

Abstract
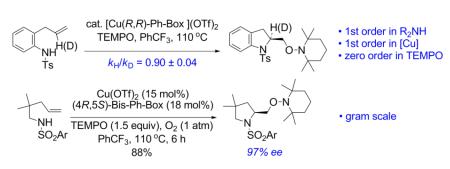
The catalytic asymmetric aminooxygenation of alkenes provides an efficient and straightforward approach to prepare chiral vicinal amino alcohols. We have reported a copper(II)-catalyzed enantioselective intramolecular alkene aminooxygenation, using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) as the oxygen source, which results in the synthesis of chiral indolines and pyrrolidines. Herein we disclose that kinetics studies indicate the reaction is first order both in substrate and the [Cu(R,R)-Ph-bis(oxazoline)]OTf2 catalyst, and zero order in TEMPO. Furthermore, kinetic isotope effect studies support that the cis aminocupration step, the addition of N-Cu across the alkene, is the rate-limiting step. Subsequent formation of a carbon radical intermediate, and direct carbon radical trapping with TEMPO is the indicated mechanism for the C-O bond formation as suggested by a deuterium labeling experiment. A ligand screen revealed that C(4)-phenyl substitution on the bis(oxazoline) is optimal for high asymmetric induction. The size of the substrate’s N-sulfonyl group also influences the enantioselectivity of the reaction. The preparative scale catalytic aminooxygenation reaction (gram scale) was demonstrated and an unexpected dependence on reaction temperature was uncovered on the larger scale reaction.
INTRODUCTION
The catalytic asymmetric aminooxygenation of alkenes provides straightforward access to chiral vicinal amino alcohols, which are valuable intermediates used in the synthesis of catalysts and ligands as well as biologically active molecules.1 Sharpless and co-workers reported the first catalytic enantioselective intermolecular aminohydroxylation, catalyzed by osmium, which has proven to be an extremely useful method, as exemplified by its use in numerous syntheses of biologically active compounds.2 More recently, Yoon and co-workers have developed copper3 and iron4 catalyzed protocols for the enantioselective intermolecular aminooxygenation of styrenes and dienes as less expensive and less toxic transition metal catalyst alternatives. Enantioselective aminooxygenation of α,β-unsaturated aldehydes catalyzed by chiral pyrrolidines have also been reported recently.5 The development of catalytic enantioselective intramolecular alkene aminooxygenation protocols, however, has been more rare.
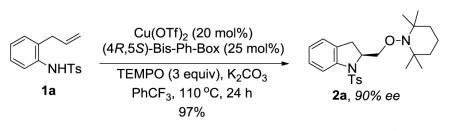
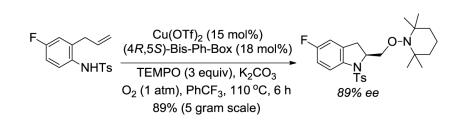
A number of regio- and diastereoselective intramolecular transition metal6-11 and non-metal promoted12 olefin aminooxygenation reactions have been reported over the years. In 2008, our group reported the first catalytic enantioselective intramolecular alkene aminooxygenation.10a,b Our method makes use of (2,2,6,6-tetramethylpiperdin-1-yl)oxyl (TEMPO) as both the oxidant and oxygen source. A preliminary report of a new enantioselective copper-catalyzed reaction that uses PhI(OAc)2 as both the stoichiometric oxidant and oxygen source has since appeared.11b A stoichiometric chiral hypervalent iodine-promoted aminooxygenation reaction has also been reported.12g Our copper-catalyzed aminooxygenation reaction provides direct access to 2-(hydroxymethyl) indoline and pyrrolidine derivatives with very good levels of enantioselectivity (Eqs 1 and 2). We have subsequently demonstrated that the chiral indoline-forming reaction could be further optimized for catalyst loading and conducted on multi-gram scale (Eq 3).10b We found, however, that we were not immediately able to scale-up the pyrrolidine-forming reaction (Eq 2) using our published method10a and the lessons we had learned in the indoline synthesis scale-up study (Eq 3).10b Herein we report our successful optimization and scale-up of the chiral pyrrolidine-forming aminooxygenation reaction, which draws on knowledge we gleaned during a detailed mechanistic analysis of the enantioselective reaction. We report herein a mechanistic analysis of the enantioselective reaction using kinetics, kinetic isotope effects and isotopic labeling experiments.
 |
(1) |
 |
(2) |
 |
(3) |
RESULTS AND DISCUSSION
Experimental Mechanistic Study Reaction Kinetics
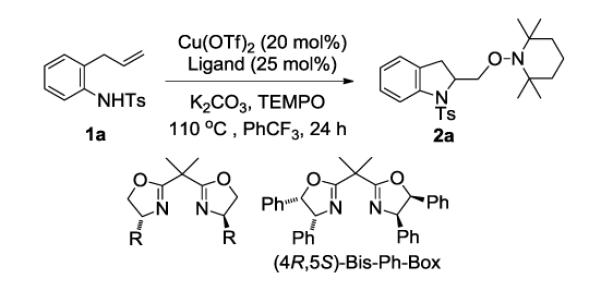
Our previously reported copper(II)-catalyzed enantioselective intramolecular alkene aminooxygenation conditions involved use of a slight excess of the (4R,5S)-Bis-Ph-Box ligand and K2CO3 as base (Table 1, entry 1).10a We reexamined the reaction conditions to identify an optimal protocol for reaction kinetics. Due to the low solubility of K2CO3 in CF3Ph, we needed to find an alternative base to enable optimal reaction kinetics studies. We also reexamined use of the commercially available (R,R)-Ph-Box ligand for further protocol simplification. Gratifyingly, we found that the use of the (R,R)-Ph-Box ligand gave us comparable yield and enantioselectivity (Table 1, entry 2). In screening soluble bases, we found that use of NBu4OAc gave high yield but no enantioselectivity (<5%, Table 1, entry 3). This base was previously used in our study of the reaction kinetics of the copper(II) 2-ethylhexanoate promoted intramolecular alkene aminooxygenation reaction.13 Use of 2,6-di-t-butyl-4-methyl-pyridine as base provided the indoline 2a in 87% ee and 75% yield (Table 1, entry 4). We next ran the reaction in the absence of additional base and found that sulfonamide 1a cyclized efficiently to form 2a in 87% yield and 88% ee (entry 5). We also found that the reaction proceeds efficiently without using a slight excess of the (R,R)-Ph-Box ligand, relative to Cu(OTf)2 (Table 1, entry 6). We hypothesized that [(R,R)-Ph-Box)Cu](OTf)2 is the active catalyst species in the aminooxygenation reaction, thus in order to keep an accurate 1:1 ratio between the Cu and the (R,R)-Ph-Box ligand we ran the kinetics experiments using the reaction conditions in Table 1, entry 6. Small-scale reactions with 2-allylaniline derived substrates such as 1a [<70 mg (ca. 0.240 mmol) 1a] do not require the use of O2 for copper turnover; the soluble TEMPO radical serves both as copper oxidant and oxygen atom source (vide infra). This simplified our kinetics study by reducing a reagent (O2) to monitor. On larger scale and with less reactive substrates, O2 (1 atm), is necessary for copper turnover as the reactions do not go to completion without it.10a,b
Table 1.
Conditions for Reaction Kinetics Optimizationa
| Entry | Ligand | Base | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | (4R,5S)-Bis-Ph-Box | K2CO3 | 97 | 90 |
| 2 | (R,R)-Ph-Box | K2CO3 | 90 | 88 |
| 3 | (R,R)-Ph-Box | NBu4OAc | 98 | <5 |
| 4 | (R,R)-Ph-Box | 2,6-di-t-butyl-4-methyl- pyridine |
75 | 87 |
| 5 | (R,R)-Ph-Box | - | 87 | 88 |
| 6d | (R,R)-Ph-Box | - | 88 | 87 |
Cu(OTf)2 (20 mol%), (R,R)-Ph-Box (25 mol%) and PhCF3 (0.07 M w/r to 1a) were combined i a pressure tube and heated to 60 °C for 2 h. Substrate 1a (1 equiv, 0.139 mmol), TEMPO (3 equiv), K2CO3 (1 equiv) were added. The reaction mixture was heated to 110 °C for 24 h.
Yield (%) refers to amount of isolated 2a after purification by flash chromatography on SiO2.
Enantiomeric excesses were determined by chiral HPLC analysis.
The reaction was run using 20 mol% of (R,R)-Ph-Box. OTf = trifluoromethanesulfonyl.
Kinetic Order of the Reaction
We determined the kinetic order of the aminooxygenation reaction by monitoring the disappearance of substrate 1a using high performance liquid chromatography (see Experimental Section for details). The kinetic order of substrate 1a was established under pseudo first-order conditions (excess TEMPO). Loss of 1a showed a linear decrease of the natural logarithm of its concentration with time, indicating first-order rate dependence (Figure 1).
Figure 1.

