baseline vs. NE, P = 0.011; baseline vs. wash, P = 0.029; NE vs. wash, P = 0.345; F = 7.588, P = 0.007; Fig. 2E). The peak value of the Gaussian distribution of the first EPSC amplitude shifted from 17.5 pA at baseline to 12.5 pA in NE and 10.0 pA in wash, and the fitted area decreased from 186 to 97 and 69, respectively as well. The failure number increased significantly from 4.86 ± 1.70 at baseline to 19.85 ± 4.17 and 21.6 ± 4.80 in NE and wash, respectively (baseline vs. NE, P = 0.003; baseline vs. wash, P = 0.009; NE vs. wash, P = 0.542 respectively; F = 14.07, P = 0.0007; Fig. 2F–H). The PPR of the first two EPSCs showed significant increase following NE administration compared with baseline (0.47 ± 0.07 in baseline vs. 0.73 ± 0.07 in NE and 0.69 ± 0.14 in wash, F = 7.125, P = 0.020), whereas the recovery ratio between the 1st and 11th EPSC showed no significant changes following NE administration compared with baseline (0.83 ± 0.23 in baseline vs.1.09 ± 0.39 in NE and 1.25 ± 0.33 in wash, P > 0.05). These results suggest that NE suppresses excitatory synaptic transmission in the P-FS connection. All of these changes, including significantly decreased EPSC amplitude and increased synaptic failure, CV, and PPR, indicated that the NE effect is likely involved in a decreased release probability in the presynaptic axon terminals that targets FS interneurons.
Figure 2.

NE significantly inhibits P-FS synapses. A, Representative traces showing the effects of NE on AMPA EPSCs in a P-FS pair. Note: The upper panels in control, NE and wash conditions represent action potentials induced by intracellular injected currents applied in the presynaptic neuron, whereas the lower panels were the corresponding postsynaptic currents (recorded in the postsynaptic neurons) in response to the individual action potentials in the presynaptic neurons. The same conventions apply in Figures 3A, 5A, and 6A. B, Scatter plot showing the depressive effect of NE on the P-FS pair in A. C, Cumulative probability plot of the first AMPA EPSCs exhibited a shift of the NE curve (n = 7). D, NE significantly decreased the first EPSC amplitude (P =0.028) and the effect was not recovered even after 10 min wash (P < 0.05). Note that three overlapping pairs were slighted shifted horizontally for graph clarification. E, CVs of the first EPSCs were also significantly increased in NE and wash (P = 0.011 and 0.028, respectively). F, G, and H, The amplitude histograms of binned first unitary EPSCs in P-FS connections from control. NE treatment and wash with Ringer’s solution with Gaussian fit, as well as significantly increased synaptic failures in both NE and wash (P < 0.05 for both).
NE had no effect on inhibitory synaptic transmission in FS-P microcircuits
Next we examined the effects of NE on monosynaptic inhibitory synapses between FS interneurons and pyramidal neurons. Luckily, we obtained a reciprocal connection between P and FS that allow us to examine NE effects on P-FS and FS-P simultaneously and respectively. In this case, three cells were recorded but connection was only found between P1 and FS without connection with P3 (Fig. 3). The morphological characteristics of this reciprocal pair (as well as those of another unconnected pyramidal neuron) were recovered with biocytin labeling, and the firing patterns of the pyramidal cell and FS interneuron were displayed in Figure 3A inset. In this reciprocal P-FS connection, we first recorded the EPSCs in the P-FS connection with the membrane potential of FS held at −70 mV; NE was bath-applied for 5-min after baseline recording in this reciprocal pair in order to offset any instability of the recording and allow both types of synapses to be studied in a limited time window. Ten minutes after NE washout, the membrane potential of the pyramidal neuron was held at −50 mV to record IPSCs in the FS-P connection and NE was re-applied for another 10-min. Interestingly, as shown in Figure 3, we found that NE selectively modulated the excitatory synapses in the P-FS but had no effects on the inhibitory synapses in the FS-P connection in a reciprocal connection between a pyramidal neuron and a FS interneuron (Fig. 3). As exhibited in the Figure 3, the amplitude of first unitary EPSCs in the P-FS connection was dramatically and significantly decreased by administration of NE (Fig. 3B, C), whereas the amplitude of the first unitary IPSCs in FS-P connection was unaltered (Fig. 3D, E). These data suggest that the effects of NE on monosynaptic synapses between layer V pyramidal neurons and FS interneurons are likely synapse-specific. However, it is also possible that the lack of NE effect on FS-P connection was derived from receptor desensitization induced by the first NE application because the suppressive NE effect on the P-FS connections persists without clear recovery (Lei et al., 2007). To test this possibility and to verify the synapse-specificity, we examined additional FS-P connections. We confirmed that the amplitude of the first IPSCs was unaltered by bath application of NE for 10 min in all of the connections exhibited in this figure (Fig. 4A) and, as noted in the scatter plot (Fig. 4B), a stable, even distribution of IPSC amplitudes was observed during baseline, NE, and washout conditions. Cumulative analysis showed that the curve generated with NE administration overlapped with those in the baseline and washout periods without significant difference (P > 0.05, K-S test; Fig. 4C). There was no significant difference between baseline and NE administration and between NE and wash periods (30.5 ± 15.5 pA at baseline vs. 27.3 ± 14.1 pA with NE, and 25.0 ± 13.6 in wash; n = 7, F = 2.44, P = 0.129; Fig. 4D) in the first IPSC amplitudes. It should be noted that 2 of the 7 pairs appeared to exhibit large IPSC amplitudes and these two pairs had few or no synaptic failures. Similarly, the CV of the first IPSCs showed no significant changes between baseline, NE application and wash conditions (baseline vs. NE, P = 0.238; baseline vs. wash, P=0.375; NE vs. wash, P = 0.216, respectively; two-way ANOVA F = 1.45, P = 0.273; Fig. 4E). The Gaussian distribution of the peak value of events per 5pA bin at baseline and with NE exhibited a slight shift to the right and the same amount of synaptic failure, without statistical significance (F = 0.852, P = 0.451; Fig. 4F–H). The PPR (0.79 ± 0.05 at baseline vs. 0.86 ± 0.13 with NE, and 0.73 ± 0.13 for wash; n = 7, F = 0.571, P = 0.579) and the recovery ratio of the 11th IPSCs (0.72 ± 0.03 at baseline vs. 0.76 ± 0.10 with NE, and 0.85 ± 0.08 of wash; n = 7, F = 1.205, P = 0.333) also exhibited no significant changes during NE administration compared with those at baseline. These data substantially reinforce the notion that the effects of NE on P-FS connections are synapse-specific and selective.
Figure 3.