A plot of ln[1a] (mM) versus time (min) showing first-order kinetics in substrate 1a
After we established the order in substrate 1a, we determined the dependence in the catalyst (Cu-L) and TEMPO concentration. We obtained the observed rate constant, kobs from plots of ln[1a] against time at varying catalyst (2.5-20.4 mM) and TEMPO (150 and 300 mM) concentrations. We found that at constant Cu-L concentration, no significant change in kobs was observed when the TEMPO concentration was doubled (see Experimental Section for the kinetic data). Thus, we concluded that the aminooxygenation reaction has zero order kinetic dependence in TEMPO.
To determine the order in catalyst, we plotted ln(kobs) against ln[Cu-L] (Figure 2). The plot gave a linear relationship with slope equal to the order of the catalyst, which is found to be 0.99±0.02, consistent with first-order kinetic behavior. In these experiments, reactions were run up to 80-90% conversion for higher catalyst loadings (20-40 mol % or 10-20 mM). We observed that at low catalyst loadings (5-10 mol % or 2.5-5 mM), the reaction tails off at 40-60% conversion, presumably due to catalyst decomposition, therefore, initial reaction rates were measured for these catalyst loadings (see Supporting Information for details).
Figure 2.

Plot of ln(kobs) against ln[Cu-L] showing first-order dependence on the catalyst concentration. The order in Cu-L was obtained from the slope of the plot, 0.99±0.02.
On the basis of the observed rate law (−d[1a]/dt = kobs [1a][Cu-L]), we proposed the catalytic cycle shown in Scheme 1. The [(R,R)-Ph-Box)Cu](OTf)2 complex is the proposed active catalyst species which coordinates with substrate 1a to form the nitrogen-copper(II) intermediate 3. The existence of an analogous R2N-Cu(II) intermediate has been previously supported by Electron Paramagnetic Resonance spectroscopy (EPR)13a and is reasonably invoked in this reaction mechanism as a heteroatom such as R2NH should readily displace a triflate anion from a copper(II) complexe. It is noteworthy that the TEMPO anion [the result of the re-oxidation of copper(I)] or trace TEMPOH14 from excess TEMPO can also serve as a base to sequester the triflic acid formed during substrate-catalyst complexation (hence the realization that K2CO3 is not always necessary). This complex then undergoes cis aminocupration via transition state A to give rise to the alkyl organocopper(II) species 4. In the proposed transition state A, the substrate’s N-substituent is trans to the nearest phenyl group of the oxazoline. This proposed transition state is consistent with the observed first-order dependence on both substrate 1a and catalyst. The cis aminocupration mechanism for this catalytic reaction is further invoked in analogy to that of the copper(II)-carboxylate promoted aminooxygenation that gives similar diastereoselectivity trends in 2,5-disubstituted pyrrolidine-forming reactions.10c,d,13 The alkyl organocopper(II) species 4 readily homolyzes to provide the carbon radical intermediate 5, giving off copper(I). The carbon radical species is quickly trapped by TEMPO to form the enantiomerically enriched aminooxygenation product 2a. Copper(I) is then reoxidized by TEMPO to form the reactive copper(II) species and complete the catalytic cycle.
Scheme 1.

Proposed mechanism for the copper(II)-catalyzed enantioselective intramolecular aminooxygenation of alkenes
Kinetic Isotope Effects (KIE)
For the rate-limiting step analysis, we investigated primary and secondary kinetic isotope effects (kH/kD). We determined the primary kinetic isotope effect by treating substrates 1a and 1a-1-[D] with our catalytic reaction conditions and comparing the rate constants of each reaction (see Supporting Information for details). The primary kinetic isotope effect that was observed (kH/kD = 0.96±0.2) is not significant compared to the maximum theoretical KIE of 8.5 based on vNH of 3100 cm−1 (25 C) (Eqs 4 and 5).15,16 This result implies that the amine deprotonation in the conversion of 1a to 3, Scheme 1 is not rate-limiting.
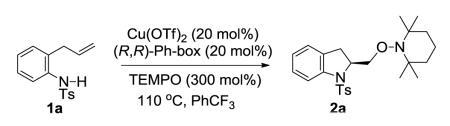 |
(4) |
 |
(5) |
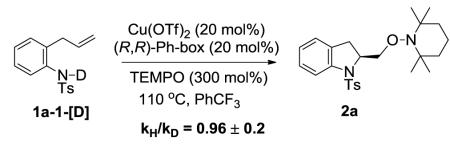
We determined the secondary kinetic isotope effect by subjecting a mixture of substrates 1a and alkene labeled 1a-2-[D] to the catalytic conditions under partial conversion and by analyzing the isotopic ratio before and after the reaction (see Supporting Information for details). An inverse secondary kinetic isotope effect was observed (kH/kD = 0.90±0.04) (Eq 6).17 An inverse secondary kinetic isotope effect is consistent with a change from C(sp2) to C(sp3) in the rate determining step of a reaction.17 This result is consistent with the inverse kinetic isotope effect we observed in the analogous copper(II) carboxylate promoted intramolecular aminooxygenation of 1a (kH/kD = 0.90±0.03).13 These studies support the cis-aminocupration step as the enantioselectivity-determining step of the reaction.
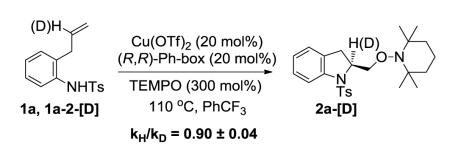 |
(6) |
Mechanism for C-O Bond Formation
When we treated the deuterioalkene 6-[D] with the catalytic aminooxygenation reaction conditions under partial conversion, 79% of the 1:1 mixture of diastereomers of the aminooxygenation product 7-[D] and 15% of the starting alkene with retained alkene geometry were isolated (Eq 7). Based on these observations we concluded that the reaction most likely proceeds via direct capture of an sp2-hybridized carbon radical intermediate with TEMPO18-21 (Scheme 1). The complete retention of the geometry of the isolated alkene indicates that the N-C bond formation is not reversible after C-Cu homolysis in this reaction. It is interesting to note that TEMPO completely out-competes O2 as the carbon radical trapping agent in this reaction. Similar relative reactivity, however, has been previously observed in reactions of carbon radicals generated from organomercury hydrides.19 It would have been difficult to predict the observed relative reactivity using independently determined rate constants reported for the reaction of carbon radicals with TEMPO and O2, respectively.18b,c It is likely the concentration of O2 at 110 ° C in PhCF3 is very low, contributing to the result. A control reaction involving the copper-catalyzed (50 mol% catalyst) aminooxygenation of N-mesyl-2-allylaniline in the presence of O2 without added TEMPO does indicate, however, that the hydroxymethylindoline can be formed directly, albeit in low yield (ca. 30% yield along with starting sulfonamide, reaction not shown).
Based on these labeling studies, we were also able to eliminate other mechanistic alternatives such as the potential formation of alkyl copper(III) complex via oxidation of an alkyl copper(II) with TEMPO followed by reductive elimination. Concerted reaction processes were also ruled out as possible mechanisms for this transformation. For example, a TEMPO-coordinated copper complex could facilitate a concerted two-electron electrophilic addition22,23 and cyclization of the sulfonamide amine to the alkene, forming an alkyl copper(III) intermediate which could subsequently undergo reductive elimination. Alternatively, an oxoammonium ion [O=NR2]+ formed from oxidation of TEMPO by CuII could also initiate a concerted electrophilic addition/cyclization mechanism. In these other mechanistic possibilities however, one diastereomer of 7-[D] would be expected.
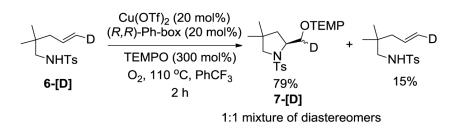 |
(7) |
Effect of Ligand Substituent on Enantioselectivity
The effect of the ligand substituents on the reaction enantioselectivity was next examined. An early screen of the bis(oxazoline) ligands revealed that cis-diphenyl substitutions on the 4- and 5- positions gave optimal ee.10a,b We found that commercially available ligands with alkyl substituents [e.g. (S,S)-t-Bu-Box, (S,S)-iPr-Box and (R,R)-Bn-Box] were less reactive and gave lower enantioselectivity (Table 2, entries 1-3). We also investigated the effect of adding a substituent on the phenyl group of the bis(oxazoline). Electron donating groups such as -OMe on the 4-position of the phenyl ring gave comparable yield and % ee to the parent phenyl substituent (Table 2, compare entry 4 to entry 9) while electron-withdrawing group (i.e. -CF3) gave a slightly lower yield and a significant decrease in % ee (entry 5). 3,5-Dimethyl phenyl substitution rendered the catalyst much less reactive as seen by the lower yield and the selectivity was also diminished (entry 6). 4-tert-Butyl-phenyl substitution, however gave comparable yield and % ee (entry 7).
Table 2.
Ligand screening, optimization with time, TEMPO and catalyst loadinga
| Entry | Ligand | yield (%)b | ee (%)c (config) |
|---|---|---|---|
| 1 | (S,S)-t-Bu-Box | 30d | 11 (R) |
| 2 | (S,S)-iPr-Box | 57d | 72 (R) |
| 3 | (R,R)-Bn-Box | 20d | 62 (S) |
| 4 | (S,S)-4-MeOPh-Box | 85 | 86 (R) |
| 5 | (R,R)-4-CF3Ph-Box | 75 | < 5 |
| 6 | (S,S)-3,5-di-Me-Ph-Box | 51d | 81 (R) |
| 7 | (S,S)-4-t-Bu-Ph-Box | 85 | 91 (R) |
| 8e | (S,S)-4-t-Bu-Ph-Box | 95 | 90 (R) |
| 9f | (R,R)-Ph-Box | 90 | 88 (S) |
| 10e | (R,R)-Ph-Box | 82d | 83 (S) |
| 11f | (4R,5S)-Bis-Ph-Box | 97 | 90 (S) |
| 12g | (4R,5S)-Bis-Ph-Box | 80d | 86 (S) |
| 13e | (4R,5S)-Bis-Ph-Box | 95 | 91 (S) |
| 14e,h | (4R,5S)-Bis-Ph-Box | 92 | 90 (S) |
Conditions: Cu(OTf)2 (20 mol%), bis(oxazoline) ligand (25 mol%), TEMPO (3 equiv), K2CO3, PhCF3 (0.07 M w/r to 1a), 110 °C, 24 h.
Yield (%) refers to amount of isolated 2a after purification by flash chromatography on SiO2.
Enantiomeric excesses were determined by chiral HPLC analysis.
The remainder of the material is the starting olefin 1a.
The reaction was run using 15 mol% of Cu(OTf)2 and 18 mol% of ligand under O2 (1 atm) for 6 h.
The reaction was run for 6 h.
The reaction was run using 15 mol% of Cu(OTf)2 and 18 mol % of ligand for 6 h.
1.5 equiv of TEMPO was used.
We further optimized the reaction with the optimal ligand as shown in Table 2, entries 8-14. When the copper and ligand [(4R,5S)-Bis-Ph-Box] loadings were further lowered to 15 and 18 mol% respectively, the starting alkene 1a was recovered (entry 12). However, in the presence of O2 the reaction goes to completion (Table 2, entries 8, 13 and 14). Advances in transition-metal catalyzed reactions that use O2 as an oxidant have been reported recently.24 O2 has been recognized as an attractive oxidant both in academic and industry due to its low expense and environmental impact. We next found that the reaction time could be reduced from 24 h to 6 h (entries 8, 13 and 14).10b Our kinetics experiments indicated that the aminooxygenation of 1a using the [Cu(R,R)-Ph-Box]OTf2 catalyst (20 mol%) was almost complete after 12 h (see Supporting Information, Figure S-2). That we were able to reduce the reaction time even further in Table 2, entry 9 indicates that the reaction goes even faster in the presence of O2 (1 atm) and K2CO3. We observed that at the lower catalyst loading the (4R,5S)-Bis-Ph-Box ligand gave higher yield and % ee than the (R,R)-Ph-Box ligand (compare entries 10 and 13). Finally, since we were no longer using TEMPO as the sole oxidant, we were also able to reduce the TEMPO loading to 1.5 equivalents (entry 14). We believe O2 serves as the terminal oxidant when it is present, oxidizing TEMPO-H back to TEMPO radical in the presence of K2CO3.25. TEMPO radical is the oxidant of [Cu(I)] to [Cu(II)] in all cases and little reaction occurs in the absence of TEMPO under O2 atmosphere under catalytic conditions (not shown). We have observed that the presence of O2 (1 atm) is necessary for the aminooxygenation reaction to go to completion when it is performed on larger scale10b and when using the less reactive N-pentenylsulfonamide substrates,10a even at lower scale. The need for O2 appears to be tied to the reactivity of the substrate and we speculate that if the aminooxygenation reaction does not occur relatively rapidly, a competitive process that diminishes the amount of TEMPO radical (e.g. autooxidation, 2TEMPO + H+ → [R2N=O]+ + TEMPOH) can occur,25 thereby impeding a productive reaction.
Effect of N-Substituent on Enantioselectivity
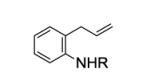
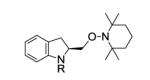
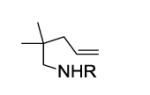
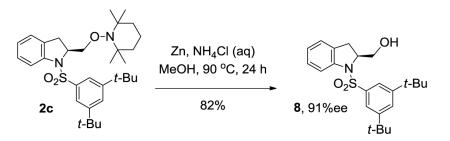
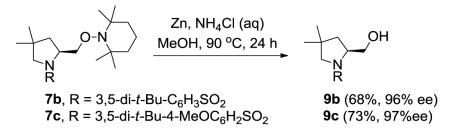
We next examined the effect of the size of the N-sulfonyl group on the level of enantioselectivity of the reaction. We have previously shown in catalytic desymmetrization reactions of meso β-substituted 4-pentenyl sulfonamides that the use of a sterically demanding aryl sulfonyl group (i.e. 3,5-di-tert-butylbenzenesulfonyl) increases the reaction selectivity.10c (Substitution ortho to the sulfonyl decreases reactivity and selectivity, not shown.) Similarly, we obtained high enantioselectivity in our catalytic enantioselective aminooxygenation reaction with the use of bulky substituents both with the 2-allyl aniline-derived substrates (up to 92%, Table 3, entries 3 and 4) and the 4-pentenylamine-derived substrates (up to 97%, Table 3, entries 6 and 7). We have previously reported that use of the more easily removable nosyl group in this reaction led to a decrease in reactivity and selectivity.10a We found the enantioselective aminooxygenation reaction efficiency and selectivity to be highest with substrates that have carbon chain alkyl substitution (mono or bis) as the parent N-tosyl-4-pentenylamine reacts poorly in this reaction (yield typically is < 50%, substrate can suffer decomposition, not shown). Enantiomeric excess for each product was determined by chiral HPLC. In the cases of products 2c, 7b and 7c, the enantiomers were inseparable by chiral HPLC. Reduction of the O-N bond of each product with zinc in the presence of saturated aqueous NH4Cl and methanol at 90 °C furnished the respective alcohols 8 and 9, whose enantiomeric ratios could be determined by chiral HPLC (Eqs 8 and 9).
Table 3.
Effect of N-substituent on enantioselectivitya
| entry | substrate | product | yield (%)b | ee (%)c |
|---|---|---|---|---|
|
|
|||
| 1 | 1a, R = Ts | 2a | 90 | 88 |
| 2 | 1b, R = Ms | 2b | 50d | 63 |
| 3 |
1c, R = 3,5-di-t-Bu- C6H3SO2 |
2c | 89 | 91 |
| 4 |
1d, R = 3,5-di-t-Bu-4- MeOC6H2SO2 |
2d | 85 | 92 |
|
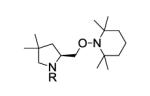
|
|||
| 5e | 6a, R = Ts | 7a | 88 | 88 |
| 6e |
6b, R = 3,5-di-t-Bu- C6H3SO2 |
7b | 92 | 96 |
| 7e |
6c, R = 3,5-di-t-Bu-4- MeOC6H2SO2 |
7c | 94 | 97 |
Conditions: Cu(OTf)2 (20 mol%), (R,R)-Ph-Box (25 mol%), TEMPO (3 equiv), K2CO3, PhCF3 (0.07 M w/r to substrate), 110 °C, 6 h.
Yield (%) refers to amount of isolated 2 after purification by flash chromatography on SiO2.
Enantiomeric excesses were determined by chiral HPLC analysis.
The remainder of the material is the starting olefin 1b. eReaction was run under O2 (1 atm, balloon).