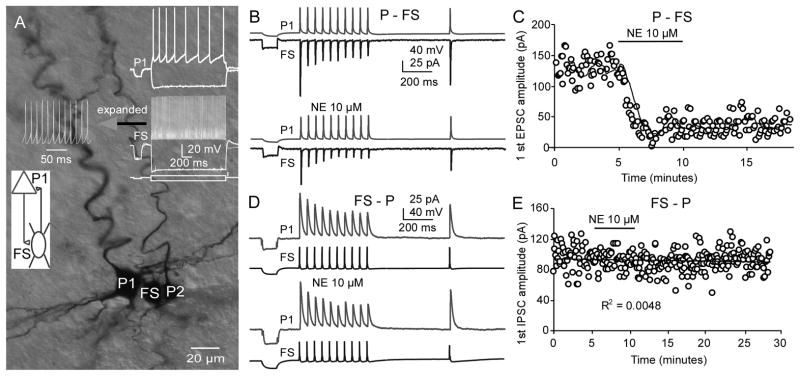
NE selectively modulates excitatory, but not inhibitory, synaptic transmission in a reciprocal P-FS connection. A, The morphological characteristics of a reciprocal pair as well as another unconnected pyramidal neuron were recovered with biocytin labeling. The firing patterns of the pyramidal cell and FS interneuron are displayed in the inset. In this reciprocal P-FS connection, we first recorded the EPSCs in the P-FS connection with the membrane potential of FS held at −70 mV. NE was bath-applied after 5-min baseline recording. Ten minutes after washing the NE, the membrane potential of pyramidal neuron was held at −50 mV and the IPSCs in the FS-P connection were recorded during re-application of NE. B and C, NE bath-application dramatically reduced the EPSC amplitudes in the P-FS connection. D and E, In contrast, NE had no clear effects on the IPSCs in the FS-P synapses.
Figure 4.

NE has no significant effect on IPSC amplitude in FS-P synapses. A, Representative traces showing the Effect of NE on IPSCs in a layer V FS-P pair in response to a 20-Hz train applied at the presynaptic neuron. B, Scatter plot showing the effect of NE on the 1st EPSC amplitude of the FS-P pair in A. C, Cumulative probability plot showing the completely overlapped curves for the first IPSCs at baseline, during NE application, and after the washout (n = 7). D, NE exhibited no effect on the amplitude of the first IPSC (P = 0.184). It should be noted that 2 of the 7 pairs appeared to exhibit large IPSC amplitudes in the FS-P connections. E, CVs of the first IPSCs were also unaltered by NE application (P= 0.238). F, G, and H, Binned first IPSC amplitude in FS-P from baseline, NE and wash with Ringer’s solution, fitted with Gaussian function. The synaptic failures were unaltered as well (P > 0.05 for both). These data confirm the findings described in Figures 3 and 4, suggesting a differential regulation of excitatory and inhibitory transmission.
NE exhibited no significant effects on monosynaptic connections between layer V pyramidal neurons
In our previous study, we reported a selective dopamine modulation of P-P excitatory synapses but not of P-FS connections in the ferret mPFC (Gao and Goldman-Rakic, 2003). Thus, we tested the effects of bath-applied NE on the monosynaptic excitatory connections between layer V pyramidal neurons. We applied the same experimental protocol that was used with P-FS and FS-P. As shown in Figure 5A, we found that there was no significant change in EPSC amplitude in these P-P connections (n = 12, Fig. 5A–D). A sample pair is shown in Figure 5A; and the changes in the 1st EPSC amplitude in this pair are shown in a scatter plot in Figure 5B. Administration of NE had no clear effect on the dynamics of the 2nd to 10th EPSCs and on the amplitudes of the unitary EPSCs. Cumulative analysis showed no difference between values obtained at the baseline and those obtained after administration of NE (P > 0.05, K-S test, Fig. 5C). The peak amplitudes of the 1st EPSC were not significantly changed by administration of NE and during the washout (n = 12, P = 0.811 and P = 0.663, respectively when compared to baseline; F = 0.806, P = 0.458 with two-way ANOVA; Fig. 5D). Consistently, the CV of the 1st EPSC was also unaltered with NE application and during wash period compared with baseline (F = 1.585, P=0.226; Fig. 5E). Further analysis indicated similar distribution patterns in the number of events (5pA/bin with Gaussian fit) and the synaptic failure with NE administration as well as in washing compared with values obtained at baseline (F = 0.696, P = 0.508; Fig. 5F–H). There were also no effects on the PPR for the first two EPSCs (PPR = 0.55 ± 0.08 at baseline, 0.76 ± 0.14 with NE, 0.63 ± 0.09 with wash; baseline vs. NE, F = 1.314, P = 0.288), and on the recovery ratio (11th EPSC/1st EPSC, F = 0.870, P = 0.432). In addition, NE had no effects on the short-term plasticity when all 10 EPSCs at baseline and during NE were compared with ANOVA analysis (F = 0.487, P = 0.963). Overall, these results indicated that NE has no significant modulatory effect on the monosynaptic recurrent excitatory transmission between layer V pyramidal neurons. These results further indicated that the actions of NE on excitatory synapses are target-cell specific, and as such, the axonal terminals derived from a single pyramidal neuron could be modulated differently by NE depending upon the identity of the cellular synaptic target.
Figure 5.

NE has no significant effects on EPSC amplitude in P-P synapses. A, Representative traces showing the effect of NE on AMPA-mediated EPSCs in a layer V P-P pair in response to a 20-Hz 10+1 train applied at the presynaptic neuron. B, Scatter plot of the data showing that NE does not affect the first EPSC amplitude of the P-P pair in A. C, Cumulative probability plot for the first AMPA EPSCs indicating no clear effect on the curves (n = 12). D, NE has no effect on the average amplitude of the first EPSC (F = 0.806, P = 0.458). E, CVs of the first EPSCs were unaltered in P-P pairs (F = 1.585, P = 0.226). F, G and H, The amplitude histograms of binned first unitary EPSCs in P-P pairs from control, NE treatment and wash with Ringer’s solution with Gaussian fit and synaptic failures (black bars), which were not altered (F = 0.696, P = 0.508).
Alpha-1 agonist phenylephrine mimicked the depressive effects of NE on the P-FS connection but had no effect on P-P synapses
We next examined the receptor specificity associated with the regulation of synaptic transmission between pyramidal neurons and GABAergic interneurons. Because the profiles of the receptors on the axonal terminals and dendrites in pyramidal neurons and GABAergic interneurons are different (Aoki et al., 1994, Aoki et al., 1998, Nakadate et al., 2006), we first tested the effects of the selective alpha-1 agonist phenylephrine on the excitatory synapses in P-P and P-FS connection, respectively.