Reaction was run under O2 (1 atm, balloon).
 |
(8) |
 |
(9) |
Scale-up of the Pyrrolidine-Forming Aminooxygenation Reaction
We next sought to establish if the enantioselective alkene aminooxygenation reaction of a 4-pentenylamine derived sulfonamide could be performed on gram scale. We recently demonstrated that the catalytic enantioselective aminooxygenation reaction of N-tosyl-2-allyl-4-fluoroaniline could be run on multigram scale using 15 mol% of Cu(OTf)2, 18 mol% of (4R,5S)-Bis-Ph-Box, 3 equivalents of TEMPO and heating the reaction in PhCF3 to 110 °C under O2 (1 atm) for 6 h (vide supra, Eq 3).10b The chiral indoline product was prepared on five gram scale and with high enantioselectivity. In that study, use of the (4R,5S)-Bis-Ph-Box ligand was essential for enabling catalyst loading reduction to 15 mol%.10b In the process of scaling up the reaction of 2,2-dimethyl-4-pentenylsulfonamide 6a, we found that the protocol we reported in 2008 for the enantioselective aminooxygenation of this substrate10a was significantly less efficient on a larger scale (250 mg, 19% yield, Table 4, entry 1). When we lowered the temperature to 110 °C, a substantial increase in isolated yield was observed (entry 2). The isolated yield of 7a was further improved when the TEMPO loading was decreased to 1.5 equivalents (entries 3 and 4). We believe the temperature effect is related to a catalyst decomposition process that can occur under the reaction conditions. We believe TEMPO may be involved in the catalyst decomposition process and also the decomposition of some substrates (e.g. the less reactive N-tosyl-4-pentenylamine, see discussion above). When [Cu(4R,5S)-Bis-Ph-Box]OTf2 was subjected to heating in the presence of TEMPO and O2 but absence of substrate, a new compound, clearly derived from the ligand, appeared in the 1H NMR spectrum of the crude mixture (see Supporting Information for spectra). We speculate the ligand may undergo oxidation at its benzylic positions. Attempted isolation/purification of the ligand mixture, unfortunately, failed. We believe, however, that the development of a more stable ligand for this aminooxygenation reaction could enable further lowering of the catalyst loading in future investigations.
Table 4.
Scaling-up of the catalytic aminooxygenation reaction of aliphatic substrates

| entry | substrate | amount substrate (mg, mmol) |
yield (%)a | ee (%)b |
|---|---|---|---|---|
| 1c | 6a, R = Ts | 50, 0.19 | 97 | 88 |
| 1c | 6a, R = Ts | 250, 0.93 | 19d | 85 |
| 2e | 6a, R = Ts | 250, 0.93 | 88f | 86 |
| 3 | 6a, R = Ts | 250, 0.93 | 95 | 90 |
| 4 |
6c, R = 3,5-di-t-Bu-4- MeOC6H2SO2 |
300, 0.76 | 89 | 95 |
| 5 |
6c, R =3,5-di-t-Bu-4- MeOC6H2SO2 |
1000, 2.53 | 88 | 97 |
Yield (%) refers to amount of isolated 7 after purification by flash chromatography on SiO2.
Enantiomeric excesses were determined by chiral HPLC analysis.
The reaction was run at 120 °C using 3 equiv of TEMPO.
80% of the starting alkene 6a was recovered.
The reaction was run at 110 °C using 3 equiv of TEMPO.
9% of the starting alkene 6a was recovered.
With the current optimized conditions in hand, we ran a gram-scale reaction using substrate 6c since it gave us the highest enantioselectivity (Table 3, entry 7). Sulfonamide 6c cyclized efficiently on gram scale to form the pyrrolidine 7c in good yield and excellent ee (Table 4, entry 5). We have demonstrated that the TEMPO adducts 7 can be reduced to the corresponding alcohol or oxidized to the corresponding aldehyde without erosion of enantiopurity.10
CONCLUSIONS
We have reported a mechanistic study of the copper(II)-catalyzed enantioselective intramolecular aminooxygenation of alkenes which involved reaction kinetics, kinetic isotope effects and deuterium labeling studies. The reaction kinetics showed first order dependence in the sulfonamide substrate and the copper-bis(oxazoline) complex and zero order in TEMPO. We also observe a secondary kinetic isotope effect for the alkene addition step. The reactions kinetics and observed secondary kinetic isotope effect are consistent with the cis-aminocupration being the rate-limiting step. Isotopic labeling studies indicate that the C-O bond formation occurs via direct carbon radical trapping with TEMPO. This deuterium labeling study also helps rule out other possible concerted reaction processes. This study is distinct from our previous study of the copper(II) 2-ethylhexanoate promoted aminooxygenation reaction where ½ order kinetics in both substrate and [copper] was observed.13 The different kinetics are explained by the structure of the starting copper complexes: a [Cu(bisoxazoline)]OTf2 complex is monomeric while Cu(II) 2-ethylhexanoate exists initially as a dimer. The copper-catalyzed enantioselective aminooxygenation mechanism (Scheme 1) also shares a number of similarities with the copper(II) carboxylate promoted aminooxygenation reaction.13 In both cases, the cis-aminocupration was the most likely rate-determining step, and the C-O bond formation was determined to occur via direct carbon radical trap. These mechanistic studies will serve as the experimental basis for follow up molecular modeling calculations that can shed light on the detailed three-dimensional transition states that determine the reaction enantioselectivity.
In this study, we were able to improve the enantioselectivity of the aminooxygenation reaction (up to 97% ee) with the use of a bulky N-substituent (i.e. 3,5-di-t-Bu-4-MeOC6H2SO2). We have shown that the reaction can be efficiently catalyzed with the use of several chiral bis(oxazoline) ligands [(S,S)-4-t-Bu-Ph-Box, (R,R)-Ph-Box, (4R,5S)-Bis-Ph-Box]. Our newly optimized reaction conditions require lower catalyst (15 mol% Cu(OTf)2 and 18 mol% bisoxazoline ligand) and TEMPO (1.5 equiv) loadings, less time (6 h), lower temperature and use of environmentally benign O2 as the stoichiometric oxidant. The practicality of this protocol was further demonstrated on a gram scale reaction. This study should aid in the practical application of this useful copper-catalyzed enantioselective alkene aminooxygenation reaction in organic synthesis and drug discovery.
EXPERIMENTAL SECTION
General Experimental Information
Reagents were used out of the bottle as purchased from the supplier without further purification unless otherwise specified. Solvents were purified using a commercial solvent filtration system. PhCF3 can generally be used out of the bottle as supplied but can be further dried by distillation over CaH2. 1H NMR spectra were recorded in CDCl3 at 500 MHz. 13C NMR spectra were recorded in CDCl3 at 75 MHz. Spectra are reported in ppm relative to residual chloroform (7.26 ppm for 1H NMRs and 77.0 ppm for 13C NMRs). IR spectra were obtained neat and characteristic peaks are reported in wavenumbers, cm−1. High resolution mass spectra were obtained using ESI (double focusing magnetic sector). Optical rotations were obtained using a polarimeter fitted with a micro cell with a 100 mm path length. Melting points are reported as uncorrected. Cu(OTf)2 was used as purchased but handled in dry atmosphere (glove box). Moisture can lower % yield and % ee. Sulfonamides 1a-1d were synthesized as previously reported.10,26 Characterization data for indolines 2a and 2b has been previously reported.10a Sulfonamides 6a and 6b were synthesized as previously reported.10,27 Characterization data for pyrrolidine 7a has been reported.10a The synthesis of sulfonamide 6-[D] and the characterization data for indolines 7-[D] have been reported.10a,13
Chiral Bis(oxazoline) Ligands
The (R,R)-Ph-Box, (S,S)-t-Bu-Box, (S,S)-i-Pr-Box, (R,R)-Bn-Box ligands were purchased from commercial sources. The (4R,5S)-di-Ph-Box ligand was synthesized as previously reported.10b The remaining ligands were synthesized from their respective chiral aminoalcohols using the procedure reported by Evans and co-workers.28,29 The aminoalcohols were prepared either from the reduction of the commercially available amino acid30 or following Sharpless’ asymmetric aminohydroxylation method31,32 from the corresponding substituted styrene. The substituted styrene was either commercially available or was readily prepared using the procedure reported by Denmark and Butler.33,34 The synthesis and characterization data of the (R,R)-4-t-Bu-Ph-Box, (R,R)-4-MeO-Ph-Box and (S,S)-4-CF3-Ph-Box ligands have been reported.35,36
(S,S)-3,5-di-Me-Ph-Box
The known (2S)-2-amino-2-[3,5-(dimethyl)phenyl]ethan-1-ol precursor was obtained using Sharpless’ method31 (96% ee). The aminoalcohol was converted to the (S,S)-3,5-di-Me-Ph-Box ligand,28,29 which was obtained as colorless oil:36 1H NMR (500 MHz, CDCl3) δ 6.89 (s, 2H), 6.86 (s, 4H), 5.16 (dd, J= 10.0, 8.0 Hz, 2H), 4.63 (dd, J= 10.0, 8.5 Hz, 2H), 4.16 (t, J= 7.5 Hz, 2H), 2.26 (s, 12H), 1.68 (s, 6H).
N-(2-allylphenyl)-3,5-di-tert-butylbenzenesulfonamide (1c)
o-Allylaniline37 (120 mg, 0.90 mmol, 1 equiv) was dissolved in dry CH2Cl2 (4.5 mL) and the solution was treated with 3,5-di-tert-butylbenzene sulfonyl chloride38,39 (286 mg, 0.99 mmol, 1.1 equiv) and Et3N (0.38 mL, 2.7 mmol, 3 equiv). The mixture was stirred at room temperature overnight, washed with 1 N HCl (5.0 mL) and extracted with CH2Cl2 (3 × 8 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. Flash chromatography of the resulting crude product on SiO2 (0-5% EtOAc in hexanes) afforded sulfonamide 1c (200 mg, 58% yield) which matches the previously reported characterization data.27 1H NMR (500 MHz, CDCl3) 7.56 (s, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.43 (s, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.13 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 7.5, 1H), 6.41 (s, 1H), 5.66 (m, 1H), 5.06 (dd, J = 10.0 Hz, 1.5 Hz, 1H), 4.87 (dd, J = 17.5 Hz, 1.5 Hz, 1H), 2.73 (d, J = 6.5 Hz, 2H), 1.25 (s, 18H).
N-(2-allylphenyl)-3,5-di-tert-butyl-4-methoxybenzenesulfonamide (1d)
The procedure for the synthesis of 1c was followed except that 3,5-di-tert-butyl-4-methoxybenzenesulfonyl chloride38,39 was used.26 Sulfonamide 1d was obtained as white solid (310 mg, 60% yield). Its spectral properties matched the reported values.26 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.5 Hz, 1H), 7.48 (s, 2H), 7.25 (t, J = 7.5, 1H), 7.13 (t, J = 7.5 Hz, 1H), 7.02 (d, J = 7.5 Hz, 1H), 6.40 (s, 1H), 5.66 (m, 1H), 5.06 (dd, J = 10.5 Hz, 1.5 Hz, 1H), 4.90 (dd, J = 17.0 Hz, 1.5 Hz, 1H), 3.66 (s, 3H), 2.80 (d, J = 6.5 Hz, 2H), 1.31 (s, 18H).
(S)-1-((3,5-Di-tert-butylphenyl)sulfonyl)-2-(((2,2,6,6-tetramethylpiperidin-1-yl)oxy)methyl)indoline (2c)
Cu(OTf)2 (7.5 mg, 0.021 mmol, 0.2 equiv) and (R,R)-Ph-Box (0.32 mL of 0.08 M solution, 0.026 mmol, 0.25 equiv) were combined and stirred in CF3Ph (0.48 mL) under Ar for 2 h at 60 °C in a 100 mL round bottom flask equipped with magnetic stir bar. The blue-green solution was cooled to rt and was treated with sulfonamide 1c (40 mg, 0.104 mmol, 1 equiv), TEMPO (48.6 mg, 0.311 mmol, 3 equiv), K2CO3 (14.3 mg, 0.104 mmol, 1 equiv) and CF3Ph (0.70 mL). The reaction mixture was heated to 110 °C for 6 h. Filtration of the cooled solution through a SiO2 plug (with Et2O washing) and removal of the solvent in vacuo afforded the crude product. Purification via flash chromatography on silica gel (0-5% Et2O in hexanes gradient) gave the TEMPO adduct 2c (50 mg, 89% yield) as white solid. m.p. 100-105 °C; [α]19D = 66.0 °(c = 1.15, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 8.0 Hz, 1H), 7.51 (s, 1H), 7.40 (s, 2H), 7.20 (m, 1H), 7.00 (d, J = 5.0 Hz, 2H), 4.25 (m, 1H), 3.99 (dd, J = 9.0 Hz, 3.5 Hz, 1H), 3.93 (dd, J = 8.0 Hz, 6.5 Hz, 1H), 2.72 (d, J = 15.5 Hz, 1H), 2.49 (dd, J = 15.5 Hz, 10.0 Hz, 1H), 1.22-1.38 (m, 6H), 1.18 (s, 18H), 1.14 (s, 3H), 1.10 (s, 3H), 0.96 (s, 3H), 0.85 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 151.8, 142.3, 136.8, 133.1, 127.2, 126.6, 124.8, 124.4, 121.0, 117.8, 78.8, 60.9, 59.9, 39.5, 35.0, 33.0, 31.6, 31.2, 31.0, 19.9, 19.7, 17.0; IR (neat, thin film) ν 2965, 2870, 1598, 1478, 1461, 1360, 1360, 1314, 1247, 1170, 1024, 788, 758, 705, 622, 600 cm−1; HRMS (ESI) calcd for [M+H]+ C32H49O3N2S1: 541.3458, found: 541.3461.
(S)-(1-((3,5-Di-tert-butylphenyl)sulfonyl)indolin-2-yl)methanol (8)
The enantiomers of the TEMPO adduct 2c were inseparable so the enantiomeric excess was determined using the free alcohol 8. Following the reported procedure for the reduction of TEMPO,10 2c (30 mg, 0.06 mmol, 1 equiv) was stirred in methanol (1.5 mL) in a pressure tube, then aqueous NH4Cl (1.5 mL) was added followed by Zn dust (157 mg, 2.4 mmol, 40 equiv). The reaction mixture was heated to 90 °C for 24 h and was then cooled to room temperature and filtered through Celite. Purification of the crude product via flash chromatography (20% ethyl acetate in hexanes) gave 82% yield of N-protected amino alcohol 8 (18.0 mg) which is a white solid. m.p. 40-45 °C; [α]19D = 140.6 °(c = 1.35, CHCl3), ee = 91%, determined by HPLC [Regis (S,S) Whelk, 5% IPA/hexane, 1.0 mL/min, λ = 254 nm, t(major) = 15.00 min, t(minor) = 13.33 min]. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 7.5 Hz, 1H), 7.53 (s, 1H), 7.39 (s, 2H), 7.24 (d, J = 6.5 Hz, 1H), 7.03 (m, 2H), 4.23 (m, 1H), 3.68 (d, J = 6.0 Hz, 2H), 2.55 (d, J = 9.5 Hz, 2H), 2.17 (s, 1H), 1.22 (s, 18H); 13C NMR (75 MHz, CDCl3) δ 152.0, 141.5, 136.2, 132.5, 127.7, 127.0, 125.3, 124.9, 121.1, 118.3, 65.3, 63.5, 35.0, 31.1, 31.0; IR (neat, thin film) ν 3541, 2963, 2869, 1593, 1476, 1461, 1349, 1243, 1171, 1095, 1039, 958, 760 cm−1; HRMS (ESI) calcd for [M+Na]+ C23H31O3N1Na1S1: 424.1917, found: 424.1925.
(S)-1-((3,5-Di-tert-butyl-4-methoxyphenyl)sulfonyl)-2-(((2,2,6,6-tetramethylpiperidin-1-yl)oxy)methyl)indoline (2d)
Sulfonamide 1d was converted to indoline 2d using the procedure described for the aminooxygenation reaction of 1c to 2c. The TEMPO adduct 2d was obtained as white solid (46 mg, 85% yield). m.p. 80-83 °C; [α]20D = 66.8 (c = 2.0, CHCl3); ee = 92%, determined by HPLC [Regis (S,S) Whelk, 0.4% IPA/hexane, 0.5 mL/min, λ = 254 nm, t(major) = 30.56 min, t(minor) = 24.74 min]; 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 8.0 Hz, 1H), 7.45 (s, 2H), 7.19 (t, J = 7.0 Hz, 1H), 7.02 (m, 2H), 4.25 (m, 1H), 4.00 (m, 1H), 3.92 (m, 1H), 3.62 (s, 3H), 2.77 (d, J = 15.5 Hz, 1H), 2.55 (dd, J = 15.5 Hz, 9.0 Hz, 1H), 1.38-145 (m, 6H), 1.26 (s, 18H), 1.16 (s, 3H), 1.13 (s, 3H), 0.95 (s, 3H), 0.85 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.4, 144.8, 142.4, 133.1, 131.5, 127.3, 125.5, 124.8, 124.4, 117.8, 78.8, 64.5, 60.8, 59.9, 39.5, 35.9, 33.1, 31.7, 31.6, 29.6, 19.8, 17.0; IR (neat, thin film) ν 2966, 2871, 1603, 1479, 1462, 1406, 1359, 1256, 1227, 1170, 1128, 1007, 883, 794, 759, 707, 665 cm−1; HRMS (ESI) calcd for [M+H]+ C33H51O4N2S1: 571.3564, found: 571.3583.
3,5-di-tert-Butyl-N-(2,2-dimethylpent-4-en-1-yl)benzenesulfonamide (6b)
2,2-Dimethylpent-4-en-1-amine41,42 (250 mg, 2.2 mmol, 1 equiv) was stirred in dry CH2Cl2 (12 mL) and 3,5-di-tert-butylsulfonyl chloride38,39 (770 mg, 2.4 mmol, 1.1 equiv) was added followed by Et3N (0.93 mL, 6.6 mmol, 3 equiv). The reaction mixture was stirred at room temperature overnight, washed with 1 N HCl (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. Purification by flash chromatography of the resulting crude product on SiO2 (0-5% EtOAc in hexanes) gave sulfonamide 6c (480 mg, 53% yield) as colorless oil.27 1H NMR (500 MHz, CDCl3) 7.70 (s, 2H), 7.59 (s, 1H), 5.66 (m, 1H), 4.91-4.96 (m, 2H), 2.69 (d, J = 6.5 Hz, 2H), 1.93 (d, J = 7.5 Hz, 2H), 1.31 (s, 18H), 0.84 (s, 3H); 13C NMR (75 Hz, CDCl3) 152.0, 139.1, 134.2, 126.5, 121.0, 117.7, 52.7, 44.0, 35.1, 34.0, 31.2, 24.7; IR (neat) ν 3279, 3076, 2964, 2873, 1639, 1598, 1466, 1410, 1359, 1324, 1313, 1247, 1161, 1070, 993, 922, 876, 831, 749, 704, 592 cm−1; HRMS (ESI) calcd for [M+Na]+ C21H35O2N1Na1S1: 388.2281, found: 388.2275.