We found that the alpha-1 adrenergic agonist phenylephrine (25 μM) had different effects on excitatory synapses targeting pyramidal neurons and FS interneurons. As shown in Figure 6, phenylephrine effectively inhibited the EPSCs in the P-FS connections, similar to the outcome observed with NE. Phenylephrine significantly decreased the EPSCs in the P-FS connections (Fig. 6A, B), with a shifted curve in cumulative analysis (P < 0.05; Fig. 6C), a significantly decreased first EPSC amplitude (19.9 ± 4.85 pA at baseline, 10.3 ± 3.58 pA in phenylephrine, 9.36 ± 4.12 pA in wash; n = 7, F = 7.846, P = 0.007; Fig. 6D). The CV of the first EPSCs were significantly increased (0.56 ± 0.07 in baseline, 0.91 ± 0.17 in phenylephrine and 0.95 ± 0.14 in wash, F = 7.482, P = 0.008; Fig. 6E). We also noted significantly increased synaptic failure (F = 5.087, P = 0.025; Fig. 6F–H). With Gaussian fit, both the peak value of EPSC in phenylephrine and fitted area of the EPSC amplitude bin exhibited clear decreases compared with those of the baseline (Fig. 6F–H). In addition, phenylephrine also significantly increased the PPR (F = 6.043, P = 0.030) although the recovery ratio was unchanged (F = 0.0036, P = 0.996).
Figure 6.

The alpha-1 agonist phenylephrine significantly inhibits P-FS synapses. A and B, Phenylephrine significantly inhibited P-FS synapses, as shown in the sample connection (A) and the scatter plot (B). C, Cumulative probability of the first AMPA EPSC amplitudes exhibited a shifted curve (n= 7). D, Phenylephrine had significant depressive effects on the first EPSC amplitude (P = 0.03) and no recovery in wash (P = 0.031). E, The CVs of the first EPSCs were significantly increased by phenylephrine (P = 0.021) and in wash (P = 0.018). F, G and H, The amplitude histograms of the binned first EPSCs in the P-FS pairs at baseline, phenylephrine, and wash, fitted with Gaussian function. The failure rates were also significantly increased by phenylephrine (P = 0.049) and remained to be increased in wash (P = 0.012).
The suppressant effects of phenylephrine on the P-FS synapses were also evident in a triple P-FS connection in which axon terminals from two layer V pyramidal neurons innervated the same FS interneuron. Phenylephrine had similar depressive effects on the two P-FS connections. As shown in Figure 7A, the morphologies of the three recorded neurons were successfully recovered; the firing patterns of these cells are displayed in the inset. To record the EPSCs in this converging connection, the membrane potential of the FS interneuron was held at −70 mV. We first recorded the EPSCs in the P1-FS connection in the presence of phenylephrine. After washout, we recorded the EPSCs again in the P2-FS connection before and after application of phenylephrine. Both unitary EPSCs in P1-FS (Fig. 7B and C) and P2-FS pairs (Fig. 7D and E) were inhibited by 25 μM phenylephrine. These data provide additional evidence that the inhibitory effect of NE on unitary EPSCs in P-FS connections is mediated through activation of the alpha-1 receptor.
Figure 7.

Phenylephrine inhibits excitatory transmissions in a triple connection in which two pyramidal neurons innervated the same FS interneurons. A, Photomicrograph of the biocytin-labeled pyramidal neurons and FS interneuron. The firing patterns of these three neurons are shown in the inset of A. In this converging connection, the membrane potential of the FS interneuron was held at −70 mV, and the EPSCs in the P1-FS connection were first recorded in the presence of phenylephrine. After washout, the EPSCs in the P2-FS connection were subsequently recorded and phenylephrine was reapplied. B and C, The sample traces and scatter plot showing the AMPA EPSCs in the P1-FS connection, which was clearly inhibited by the alpha-1 agonist phenylephrine (25 μM). D and E, The sample traces and scatter plot of the AMPA EPSCs in the P2-FS connection that were similarly inhibited by phenylephrine. These data further verified the results shown in Figure 6.
In contrast, as expected, bath application of phenylephrine had no effect on the P-P connections. The peak amplitude of the first EPSCs were similar without significant changes during alpha-1 agonist administration (Fig. 8A, B). Cumulative analysis exhibited identical curves at baseline and with phenylephrine (Fig. 8C). Student paired t-test analysis displayed no significant changes in the amplitude and CV of the first EPSCs (n = 8, amplitude: F = 0.414, P = 0.669; CV: F = 0.310, P = 0.738; Fig. 8D and E), as well as in the PPR (F = 0.327, P = 0.727) or in the recovery ratio (F = 1.079, P = 0.367). Measures of Gaussian fit (fitted area 106, 97, and 94 for baseline, phenylephrine and wash), peak value of EPSC events for baseline, phenylephrine and wash was 6.16, 6.43, and 5.96, respectively. The synaptic failure rates in baseline, phenylephrine, and wash were 18.6 ± 4.24, 20.0 ± 5.23 and 18.9 ± 5.38, respectively, without significance (F = 0.164, P = 0.851; Fig. 8F–H).
Figure 8.

The alpha-1 agonist phenylephrine exhibits no significant effect in P-P synapses. A and B, Phenylephrine (25 μM) exhibited no significant effect on a P-P pair. C, Cumulative probability of the first AMPA EPSC amplitudes showed overlapped curves (n = 8). D, Effects of phenylephrine on the first EPSC amplitude were unaltered (P = 0.883). E, The CVs of the first EPSC amplitudes were not significant (P = 0.928). F, G, and H, Binned first EPSC amplitudes in P-P pairs in baseline, phenylephrine, and wash, fitted with Gaussian function. The failure rates were slightly increased but not significant in phenylephrine (P = 0.431) and continuously unaltered in wash (P > 0.05).
Alpha-1 antagonist prazosin prevented the depressive effects of NE on the P-FS connections
To further confirm the depressive effects of NE and of alpha-1 agonist phenylephrine on the P-FS connections, we applied the alpha-1 antagonist prazosin (1 μM) prior to the NE (10 μM) application. As shown in Figure 9, prazosin itself slightly decreased the EPSC amplitude, but the decrease was not significant (13.3 ± 2.79 pA in baseline vs. 10.1 ± 3.33 pA in prazosin, n = 7, P = 0.140; Fig. 9A–D). NE application induced slightly more decrease in EPSC amplitude but the change was not significant as well (10.1 ± 3.33 pA in prazosin vs. 8.41 ± 3.46 pA, P = 0.295). Overall evaluation with two-way ANOVA analysis also did not exhibit significance (F = 0.883, P = 0.439), indicating that prazosin prevented the depressive effects of NE on the P-FS connections (Fig. 9A–D). Indeed, cumulative analysis showed no significant curve shift between prazosin and prazosin + NE although the baseline curve was slightly right shifted (P > 0.05 for all; Fig. 9C). The CV of the first EPSC in prazosin and prazosin + NE also exhibited no significant changes compared with that of baseline (P = 0.769 and P = 0.085, respectively; Fig. 9E) although the CV in wash was significantly increased compared with prazosin (P = 0.04). Similarly, both prazosin and prazosin + NE did not change the PPR (P = 0.303 and P = 0.152, respectively) and the recovery ratio (P = 0.774 and P = 0.289, respectively) compared to the baseline. We also noted that the synaptic failures during prazosin and prazosin + NE administration were slightly increased but overall these changes were not significant (P = 0.570 and P=0.132 respectively when compared with baseline, and P = 0.147 between prazosin and prazosin + NE; two-way ANOVA F = 2.239, P = 0.149; Fig. 9F, G, H and I). Gaussian fit showed that the peak values of EPSC in prazosin and prazosin + NE shifted from 20 pA in baseline to 15 and 10 pA, respectively, but returned to 15 pA during washout.. These results further suggest that the depressive effects of NE on the P-FS synapses are mediated by alpha-1 receptors.