3,5-Di-tert-butyl-N-(2,2-dimethylpent-4-en-1-yl)-4-methoxybenzenesulfonamide (6c)
2,2-Dimethyl-4-pentenamine (1.00 g, 8.83 mmol) was dissolved in CH2Cl2 (44 mL) and was treated with 3,5-di-tert-butyl-4-methoxybenzenesulfonyl chloride 38,40 (2.95 g, 9.27 mmol) and Et3N (3.7 mL, 26.5 mmol). The solution was stirred for 2 days at rt, then was quenched with H2O and extracted with CH2Cl2 (2x). The combined organic extracts were washed with 1 N HCl and brine, and then were dried over MgSO4. The solution was filtered and the volatiles were removed in vacuo. Chromatography on silica gel (0-10% EtOAc in hexanes gradient) afforded sulfonamide 6c as white solid (2.5 g, 72% yield). m.p. 95-98 C; 1H NMR (500 MHz, CDCl3)δ 7.71 (s, 2H), 5.68 (m, 1H), 4.96-5.01 (m, 2H), 4.37 (s, 1H), 3.70 (s, 3H), 2.73 (d, J = 6.5 Hz, 2H), 1.93 (d, J = 7.0 Hz, 2H), 1.43 (s, 18H), 0.85 (s, 6H); 13C NMR (75 Hz, CDCl3) 163.2, 145.1, 134.2, 133.6, 125.6, 117.8, 64.5, 52.7, 44.1, 36.1, 34.0, 31.7, 24.8; IR (neat) ν 3266, 2963, 2871, 1639, 1453, 1407, 1327, 1255, 1227, 1163, 1116, 1066, 1004, 928, 886, 756, 710, 592 cm−1; HRMS (ESI) calcd for [M+H]+ C22H38O3N1S1: 396.2567, found: 396.2569.
(S)-1-((1-((3,5-Di-tert-butylphenyl)sulfonyl)-4,4-dimethylpyrrolidin-2-yl)methoxy)-2,2,6,6-tetramethylpiperidine (7b)
In a 100 mL round bottom flask equipped with magnetic stir bar were added Cu(OTf)2 (7.9 mg, 0.022 mmol, 0.2 equiv), (R,R)-Ph-Box (0.34 mL of 0.08 M solution, 0.027 mmol, 0.25 equiv) and CF3Ph (0.46 mL). The mixture was heated and stirred under Ar for 2 h at 60 °C. The blue-green solution was then cooled to room temperature and sulfonamide 6b (40 mg, 0.109 mmol, 1 equiv), TEMPO (48.6 mg, 0.327 mmol, 3 equiv), K2CO3 (14.3 mg, 0.109 mmol, 1 equiv) and CF3Ph (0.8 mL) were added. The flask was then fitted with a glass side-arm adapter tapered to connect a short rubber vacuum hose through which the O2 balloon was connected. The reaction mixture was heated to 110 °C for 6 h and then was cooled to room temperature and filtered through a SiO2 plug (with Et2O washing). The solvent was removed in vacuo to afford the crude product which was then purification by flash chromatography on silica gel (0-5% Et2O in hexanes gradient). The TEMPO adduct 7b was obtained as white solid (52.1 mg, 92% yield). m.p. 90-95 C; [α]21D = −68.4 (c = 1.0, CHCl ); 1 3 H NMR (500 MHz, CDCl3) δ 7.65 (s, 2H), 7.60 (s, 1H), 4.25 (dd, J = 8.5 Hz, 4.0 Hz, 1H), 3.86 (t, J = 8.5 Hz, 1H), 3.62 (m, 1H), 3.14 (AB quartet, J = 10.5 Hz, Δν = 20.6 Hz, 2H), 1.73 (d, J = 7.5 Hz, 2H), 1.46 (m, 6H), 1.34 (s, 18H), 1.25 (s, 3H), 1.14 (s, 3H), 1.06 (s, 6H), 1.04 (s, 3H), 0.40 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 151.9, 137.0, 126.4, 121.5, 79.8, 61.6, 59.7, 58.0, 44.4, 39.6, 37.4, 35.0, 33.0, 31.2, 26.4, 25.8, 20.1, 17.1; IR (neat, thin film) ν 2963, 2873, 1598, 1466, 1351, 1246, 1165, 1110, 1039, 755, 704, 625 cm−1; HRMS (ESI) calcd for [M+H]+ C30H53O3N2S1: 521.3771, found: 521.3784.
(S)-(1-((3,5-Di-tert-butylphenyl)sulfonyl)-4,4-dimethylpyrrolidin-2-yl)methanol (9b)
The enantiomeric excess of 7b was determined using the free alcohol. Compound 9b was obtained as colorless oil using the same method as in the conversion of 2c to 8 (17 mg, 68% yield). [α]22D = −20.6 °(c = 1.25, CHCl3), ee = 96%, determined by HPLC [Chiralpak AD-RH, 50% H2O/CH3CN, 0.5 mL/min, λ = 254 nm, t(major) = 20.58 min, t(minor) = 17.82 min]; 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 2H), 7.63 (s, 1H), 3.80 (dd, J = 12.0 Hz, 3.0 Hz, 1H), 3.69 (dd, J = 12.0 Hz, 4.5 Hz, 1H), 3.57 (m, 1H), 3.16 (AB quartet, J = 10.5 Hz, Δν = 22.7 Hz, 2H), 1.61 (dd, J = 7.5 Hz, 1.5 Hz, 2H), 1.34 (s, 18H), 0.99 (s, 3H), 0.31 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 152.2, 136.3, 126.8, 121.5, 65.8, 62.3, 62.2, 43.3, 36.9, 35.1, 31.2, 26.0, 25.3; IR (neat, thin film) ν 3504, 2961, 2873, 1598, 1466, 1340, 1247, 1162, 1114, 1066, 1036, 968, 791, 756, 707, 631, 592 cm−1; HRMS (ESI) calcd for [M+Na]+ C21H35O3N1Na1S1: 404.2230, found: 404.2226.
(S)-1-((1-((3,5-Di-tert-butyl-4-methoxyphenyl)sulfonyl)-4,4-dimethylpyrrolidin-2-yl)methoxy)-2,2,6,6-tetramethylpiperidine (7c)
Sulfonamide 6c was converted to pyrrolidine 7c using the procedure described for the conversion of 6b to 7b. The TEMPO adduct 7c was obtained as white solid (52 mg, 94% yield). m.p. 140-145 °C; [α]21D = −64.1 °(c = 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 2H), 4.24 (dd, J = 12.5 Hz, 3.0 Hz, 1H), 3.84 (t, J = 8.0 Hz, 1H), 3.67 (s, 3H), 3.63 (m, 1H), 3.12 (AB quartet, J = 10.5 Hz, Δν = 20.6 Hz, 2H), 1.74 (m, 2H), 1.50-1.58 (m, 6H), 1.43 (s, 18H), 1.15 (s, 3H), 1.06 (s, 6H), 1.04 (s, 3H), 0.41 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.2, 144.8, 131.6, 125.9, 79.9, 64.6, 61.5, 59.7, 58.1, 44.5, 39.6, 37.4, 36.0, 33.2, 31.7, 26.4, 25.7, 20.0, 17.1; IR (neat, thin film) ν 2955, 2874, 1471, 1453, 1405, 1349, 1253, 1225, 1165, 1125, 1042, 1008, 967, 920, 885, 812, 757, 710, 632, 595 cm−1; HRMS (ESI) calcd for [M+H]+ C31H55O4N2S1: 551.3877, found: 551.3887.
(S)-(1-((3,5-Di-tert-butyl-4-methoxyphenyl)sulfonyl)-4,4-dimethylpyrrolidin-2-yl)methanol (9c)
The enantiomeric excess of 7c was determined using the free alcohol. Compound 9c was obtained as colorless oil using the same procedure as in the conversion of 2c to 8 (16 mg, 73% yield). [α]22D = −19.3 °(c = 1.0, CHCl3), ee = 97%, determined by HPLC [Chiralpak AD-RH, 40% H2O/CH3CN, 0.75 mL/min, λ = 254 nm, t(major) = 9.61 min, t(minor) = 8.35 min]; 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 2H), 3.80 (dd, J = 12.0 Hz, 3.0 Hz, 1H), 3.69 (m, 1H), 3.67 (s, 3H), 3.58 (m, 1H), 3.19 (AB quartet, J = 11.0 Hz, Δν = 24.4 Hz, 2H), 1.60-1.63 (m, 2H), 1.44 (s, 18H), 1.00 (s, 3H), 0.34 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.6, 145.2, 130.9, 125.9, 65.8, 64.6, 62.4, 62.2, 43.3, 36.9, 36.0, 31.7, 29.6, 26.0, 25.3; IR (neat, thin film) ν 3503, 2961, 2873, 1576, 1465, 1406, 1337, 1254, 1226, 1162, 1123, 1065, 1034, 1006, 967, 885, 795, 756, 632, 596 cm−1; HRMS (ESI) calcd for [M+H]+ C22H38O4N1S1: 412.2516, found: 412.2517.