Figure 9.

The alpha 1 antagonist prazosin prevented the depressive effects of NE on P-FS synapses. A and B, Representative samples show the effects of prazosin and prazosin + NE on EPSC amplitude and synaptic failure. Prazosin (1 μM) itself slightly decreased the amplitude of EPSC and NE also exhibited slightly depressive effect on the EPSC amplitude when co-applied with prazosin. There was a trend of decreasing EPSC amplitudes in prazosin + NE and in wash but the number of synaptic failures did not show clear change. C, Cumulative probabilities of the first EPSC amplitudes in prazosin, prazosin + NE, and wash exhibited small shifts to the left but not significant compared to baseline (P > 0.05 for all). D, Prazosin and prazosin + NE slightly decreased the first EPSC amplitudes but not significant compared to baseline (n = 7, P > 0.05 for both). E, The CVs of the first EPSC amplitudes of prazosin and prazosin + NE were also slightly increased but not significant compared to baseline EPSC (P = 0.769 and P = 0.085, respectively). F, G, H and I, The first EPSC amplitudes in the P-FS pairs in baseline, prazosin, prazosin + NE, and wash period were binned and fitted with Gaussian function. The synaptic failures in prazosin and prazosin + NE were slightly increased during the drug application but overall not significant (P = 0.570 and P=0.132, respectively, compared with baseline and P = 0.147 between prazosin and prazosin + NE; two-way ANOVA F = 2.239, P = 0.149). Gaussian fit showed that the peak values of EPSC in prazosin and prazosin + NE shifted from 20 pA in baseline to 15 and 10 pA, respectively and returned to 15 pA in wash.
Discussion
We have characterized the actions of NE on individual monosynaptic connections between layer V pyramidal neurons and FS interneurons in the juvenile (PD20-23) rat PFC local circuitry. We found that NE selectively depresses excitatory synaptic transmission in P-FS connections but has no apparent effect on the excitatory synapses in the P-P connection and on the inhibitory synapses in the FS-P connections in this age group. These results indicate that the effects of NE are synapse-specific and that the actions of axon collaterals from single pyramidal neurons can be selectively regulated by NE at the level of individual synapses depending upon the identity of the postsynaptic neurons. In particular, NE exerts depressant actions on identified excitatory synapses that target GABAergic FS interneurons but has no significant effect on synapses onto pyramidal neurons at the concentration and age tested.
These results are novel and interesting with respect to NE’s actions in the PFC but are generally consistent with the results of previous studies that focused on the actions of dopamine (Urban et al., 2002, Gao and Goldman-Rakic, 2003) and acetylcholine (Xiang et al., 1998, Porter et al., 1999) in the cortex. Taken together, this work suggests that the effects of ascending modulatory systems on monosynaptic connections between pyramidal neurons and GABAergic interneurons are target cell- and synapse-specific (Toth and McBain, 2000, Bacci et al., 2005). In fact, target-specific expression of pre- and postsynaptic mechanisms of synaptic transmission has been shown in a variety of central neurons by a number of laboratories, as summarized in a previous review (Toth and McBain, 2000). Collectively, these data have demonstrated that synaptic transmission between single axons diverging onto distinct target neurons can behave independently, differentially influencing activity in the target neuron, as we reported in a previous study (Gao and Goldman-Rakic, 2003). This target specificity adds another level of complexity to un-ravel the roles played by individual neurons within a local neural circuits. Whether presynaptic adrenoceptors are different in the presynaptic axon terminals of pyramidal neurons that target different types of cells remains unknown. However, the effects of NE on the individual excitatory and inhibitory synapses in the prefrontal local circuitry are different from those of dopamine, as least at the concentration of 10 μM. For example, NE exhibited no significant effect on P-P connections whereas dopamine at the same dose range exerts a depressive effect on P-P connections via a presynaptic mechanism (Gao et al., 2001). In contrast, NE significantly suppressed excitatory synaptic transmission between P-FS cells whereas dopamine has no effect on these connections (Gao and Goldman-Rakic, 2003). In addition, NE exhibited no significant effects on GABAergic synapses between FS-P whereas dopamine differentially modulated perisomatic (FS-P) and peridendritic (non-FS-P) synapses in a cell-specific manner (Gao et al., 2003).
The second major finding from the present study is that the NE-mediated suppressant effect on excitatory transmission between pyramidal neurons and FS interneurons is mimicked by activation of alpha-1 receptors. Although alpha-1 receptors regulate both EPSCs (Law-Tho et al., 1993, Marek and Aghajanian, 1999, Chen et al., 2006, Kobayashi, 2007, Dinh et al., 2009, Kobayashi et al., 2009) and IPSCs (Bergles et al., 1996, Kawaguchi and Shindou, 1998, Han et al., 2002, Braga et al., 2004, Herold et al., 2005, Chen et al., 2006, Hirono and Obata, 2006, Lei et al., 2007, Salgado et al., 2011) in cortical pyramidal neurons, there have been no reports of alpha-1 receptor regulation of EPSCs on identified GABAergic interneurons.
The existing data on NE modulation of synaptic transmission from various brain regions are inconsistent or contradictory (Lei et al., 2007). For example, several studies emphasize the importance of alpha-2 receptors in cognitive functions (Li et al., 1999, Wang et al., 2007, Ji et al., 2008b), whereas others have found that alpha-1 or beta receptors mediate enhancement of glutamate or GABA transmission (Law-Tho et al., 1993, Bergles et al., 1996, Kawaguchi and Shindou, 1998, Marek and Aghajanian, 1999, Devilbiss and Waterhouse, 2000, Braga et al., 2004, Kobayashi, 2007, Lei et al., 2007, Kobayashi et al., 2009, Salgado et al., 2011). In fact, NE has a variety of effects at alpha- as well as beta- receptors in central neurons. Previous studies indicate that NE at 10–20 μM range consistently decreases AMPA-induced current (Dinh et al., 2009), AMPA-EPSC (Law-Tho et al., 1993, Kobayashi, 2007, Kobayashi et al., 2009), or NMDA-EPSC (Liu et al., 2006) via either postsynaptic activation of alpha-1 receptor in the PFC or presynaptic alpha-2 receptor in amygdala (Delaney et al., 2007), or even postsynaptic alpha-2 receptor in PFC (Ji et al., 2008b). Whatever the receptor-specificity and pre- or postsynaptic action, the consistent finding of a depressive effect of NE on excitatory synaptic transmission is contradictory to the insignificant effect of NE in P-P monosynaptic connections as observed in the current study. The reason for this discrepancy is not clear but it may attributable to different technical approaches. It is possible that we recorded from and stimulated synaptic connections from a specific subset of layer V pyramidal neurons using multiple whole-cell patch clamp whereas the extracellular mode of stimulation used in other studies may activate multiple excitatory afferents to layer V cells. Our results are in agreement with the idea that NE effects on cortical synapses depend on the cellular location of the synapse (Salgado et al., 2011), cell type (Kawaguchi and Shindou, 1998, Devilbiss and Waterhouse, 2000, Dembrow et al., 2010), or the specificity of the receptors (Devilbiss and Waterhouse, 2000). Indeed, alpha-1 receptors have been localized in cell soma and dendrites in the rat primary visual cortex (Nakadate et al., 2006) but have been identified as primarily presynaptic elements in the striatum and midbrain (Rommelfanger et al., 2009). The distribution of alpha-1 receptors on prefrontal neurons is not clear; but as our results suggested, it is likely that activation of alpha-1 receptors exhibits distinct effects on regulation of presynaptic glutamate release in the excitatory synapses targeting FS interneurons compared to those synapsing on pyramidal neurons, at least in the short age range (PD20-23) tested in this study.