(S)-1-((1-((3,5-Di-tert-butyl-4-methoxyphenyl)sulfonyl)-4,4-dimethylpyrrolidin-2-yl)methoxy)-2,2,6,6-tetramethylpiperidine (7c) (Gram Scale)
To an oven-dried 250 mL round-bottom flask equipped with magnetic stir bar were added Cu(OTf)2 (137 mg, 0.38 mmol, 0.15 equiv), (4R,5S)-Bis-Ph-Box (222 mg, 0.46 mmol, 0.18 equiv) and CF3Ph (30.0 mL). The solution was heated and stirred for 2 h at 60 °C under Ar atmosphere. The blue-green solution was cooled to room temperature and was treated with sulfonamide 6c (1.0 g, 2.53 mmol, 1 equiv), TEMPO (593 mg, 3.8 mmol, 1.5 equiv), K2CO3 (349 mg, 2.53 mmol, 1 equiv) and an additional 6.0 mL of CF3Ph. The reaction mixture was heated to 110 °C for 6 h under O2 (1 atm) and was then cooled to room temperature and filtered through a SiO2 plug (with Et2O washing). Removal of the solvent in vacuo afforded the crude product. Purification via flash chromatography on SiO2 (0-5% Et2O in hexanes) gave the TEMPO adduct 7c (1.23 g, 88% yield). Enantioselectivity (97% ee) was determined using the amino alcohol 9c.
Mechanistic Analysis of C-O Bond Formation
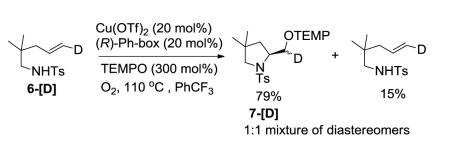
Cu(OTf)2 (5.4 mg, 0.015 mmol, 0.20 equiv) and (R,R)-Ph-Box (0.20 mL of 0.08 M solution, 0.016 mmol, 0.20 equiv) were heated and stirred in CF3Ph (0.31 mL) for 2 h at 60 °C in a 100 mL round bottom flask equipped with magnetic stir bar. Upon cooling to room temperature the reaction mixture was treated with deuterated substrate 6-[D]13b (20 mg, 0.075 mmol, 1 equiv), TEMPO (35.2 mg, 0.23 mmol, 3 equiv) and CF3Ph (0.60 mL). An O2 balloon was connected to the flask with the use of a glass side-arm adapter connected to a short rubber vacuum hose. The reaction mixture was heated in an oil bath at 110 °C for 2 h, then was cooled to room temperature, diluted with Et2O and filtered through a plug of silica gel with Et2O. Removal of the solvent in vacuo afforded the crude product. Purification by flash chromatography on silica gel (5-10% EtOAc in hexanes) gave compound 7-[D] (22 mg, 70% yield) and the unscrambled starting material (4.5 mg, 22% yield). The crude 1H NMR spectrum showed two –CHD-OTEMP protons at 4.20 and 3.87 ppm, respectively, each integrating as 0.5 H, which indicates the presence of a 1:1 mixture of diastereomers. This result is consistent with the analysis of the previously reported C-O bond formation of the copper(II)-promoted aminooxygenation reaction of alkene.13 Data for compound 7-[D] matches the previously reported characterization:10a,13 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 4.17 (m, 0.5H), 3.85 (m, 0.5H), 3.68 (m, 1H), 3.13 (ABq, ΔνAB = 18.5 Hz, J = 10.4 Hz, 2H), 2.43 (s, 3H), 1.78-1.75 (m, 2H), 1.45-1.42 (m, 4H), 1.20 (s, 3H), 1.16 (s, 3H), 1.08 (s, 3H), 1.06 (s, 6H), 0.55 (s, 3H).
Catalyst Decomposition Study
Three separate solutions were prepared following the procedure below. Cu(OTf)2 (10 mg, 0.028 mmol, 1 equiv) and (4R,5S)-Bis-PhBox (16 mg, 0.033 mmol, 1.2 equiv) were heated and stirred in CF3Ph (2.7 mL) for 2 h at 60 °C in a 10 mL round bottom flask equipped with magnetic stir bar. The solution was cooled to room temperature and was treated with TEMPO (88 mg, 0.56 mmol, 20 equiv). The first solution was stirred at room temperature for about 15 min, the second and third solution was heated to 110 °C and 120 °C for 6 h respectively. The cooled solution was filtered through Celite (with Et2O washing) and the filtrate was washed with saturated aqueous Na2EDTA (2 × 3 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo to afford the crude product. 1H NMR spectra of the three crude materials were obtained.
General Procedure for Kinetics Experiments
Cu(OTf)2 and (R,R)-Ph-Box were heated and stirred in CF3Ph under argon for 2 h at 60 °C in an oven-dried 10 mL round bottom flask equipped with magnetic stir bar and sealed with septa. The solution was cooled to rt and treated with substrate 1a and TEMPO. The reaction mixture was heated to 110 °C using a temperature regulated oil bath. It was taken out of the oil bath every hour and a 20 μL aliquot was collected using a gas-tight syringe. The aliquots collected were dried under vacuum and the residue was dissolved in 200 μL acetonitrile. The samples were analyzed using HPLC in a Microsorb-MV 100 C8 column by gradient elution (65-100% CH3CN in H2O). Calibration plots for substrate 1a and the TEMPO adduct 2a were used to calculate the concentrations of 1a and 2a as a function of time.
Reactions were run up to 80-90% conversion for higher catalyst loading (20-40 mol% or 10-20 mM). We observed that at low catalyst loading (5-10 mol% or 2.5-5 mM), reactions do not go to completion and the rate tails off at 40-60% conversion presumably due to catalyst decomposition. Therefore, initial reaction rates were measured for these catalyst loadings.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by the National Institutes of Health, NIGMS (GM-078383). We are also grateful to Dr. Alice Bergmann and Ying Long for assistance with mass spectrometry.
Footnotes
Supporting Information Experimental procedures, kinetic studies, kinetic isotope effect studies and characterization data for all reactions and products, including 1H NMR and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1) (a).Bergmeier SC. Tetrahedron. 2000;56:2561–2576. [Google Scholar]; (b) Donohoe TJ, Callens CKA, Flores A, Lacy AR, Rathi AH. Chem. Eur. J. 2011;17:58–76. doi: 10.1002/chem.201002323. [DOI] [PubMed] [Google Scholar]
- (2) (a).Nilov D. Reiser, O. Adv. Synth. Catal. 2002;344:1169–1173. [Google Scholar]; (b) Li G, Chang H-T, Sharpless KB. Angew. Chem. Int. Ed. 1996;35:451–454. [Google Scholar]; (c) Reiser O. Angew. Chem. Int. Ed. 1996;35:1308–1309. [Google Scholar]; (d) O’Brien P. Angew. Chem. Int. Ed. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; (e) Bodkin JA, McLeod MD. J. Chem. Soc., Perkin Trans. 2002;1:2733–2746. [Google Scholar]; (f) Muniz K. Chem. Soc. Rev. 2004;33:166–174. doi: 10.1039/b307102m. [DOI] [PubMed] [Google Scholar]
- (3).Michaelis DJ, Williamson KS, Yoon TP. Tetrahedron. 2009;65:5118–5124. doi: 10.1016/j.tet.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Williamson KS, Yoon TP. J. Am. Chem. Soc. 2012;134:12370–12373. doi: 10.1021/ja3046684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Simmons B, Walji AM, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Osmium:Donohoe TJ, Callens CKA, Lacy AR, Winter C. Eur. J. Org. Chem. 2012:655–663. Donohoe TJ, Bataille CJR, Gattrell W, Kloesges J, Rossignol E. Org. Lett. 2007;9:1725–1728. doi: 10.1021/ol070430v. Donohoe TJ, Chughtai MJ, Klauber DJ, Griffin D, Campbell AD. J. Am. Chem. Soc. 2006;128:2514–2515. doi: 10.1021/ja057389g. Donohoe TJ, Churchill GH, Wheelhouse KMP, Glossop PA. Angew. Chem., Int. Ed. 2006;45:8025–8028. doi: 10.1002/anie.200603240. Donohoe TJ, Johnson PD, Pye RJ, Keenan M. Org. Lett. 2004;6:2583–2585. doi: 10.1021/ol049136i. Donohoe TJ, Johnson PD, Pye RJ. Org. Biomol. Chem. 2003;1:2025–2028. doi: 10.1039/b305189g. Donohoe TJ, Johnson PD, Cowley A, Keenan M. J. Am. Chem. Soc. 2002;124:12934–12935. doi: 10.1021/ja0276117. Donohoe TJ, Johnson PD, Helliwell M, Keenan M. Chem. Comm. 2001:2078–2079. doi: 10.1039/b107253f. Donohoe TJ, Lindsay-Scott PJ, Parker JS, Callens CKA. Org. Lett. 2010;12:1060–1063. doi: 10.1021/ol100046a.