Another possible explanation for these disparate findings is that the effects of NE are dose-dependent. NE at a concentration of 10 μM is sufficient to achieve plateau effects on excitatory synapses in the amygdala (Delaney et al., 2007) and near plateau effects in inhibitory synapses in the entorhinal cortex (EC50 = 4–5 μM) (Lei et al., 2007). However, there is general agreement that NE can modulate neuronal responses to synaptic inputs in an inverted “U” concentration-dependent manner (Berridge and Waterhouse, 2003, Aston-Jones and Cohen, 2005). Additional studies also suggest that moderate levels of NE improve PFC function, possibly via alpha-2a-adrenoceptors, whereas high levels of NE engage low affinity alpha-1 receptors (Arnsten et al., 1988, Tanila et al., 1996, Jakala et al., 1999, Li et al., 1999, Arnsten, 2000, Franowicz et al., 2002, Lapiz and Morilak, 2006, Wang et al., 2007). A recent study also reported that elevating noradrenergic activity at alpha-1 receptors in mPFC facilitates cognitive performance of rats in both sustained (Berridge et al., 2012) and flexible attention tasks, an observation which provides general support for the role of NE in behavioral state changes such as arousal and a plausible explanation for the beneficial cognitive effects of psychotherapeutic drugs that target noradrenergic neurotransmission (Lapiz and Morilak, 2006). It is possible and likely that NE at either lower or higher concentrations, or under condition of selective activation of specific receptor subtypes, also regulates P-P or FS-P synaptic connections. Indeed, at higher concentration of 100 μM NE appears to enhance EPSC by activation of the alpha 1 receptor in PFC neurons (Marek and Aghajanian, 1999). Similarly, activation of beta receptors consistently enhances EPSC in layer V pyramidal neurons of PFC by postsynaptic mechanisms via cAMP/PKA signaling (Kobayashi, 2007, Ji et al., 2008a) or presynaptic mechanism in PFC (Ji et al., 2008a, Kobayashi et al., 2009) or pyramidal neurons in CA1 (Gereau and Conn, 1994). Moreover, activation of alpha-2 receptors improves working memory function (Wang et al., 2007), possibly by increasing neuronal excitability via inhibition of cAMP-dependent HCN channels in the PFC (Carr et al., 2007). However, further study is needed to bridge the gap between behavioral outcomes, neuronal response properties, and actions at the level of individual synapse.
Our data also indicate that the effects of NE on the excitatory synapses at FS interneurons are long-lasting without recovery even after 15–20 min wash. This is in agreement with a previous study that, despite the transient (< 1 min) nature of the depolarizing effects on FS cells (Kawaguchi and Shindou, 1998), NE robustly decreased excitatory drive in GABAergic interneurons. These actions would be expected to reduce inhibition and increase excitability in the PFC circuitry, and consequently, such actions at the cellular level might improve attention and working memory function as previously reported (Lapiz and Morilak, 2006, Berridge et al., 2012). Our study thus provides a putative mechanism for fine-tuning of the flow of information in PFC. However, it should be noted that the selective effects of NE on P-FS implication at the concentration tested in this study should be interpreted cautiously because the recordings were performed in layer V cells of PFC only. Whether this unique finding can be applied to layer II/III cortical neurons, an area engaged in further higher cortico-cortical information processing, is unknown but could be different based on the results of a previous study (Lei et al., 2007). In fact, a previous study has shown that NE, in the same dose range as that used here, induces long-term depression (LTD) of glutamatergic synaptic transmission in layer I–II to layer V PFC pyramidal neurons (Marzo et al., 2010). Several others studies also showed that NE decreases the amplitude of AMPA-mediated glutamatergic responses (Dodt et al., 1991, Law-Tho et al., 1993, Pralong and Magistretti, 1994, Hasselmo et al., 1997, Dinh et al., 2009). At first glance, our result seems to be in contrast to the depressive effect of NE on layer I–II to layer V PFC pyramidal neurons (Marzo et al., 2010) and on excitatory cortical synaptic transmission between pyramidal neurons in general. In fact, our finding is in agreement with Marzo et al’s report (Marzo et al., 2010), in which NE at the concentration that induced LTD in layer I–II to layer V pyramidal neuron synapses did not induce clear LTD in layer VI to layer V pyramidal neuron synapses. Therefore, it is likely that distribution of adrenergic receptors in the cerebral cortex may exhibit laminar or afferent specificity. Indeed, as Marzo et al explained (Marzo et al., 2010), the deep layer afferents contain a large number of projection fibers from other brain regions, for example, the hippocampus (Jay and Witter, 1991) and the mediodorsal nucleus of thalamus (Giguere and Goldman-Rakic, 1988). The lack of clear NE effects in the layer V synapses indicates a possible difference of the sensitivity to NE between the upper-layer cortico-cortical inputs and the deep-layer subcortical projecting inputs. This difference might be attributable to a relatively less pronounced distribution of alpha2-adrenoceptors in deep layers (Aoki et al., 1998). Apparently, additional studies are needed to account for cellular mechanisms underlying this disparity between sub-populations of PFC synapses.
Another point that should be also considered is the existence of a variety of GABAergic neurons in the neocortex (Kawaguchi, 1995, Galarreta and Hestrin, 1999, Gibson et al., 1999, Gao et al., 2003, Wang and Gao, 2009). In this study, we only tested the effects of NE on FS interneurons. NE actions on other GABAergic cell types, such as low-threshold spiking and regular spiking interneurons, might be different as their receptor distributions are distinct from FS interneurons (Wang and Gao, 2009). Furthermore, medial PFC is one of a few cortical area receiving both dopaminergic and noradrenergic inputs compared to other neocortical areas. Thus, the observed findings may be due to strong dual inputs compared to other cortical areas such as visual, auditory and somatosensory cortex which receive rather limited or no direct dopaminergic influence. This could partially explain the different or contradictory results obtained from various brain regions as described above. Finally, our recordings were conducted in prefrontal neurons of normal young animals; whether NE exhibits similar or different effects on adult neurons or on cells recorded from brains that model psychiatric disorders such as the spontaneously hypertensive rat model of attention deficit hyperactivity disorder (Sagvolden et al., 2005a, Sagvolden et al., 2005b) is another intriguing question that needs further exploration. Therefore, the overall actions of NE in vivo may be very different from what we have discussed here.