- (7).Palladium: Desai LV, Sanford MS. Angew. Chem., Int. Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. Alexanian EJ, Lee C, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. Szolcsanyi P, Gracza T. Chem. Comm. 2005:3948–3950. doi: 10.1039/b506731f. Liskin DV, Sibbald PA, Rosewall CF, Michael FE. J. Org. Chem. 2010;75:6294–6296. doi: 10.1021/jo101171g.
- (8).Gold: de Haro T, Nevado C. Angew. Chem. Int. Ed. 2011;50:906–910. doi: 10.1002/anie.201005763.
- (9).Copper: Mancheno DE, Thornton AR, Stoll AH, Kong A, Blakey SB. Org. Lett. 2010;10:4110–4113. doi: 10.1021/ol101702w. Noack M, Gottlich R. Chem. Comm. 2002:536–537. doi: 10.1039/b111656h.
- (10).Copper: Fuller PH, Kim J-W, Chemler SR. J. Am. Chem. Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. Sequeira FC, Bovino MT, Chipre AJ, Chemler SR. Synthesis. 2012;44:1481–1484. doi: 10.1055/s-0031-1289762. Paderes MC, Chemler SR. Eur. J. Org. Chem. 2011;2011:3679–3684. doi: 10.1002/ejoc.201100444. Paderes MC, Chemler SR. Org. Lett. 2009;11:1915–1918. doi: 10.1021/ol9003492. Sherman ES, Chemler SR. Adv. Syn. Cat. 2009;351:467–471. doi: 10.1002/adsc.200800705. Karyakarte SD, Smith TP, Chemler SR. J. Org. Chem. 2012;77:7755–7760. doi: 10.1021/jo3013226.
- (11).Copper: Sanjaya S, Chua SH, Chiba S. Synlett. 2012;23:1657–1661. Sanjaya S, Chiba S. Org. Lett. 2012;14:5342–5345. doi: 10.1021/ol302525m.
- (12).Metal-free aminooxygenations: Wardrop DJ, Bowen EG, Forslund RE, Sussman AD, Weerasekera SL. J. Am. Chem. Soc. 2010;132:1188–1189. doi: 10.1021/ja9069997. Schmidt VA, Alexanian EJ. J. Am. Chem. Soc. 2011;133:11402–11405. doi: 10.1021/ja204255e. Lovick HM, Michael FE. J. Am. Chem. Soc. 2010;132:1249–1251. doi: 10.1021/ja906648w. Cochran BM, Michael FE. Org. Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165. Correa A, Tellitu I, Dominguez E, SanMartin R. J. Org. Chem. 2006;71:8316–8319. doi: 10.1021/jo061486q. Li H, Widenhoefer RA. Tetrahedron. 2010;66:4827–4831. doi: 10.1016/j.tet.2010.03.082. Tellitu I, Serna S, Moreno I, Herrero MT, Dominguez E, SanMartin R, Correa A. Eur. J. Org. Chem. 2006:437–444. Enantioselective aminooxygenation with chiral hypervalent iodine reagent: Farid U, Wirth T. Angew. Chem. Int. Ed. 2012;51:3462–3465. doi: 10.1002/anie.201107703.
- (13) (a).Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem. – Eur. J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sherman ES, Fuller PH, Kasi D, Chemler SR. J. Org. Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rappoport Z, Liebman JF. Editors The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, Part 1. John Wiley & Sons Ltd.; 2009. [Google Scholar]
- (15).Bell RP. The Proton in Chemistry. 2nd ed Chapman and Hall; 1973. [Google Scholar]
- (16).Melander L, Saunders WH., Jr. Reaction Rates of Isotopic Molecules. John Wiley and Sons; 1979. [Google Scholar]
- (17).Carey FA, Sundberg RJ. Editors Advanced Organic Chemistry, Part A: Structure and Mechanisms. Fourth Edition Kluwer Academic/Plenum Publishers; 2000. [Google Scholar]
- (18) (a).Root KS, Hill CL, Lawrence LM, Whitesides GM. J. Am. Chem. Soc. 1989;111:5405–5412. [Google Scholar]; (b) Chateauneuf J, Lusztyk J, Ingold KU. J. Org. Chem. 1988;53:1629–1632. [Google Scholar]; (c) Maillard B, Ingold KU, Scaiano JC. J. Am. Chem. Soc. 1983;105:5095–5099. [Google Scholar]
- (19).Hayes P, Suthers BD, Kitching W. Tetrahedron Lett. 2000;41:6175–6179. [Google Scholar]
- (20).Gilbert BC, Kalz W, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. Tetrahedron Lett. 1999;40:6095–6098. [Google Scholar]
- (21).Blank O, Wetzel A, Ullrich D, Heinrich MR. Eur. J. Org. Chem. 2008;2008:3179–3189. [Google Scholar]
- (22).Van Humbeck JF, Simonovich SP, Knowles RR, MacMillan DWC. J. Am. Chem. Soc. 2010;132:10012–10014. doi: 10.1021/ja1043006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Michel C, Belanzoni P, Gamez P, Reedijk J, Baerends EJ. Inorg. Chem. 2009;48:11909–11920. doi: 10.1021/ic902155m. [DOI] [PubMed] [Google Scholar]
- (24).Shi Z, Zhang C, Tang C, Jiao N. Chem. Soc. Rev. 2012;41:3381–3430. doi: 10.1039/c2cs15224j. [DOI] [PubMed] [Google Scholar]
- (25).Vogler T, Studer A. Synthesis. 2008:1979–1993. [Google Scholar]
- (26).Turnpenny BW, Hyman KL, Chemler SR. Organometallics. 2012;31:7819–7822. doi: 10.1021/om300744m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liwosz TW, Chemler SR. J. Am. Chem. Soc. 2012;134:2020–2023. doi: 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Evans DA, Johnson JS, Olhava EJ. J. Am. Chem. Soc. 2000;122:1635–1649. [Google Scholar]
- (29).Evans DA, Peterson GS, Johnson JS, Barnes DM, Campos KR, Woerpel KA. J. Org. Chem. 1998;63:4541–4544. [Google Scholar]
- (30).Wei Z-L, Duncton MAJ, Kincaid J, Kelly MG, O’Mahony D, Wang Z. PCT Int. App. 2008 Oct 30;2008130481 [Google Scholar]
- (31) (a).Reddy KL, Sharpless KB. J. Am. Chem. Soc. 1998;120:1207–1217. [Google Scholar]; (b) O’Brien P, Osborne SA, Parker DD. J. Chem. Soc., Perkin Trans. 1998;1:2519–2526. [Google Scholar]
- (32).Nolin KA, Ahn RW, Kobayashi Y, Kennedy-Smith JJ, Toste FD. Chem. Eur. J. 2010;16:9555–9562. doi: 10.1002/chem.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Denmark SE, Butler CR. Org. Synth. 2009;86:274–286. [Google Scholar]
- (34).Odell LR, Lindh J, Gustafsson T, Larhed M. Eur. J. Org. Chem. 2010:2270–2274. [Google Scholar]
- (35).Kato K, Matsuba C, Kusakabe T, Takayama H, Yamaura S, Mochida T, Akita H, Peganova TYA, Vologdin NV, Gusev OV. Tetrahedron. 2006;62:9988–9999. [Google Scholar]
- (36).Kusakabe T, Kato K, Takaishi S, Yamamura S, Mochida T, Akita H, Peganova TYA, Vologdin NV, Gusev OV. Tetrahedron. 2008;64:319–327. [Google Scholar]
- (37).Bennasar ML, Roca T, Monerris M, Garcia-Diaz D. J. Org. Chem. 2006;71:7028–7034. doi: 10.1021/jo061180j. [DOI] [PubMed] [Google Scholar]
- (38).Huntress EH, Autenrieth JS. J. Am. Chem. Soc. 1941;63:3446–3448. [Google Scholar]
- (40).Knorr R, Rossmann EC, Knittl M. Synthesis. 2010:2124–2128. [Google Scholar]
- (41).Michael FE, Cochran BM. J. Am. Chem. Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]
- (42).Bender CF, Widenhoefer RA. J. Am. Chem. Soc. 2005;127:1070–1071. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.