Nevertheless, our data have provided novel evidence that synaptic connections between individual neurons in the local prefrontal circuitry are differentially regulated by noradrenergic neurotransmission and these effects are complementary to the actions of dopamine, at least in juvenile animals. It is known that NE systems are required for normal baseline operations of the PFC. For example, selective impairment of NE transmission in the PFC disrupts working memory (Arnsten et al., 1999, Friedman et al., 1999, Arnsten, 2004). Other studies indicate that complementary levels of catecholamine receptor stimulation are needed to optimize PFC cognitive function (Arnsten and Li, 2005, Pascucci et al., 2007). In the PFC of behaving monkeys, Sawaguchi et al showed that NE strongly decreased the overall background neural activity, particularly in the pre-cue period of a working memory task, thus increasing the signal-to-noise ratio of the delay response (Sawaguchi et al., 1990). In addition, this study reported differential effects of NE and dopamine on prefrontal neural activity related to the delay response task. The differential nature of dopamine and NE effects on different synapses in vitro, as we reported here, is in support of this in vivo finding. The results of the present study thus underscore the heterogeneous and, perhaps, complementary nature of catecholamine transmitter/modulator actions in PFC functions. Dopamine and NE are capable of jointly affecting individual synapses in local PFC circuits and, consequently, influencing network activity and behavioral outcomes mediated by the PFC (Arnsten et al., 2010). A major challenge for future investigation is to account for all of the cellular synaptic actions of NE and dopamine in the context of dynamic neural circuit operations with the goal of developing a comprehensive understanding of the cellular basis of PFC function in adaptive and maladaptive behaviors.
Highlights.
The actions of NE on monosynaptic connections in the PFC circuitry were characterized.
NE selectively depresses excitatory synaptic transmission in P-FS connections.
NE has no detectable effect on P-P and FS-P connections.
NE apparently exerts distinctly different modulatory actions on identified synapses.
Depending on the postsynaptic targets, the effects of NE in PFC are synapse-specific.
Acknowledgments
We thank Dr. Melissa A. Snyder at Drexel University and Dr. Rick C. S. Lin at the University of Mississippi Medical Center for comments on the manuscript and Ms. Pamela Fried from DUCOM Academic Publishing Services for editorial assistance. This work was supported by a Translational Grant (HCEP) from Drexel University, a CURE grant from Drexel University College of Medicine, NARSAD young investigator award, NIMH R21 MH232307, and R01MH232395 to W-J Gao; and a Translational Grant from Drexel University, Pennsylvania Department of Health, and NIDA DA017960 to B.D. Waterhouse.
Abbreviations
- AHP
afterhyperpolarization
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CVs
coefficients of variation
- EPSC
excitatory postsynaptic current
- FS
fast-spiking
- GABA
gamma-aminobutyric acid
- IPSC
inhibitory postsynaptic current
- K-S
Kolmogorov-Smirnov
- NE
norepinephrine
- P
pyramidal neuron
- PFC
prefrontal cortex
- PPR
paired-pulse ratio
Footnotes
Conflict of interest
The authors claim no conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki C, Go CG, Venkatesan C, Kurose H. Perikaryal and synaptic localization of alpha 2A-adrenergic receptor-like immunoreactivity. Brain Res. 1994;650:181–204. doi: 10.1016/0006-8993(94)91782-5. [DOI] [PubMed] [Google Scholar]
- Aoki C, Venkatesan C, Go CG, Forman R, Kurose H. Cellular and subcellular sites for noradrenergic action in the monkey dorsolateral prefrontal cortex as revealed by the immunocytochemical localization of noradrenergic receptors and axons. Cereb Cortex. 1998;8:269–277. doi: 10.1093/cercor/8.3.269. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Stress impairs prefrontal cortical function in rats and monkeys: role of dopamine D1 and norepinephrine alpha-1 receptor mechanisms. Prog Brain Res. 2000;126:183–192. doi: 10.1016/S0079-6123(00)26014-7. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Adrenergic targets for the treatment of cognitive deficits in schizophrenia. Psychopharmacology (Berl) 2004;174:25–31. doi: 10.1007/s00213-003-1724-3. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Cai JX, Goldman-Rakic PS. The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for alpha-2 receptor subtypes. J Neurosci. 1988;8:4287–4298. doi: 10.1523/JNEUROSCI.08-11-04287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Li BM. Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry. 2005;57:1377–1384. doi: 10.1016/j.biopsych.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Mathew R, Ubriani R, Taylor JR, Li BM. Alpha-1 noradrenergic receptor stimulation impairs prefrontal cortical cognitive function. Biol Psychiatry. 1999;45:26–31. doi: 10.1016/s0006-3223(98)00296-0. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Paspalas CD, Gamo NJ, Yang Y, Wang M. Dynamic Network Connectivity: A new form of neuroplasticity. Trends Cogn Sci. 2010 doi: 10.1016/j.tics.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA. Modulation of neocortical interneurons: extrinsic influences and exercises in self-control. Trends in Neurosci. 2005;28:602–610. doi: 10.1016/j.tins.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Huguenard JR, Prince DA. Adrenergic modulation of GABAA receptor-mediated inhibition in rat sensorimotor cortex. J Neurophysiol. 1998;79:937–946. doi: 10.1152/jn.1998.79.2.937. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci. 1996;16:572–585. doi: 10.1523/JNEUROSCI.16-02-00572.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AF, Kelley AE, Schmeichel B, Hamilton C, Spencer RC. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry. 2006;60:1111–1120. doi: 10.1016/j.biopsych.2006.04.022. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Shumsky JS, Andrzejewski ME, McGaughy JA, Spencer RC, Devilbiss DM, Waterhouse BD. Differential sensitivity to psychostimulants across prefrontal cognitive tasks: differential involvement of noradrenergic alpha -1 and alpha-2 receptors. Biol Psychiatry. 2012;71:467–473. doi: 10.1016/j.biopsych.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Research Reviews. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. Stress impairs alpha(1A) adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology. 2004;29:45–58. doi: 10.1038/sj.npp.1300297. [DOI] [PubMed] [Google Scholar]
- Carr DB, Andrews GD, Glen WB, Lavin A. alpha2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J Physiol. 2007;584:437–450. doi: 10.1113/jphysiol.2007.141671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Li DP, Pan HL. Presynaptic alpha1 adrenergic receptors differentially regulate synaptic glutamate and GABA release to hypothalamic presympathetic neurons. J Pharmacol Exp Ther. 2006;316:733–742. doi: 10.1124/jpet.105.094797. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline Modulates Transmission at a Central Synapse by a Presynaptic Mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D. Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci. 2010;30:16922–16937. doi: 10.1523/JNEUROSCI.3644-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilbiss DM, Waterhouse BD. Norepinephrine exhibits two distinct profiles of action on sensory cortical neuron responses to excitatory synaptic stimuli. Synapse. 2000;37:273–282. doi: 10.1002/1098-2396(20000915)37:4<273::AID-SYN4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Dinh L, Nguyen T, Salgado H, Atzori M. Norepinephrine homogeneously inhibits alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate- (AMPAR-) mediated currents in all layers of the temporal cortex of the rat. Neurochem Res. 2009;34:1896–1906. doi: 10.1007/s11064-009-9966-z. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Pawelzik H, Zieglgansberger W. Actions of noradrenaline on neocortical neurons in vitro. Brain Res. 1991;545:307–311. doi: 10.1016/0006-8993(91)91303-i. [DOI] [PubMed] [Google Scholar]
- Elliott JM, Beveridge TJ. Psychostimulants and monoamine transporters: upsetting the balance. Curr Opin Pharmacol. 2005;5:94–100. doi: 10.1016/j.coph.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Franowicz JS, Kessler LE, Borja CM, Kobilka BK, Limbird LE, Arnsten AF. Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci. 2002;22:8771–8777. doi: 10.1523/JNEUROSCI.22-19-08771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JI, Stewart DG, Gorman JM. Potential noradrenergic targets for cognitive enhancement in schizophrenia. CNS Spectr. 2004;9:350–355. doi: 10.1017/s1092852900009330. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Temporini H, Davis KL. Pharmacologic strategies for augmenting cognitive performance in schizophrenia. Biol Psychiatry. 1999;45:1–16. doi: 10.1016/s0006-3223(98)00287-x. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature. 1999;402:72–75. doi: 10.1038/47029. [DOI] [PubMed] [Google Scholar]
- Gao WJ. Acute clozapine suppresses synchronized pyramidal synaptic network activity by increasing inhibition in the ferret prefrontal cortex. J Neurophysiol. 2007;97:1196–1208. doi: 10.1152/jn.00400.2006. [DOI] [PubMed] [Google Scholar]
- Gao WJ, Goldman-Rakic PS. Selective modulation of excitatory and inhibitory microcircuits by dopamine. Proc Natl Acad Sci U S A. 2003;100:2836–2841. doi: 10.1073/pnas.262796399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J Neurosci. 2003;23:1622–1630. doi: 10.1523/JNEUROSCI.23-05-01622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RWt, Conn PJ. Presynaptic enhancement of excitatory synaptic transmission by beta-adrenergic receptor activation. J Neurophysiol. 1994;72:1438–1442. doi: 10.1152/jn.1994.72.3.1438. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Giguere M, Goldman-Rakic PS. Mediodorsal nucleus: areal, laminar, and tangential distribution of afferents and efferents in the frontal lobe of rhesus monkeys. J Comp Neurol. 1988;277:195–213. doi: 10.1002/cne.902770204. [DOI] [PubMed] [Google Scholar]
- Han SK, Chong W, Li LH, Lee IS, Murase K, Ryu PD. Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J Neurophysiol. 2002;87:2287–2296. doi: 10.1152/jn.2002.87.5.2287. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Linster C, Patil M, Ma D, Cekic M. Noradrenergic suppression of synaptic transmission may influence cortical signal-to-noise ratio. J Neurophysiol. 1997;77:3326–3339. doi: 10.1152/jn.1997.77.6.3326. [DOI] [PubMed] [Google Scholar]
- Herold S, Hecker C, Deitmer JW, Brockhaus J. alpha1-Adrenergic modulation of synaptic input to Purkinje neurons in rat cerebellar brain slices. J Neurosci Res. 2005;82:571–579. doi: 10.1002/jnr.20660. [DOI] [PubMed] [Google Scholar]
- Hirono M, Obata K. Alpha-adrenoceptive dual modulation of inhibitory GABAergic inputs to Purkinje cells in the mouse cerebellum. J Neurophysiol. 2006;95:700–708. doi: 10.1152/jn.00711.2005. [DOI] [PubMed] [Google Scholar]
- Jakala P, Sirvio J, Riekkinen M, Koivisto E, Kejonen K, Vanhanen M, Riekkinen P., Jr Guanfacine and clonidine, alpha 2-agonists, improve paired associates learning, but not delayed matching to sample, in humans. Neuropsychopharmacology. 1999;20:119–130. doi: 10.1016/S0893-133X(98)00055-4. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Ji X-H, Cao X-H, Zhang C-L, Feng Z-J, Zhang X-H, Ma L, Li B-M. Pre- and postsynaptic {beta}-adrenergic activation enhances excitatory synaptic transmission in layer V/VI pyramidal neurons of the medial prefrontal cortex of rats. Cereb Cortex. 2008a;18:1506–1520. doi: 10.1093/cercor/bhm177. [DOI] [PubMed] [Google Scholar]
- Ji XH, Ji JZ, Zhang H, Li BM. Stimulation of alpha2-adrenoceptors suppresses excitatory synaptic transmission in the medial prefrontal cortex of rat. Neuropsychopharmacology. 2008b;33:2263–2271. doi: 10.1038/sj.npp.1301603. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J Neurosci. 1995;15:2638–2655. doi: 10.1523/JNEUROSCI.15-04-02638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. J Neurosci. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M. Differential regulation of synaptic transmission by adrenergic agonists via protein kinase A and protein kinase C in layer V pyramidal neurons of rat cerebral cortex. Neuroscience. 2007;146:1772–1784. doi: 10.1016/j.neuroscience.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kojima M, Koyanagi Y, Adachi K, Imamura K, Koshikawa N. Presynaptic and postsynaptic modulation of glutamatergic synaptic transmission by activation of alpha(1)- and beta-adrenoceptors in layer V pyramidal neurons of rat cerebral cortex. Synapse. 2009;63:269–281. doi: 10.1002/syn.20604. [DOI] [PubMed] [Google Scholar]
- Koyanagi Y, Yamamoto K, Oi Y, Koshikawa N, Kobayashi M. Presynaptic interneuron subtype- and age-dependent modulation of GABAergic synaptic transmission by beta-adrenoceptors in rat insular cortex. J Neurophysiol. 2010;103:2876–2888. doi: 10.1152/jn.00972.2009. [DOI] [PubMed] [Google Scholar]
- Lapiz MD, Morilak DA. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137:1039–1049. doi: 10.1016/j.neuroscience.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Law-Tho D, Crepel F, Hirsch JC. Noradrenaline decreases transmission of NMDA- and non-NMDA-receptor mediated monosynaptic EPSPs in rat prefrontal neurons in vitro. Eur J Neurosci. 1993;5:1494–1500. doi: 10.1111/j.1460-9568.1993.tb00217.x. [DOI] [PubMed] [Google Scholar]
- Lei S, Deng P-Y, Porter JE, Shin H-S. Adrenergic facilitation of GABAergic transmission in rat entorhinal cortex. J Neurophysiol. 2007;98:2868–2877. doi: 10.1152/jn.00679.2007. [DOI] [PubMed] [Google Scholar]
- Li BM, Mao ZM, Wang M, Mei ZT. Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropsychopharmacology. 1999;21:601–610. doi: 10.1016/S0893-133X(99)00070-6. [DOI] [PubMed] [Google Scholar]
- Liu W, Yuen EY, Allen PB, Feng J, Greengard P, Yan Z. Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proc Natl Acad Sci U S A. 2006;103:18338–18343. doi: 10.1073/pnas.0604560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Aghajanian GK. 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol. 1999;367:197–206. doi: 10.1016/s0014-2999(98)00945-5. [DOI] [PubMed] [Google Scholar]
- Marzo A, Bai J, Caboche J, Vanhoutte P, Otani S. Cellular mechanisms of long-term depression induced by noradrenaline in rat prefrontal neurons. Neuroscience. 2010;169:74–86. doi: 10.1016/j.neuroscience.2010.04.046. [DOI] [PubMed] [Google Scholar]
- Milstein JA, Lehmann O, Theobald DE, Dalley JW, Robbins TW. Selective depletion of cortical noradrenaline by anti-dopamine beta-hydroxylase-saporin impairs attentional function and enhances the effects of guanfacine in the rat. Psychopharmacology (Berl) 2007;190:51–63. doi: 10.1007/s00213-006-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner LH, Jedema HP, Moore FW, Blakely RD, Grace AA, Sesack SR. Chronic stress increases the plasmalemmal distribution of the norepinephrine transporter and the coexpression of tyrosine hydroxylase in norepinephrine axons in the prefrontal cortex. J Neurosci. 2006;26:1571–1578. doi: 10.1523/JNEUROSCI.4450-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO. Role of brain norepinephrine in the behavioral response to stress. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2005;29:1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Nakadate K, Imamura K, Watanabe Y. Cellular and subcellular localization of alpha-1 adrenoceptors in the rat visual cortex. Neuroscience. 2006;141:1783–1792. doi: 10.1016/j.neuroscience.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Newman LA, Darling J, McGaughy J. Atomoxetine reverses attentional deficits produced by noradrenergic deafferentation of medial prefrontal cortex. Psychopharmacology (Berl) 2008;200:39–50. doi: 10.1007/s00213-008-1097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascucci T, Ventura R, Latagliata EC, Cabib S, Puglisi-Allegra S. The medial prefrontal cortex determines the accumbens dopamine response to stress through the opposing influences of norepinephrine and dopamine. Cereb Cortex. 2007;17:2796–2804. doi: 10.1093/cercor/bhm008. [DOI] [PubMed] [Google Scholar]
- Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999;19:5228–5235. doi: 10.1523/JNEUROSCI.19-13-05228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong E, Magistretti PJ. Noradrenaline reduces synaptic responses in normal and tottering mouse entorhinal cortex via alpha 2 receptors. Neurosci Lett. 1994;179:145–148. doi: 10.1016/0304-3940(94)90955-5. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Mitrano DA, Smith Y, Weinshenker D. Light and electron microscopic localization of alpha-1 adrenergic receptor immunoreactivity in the rat striatum and ventral midbrain. Neuroscience. 2009;158:1530–1540. doi: 10.1016/j.neuroscience.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagvolden T, Johansen EB, Aase H, Russell VA. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behav Brain Sci. 2005a;28:397–419. doi: 10.1017/S0140525X05000075. discussion 419-368. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Russell VA, Aase H, Johansen EB, Farshbaf M. Rodent models of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005b;57:1239–1247. doi: 10.1016/j.biopsych.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Salgado H, Garcia-Oscos F, Patel A, Martinolich L, Nichols JA, Dinh L, Roychowdhury S, Tseng KY, Atzori M. Layer-specific noradrenergic modulation of inhibition in cortical layer II/III. Cereb Cortex. 2011;21:212–221. doi: 10.1093/cercor/bhq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K. Catecholaminergic effects on neuronal activity related to a delayed response task in monkey prefrontal cortex. J Neurophysiol. 1990;63:1385–1400. doi: 10.1152/jn.1990.63.6.1385. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Sessler FM, Liu W, Kirifides ML, Mouradian RD, Lin RC, Waterhouse BD. Noradrenergic enhancement of GABA-induced input resistance changes in layer V regular spiking pyramidal neurons of rat somatosensory cortex. Brain Res. 1995;675:171–182. doi: 10.1016/0006-8993(95)00060-4. [DOI] [PubMed] [Google Scholar]
- Tanila H, Rama P, Carlson S. The effects of prefrontal intracortical microinjections of an alpha-2 agonist, alpha-2 antagonist and lidocaine on the delayed alternation performance of aged rats. Brain Res Bull. 1996;40:117–119. doi: 10.1016/0361-9230(96)00026-3. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Toth K, McBain CJ. Target-specific expression of pre- and postsynaptic mechanisms. J Physiol. 2000;525(Pt 1):41–51. doi: 10.1111/j.1469-7793.2000.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. PNAS. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NN, Gonzalez-Burgos G, Henze DA, Lewis DA, Barrionuevo G. Selective reduction by dopamine of excitatory synaptic inputs to pyramidal neurons in primate prefrontal cortex. J Physiol. 2002;539:707–712. doi: 10.1113/jphysiol.2001.015024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viggiano D, Ruocco LA, Arcieri S, Sadile AG. Involvement of norepinephrine in the control of activity and attentive processes in animal models of attention deficit hyperactivity disorder. Neural Plast. 2004;11:133–149. doi: 10.1155/NP.2004.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacol. 2009;34:2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J Physiol. 2010;588:2823–2838. doi: 10.1113/jphysiol.2010.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AFT. alpha-2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Waterhouse BD, Mouradian R, Sessler FM, Lin RC. Differential modulatory effects of norepinephrine on synaptically driven responses of layer V barrel field cortical neurons. Brain Res. 2000;868:39–47. doi: 10.1016/s0006-8993(00)02261-7. [DOI] [PubMed] [Google Scholar]
- Wojtowicz AM, Fidzinski P, Heinemann U, Behr J. Beta-adrenergic receptor activation induces long-lasting potentiation in burst-spiking but not regular-spiking cells at CA1-subiculum synapses. Neuroscience. 2010;171:367–372. doi: 10.1016/j.neuroscience.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. GABAA receptor-mediated currents in interneurons and pyramidal cells of rat visual cortex. J Physiol. 1998;506 ( Pt 3):715–730. doi: 10.1111/j.1469-7793.1998.715bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]