

Abstract
The protein Pspto_3016 is a 117-residue member of the protein domain family PF04237 (DUF419), which is to date a functionally uncharacterized family of proteins. In this report, we describe the structure of Pspto_3016 from Pseudomonas syringae solved by both solution NMR and X-ray crystallography at 2.5 Å resolution. In both cases, the structure of Pspto_3016 adopts a “double wing” α/β sandwich fold similar to that of protein YjbR from Escherichia coli and to the C-terminal DNA binding domain of the MotA transcription factor (MotCF) from T4 bacteriophage, along with other uncharacterized proteins. Pspto_3016 was selected by the Protein Structure Initiative of the National Institutes of Health and the Northeast Structural Genomics Consortium (NESG ID PsR293).
Keywords: Pspto_3016, PF04237, DUF419, structural genomics, 2KFP, 3H9X, double wing, NMR, X-ray crystallography
INTRODUCTION
We have determined the solution NMR (PDB ID 2KFP) and X-ray crystal structures (PDB ID 3H9X) of the 117-residue protein Pspto_3016 (NESG Target ID PsR293) from the plant pathogen Pseudomonas syringae (UniProtKB/TrEMBL ID Q880Y4_PSESM), a member of the protein domain family Pfam [5] PF04237 (DUF419). P. syringae is a well-known plant pathogen capable of infecting several species ranging from Arabidopsis thaliana to the tomato plant [7]. The Pspto_3016 protein from P. syringae was selected by the Northeast Structural Genomics Consortium (NESG) as part of the National Institutes of Health Protein Structure Initiative (PSI-2 and PSI-Biology) programs. The goals of PSI-2/PSI-Biology include the “structural coverage” of broadly conserved protein domain families by determination of one or more structures from each of several hundred domain families using NMR and X-ray crystallography [9, 23]. The Pfam family PF04237 remains an uncharacterized family of bacterial proteins lacking functional annotation. Pfam family PF04237 also includes domains of proteins that have been identified as human bacterial gut metagenomic targets of the PSI-Biology program [9]. Protein YjbR from Escherichia coli (NESG target ID ER226; PDB ID 2FKI) and protein DR2400 from Deinococcus radiodurans, (PDB ID 2A1V), for which 3D structures have been determined by solution NMR [30] and X-ray crystallography (U.A. Ramagopal, Y.V. Patskovsky, S.C. Almo, New York Structural GenomiX Research Consortium). Crystal structure of Deinococcus radiodurans protein DR2400, Pfam domain DUF419) respectively, are also members of Pfam PF04237. Based on structural similarities to the C-terminal DNA binding domain [12] of the MotA transcription factor (MotCF) from T4 bacteriophage, outlined below, we propose that Pspto_3016 functions as a sequence-specific DNA-binding protein.
MATERIALS AND METHODS
Protein sample preparation
Pspto_3016 was cloned with a C-terminal His6 affinity tag (LEHHHHHH), expressed, and purified following standard protocols of the NESG consortium [1], providing uniform [U-13C,15N] and uniform 100% 15N, 5% biosynthetically directed 13C (NC5) isotopically enriched samples for NMR. The expression vector plasmid for Pspto_3016 (PsR293-21) is available from the PSI Material Repository (http://psimr.asu.edu/) and from the Structural Biology Knowledgebase [14]. Pspto_3016 behaved as a monomeric protein according to static light scattering in-line with gel filtration, and based on an isotropic overall rotational correlation time (τc) of 11.2 ns that was estimated from one-dimensional 15N T1 and T1ρ relaxation data under the conditions used for NMR (1.1 mM Pspto_3016 protein, 10% v/v D2O, 100 mM NaCl, 10 mM DTT, 5 mM CaCl2, 0.02% NaN3, 20 mM ammonium acetate pH 4.5 at 20°C) (Supplementary Figure S1).
Protein NMR studies
Resonance assignments for the amide backbone 1HN, 15N, 13Cα13C' and side chain 13Cβ atoms were made automatically using AutoAssign 2.3.0 [24] after manual peak picking of the 15N-HSQC, HNCACB, CBCA(CO)NH, HNCO, HNCA, and HN(CO)CA, spectra. Additional experiments used for backbone assignments but not submitted to AutoAssign include: 15N-edited NOESY-HSQC, HBHA(CO)NH, H(CC)(CO)NH-TOCSY, and (H)CC(CO)NH-TOCSY. Side chain resonance assignments were completed manually using the aforementioned experiments together with the 13C-edited NOESY-HSQC, H(C)CH-TOCSY, H(C)CH-COSY, in addition to (H)CCH-TOCSY, and 4D 13C-13C-HMQC-NOESY-HMQC experiments collected on NC samples in 99.9% D2O. Stereospecific assignments of isopropyl side chain methyl groups of Leu and Val residues were performed using constant and non-constant time 13C-HSQC experiments collected on an NC5 sample. Dihedral angle constraints were computed by TALOS [8] based on the chemical shift assignments for backbone resonances. Preliminary structures of Pspto_3016 were calculated using AutoStructure 2.2.1 [18] before final structures were calculated using CYANA 2.1 [15, 16] and XPLOR-NIH-2.20 [28] and refined in explicit water using CNS version 1.1 [6]. The final constraints and ensemble of 20 models sorted by the lowest energy was deposited to the Protein Data Bank (PDB ID 2KFP) and the chemical shifts, NOESY FIDS, and peaklists were deposited to the database BioMagResDB (BMRB accession number, 16186).
Protein crystallization and crystallography
Triclinic crystals of Se-Met-labeled Pspto_3016 from P. syringae were obtained by hanging drop vapor diffusion at 18°C from drops containing a 1:1 mixture of protein solution (7.5 mg/mL) and well-precipitant solution. The protein solutions contained 100 mM NaCl, 5 mM DTT, 0.02% NaN3, 10 mM Tris-HCl (pH 7.5) and the reservoir solution contained 100 mM monosodium citrate (pH 4.2), 20% PEG 8000 and 60 mM NaNO3. The crystal was cryo-protected with 20% glycerol and flash-cooled in liquid nitrogen. Diffraction data sets were collected on a single crystal using the beam line X4A with a Quantum 4R detector at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory. Data were integrated and scaled with HKL2000 [25]. The crystal structure of Pspto_3016 was solved by SAD using SHELX [29]. The locations of three selenium sites per molecule were identified from 2.9 Å resolution data. Matthews coefficient calculations indicated four monomers per asymmetric unit. Initial phases obtained using SHELXD, were improved by density modification and solvent flattening (using a solvent content of 46%). After phase refinement, an initial model constructed with RESOLVE [31] was extended by ARP/wARP [26] to 2.5 Å and refined with CNS [6]. Model building was performed using Coot [10]. Several cycles of simulated annealing and minimization were carried out using the CNS program package [6]. Atomic coordinates were deposited in the Protein Data Bank (PDB ID 3H9X).
RESULTS AND DISCUSSION
The solution NMR structural ensemble of Pspto_3016 (PDB ID 2KFP) is shown in Figure 1A. The structure adopted a characteristic α/β sandwich fold similar to that of the two other proteins with known structures from Pfam PF04237, revealing the putative “double wing” DNA binding motif [21, 30]. The secondary structure is composed of three α-helices (α1–3), a solvent exposed β-sheet consisting of six anti-parallel strands (β1–6), and several loops with a secondary structure arrangement in the order N-α1-β1-β2-β3-β4-α2-β5-β6-α3-C (Figure 1B). NMR structure quality statistics are given in Table 1.
Figure 1.
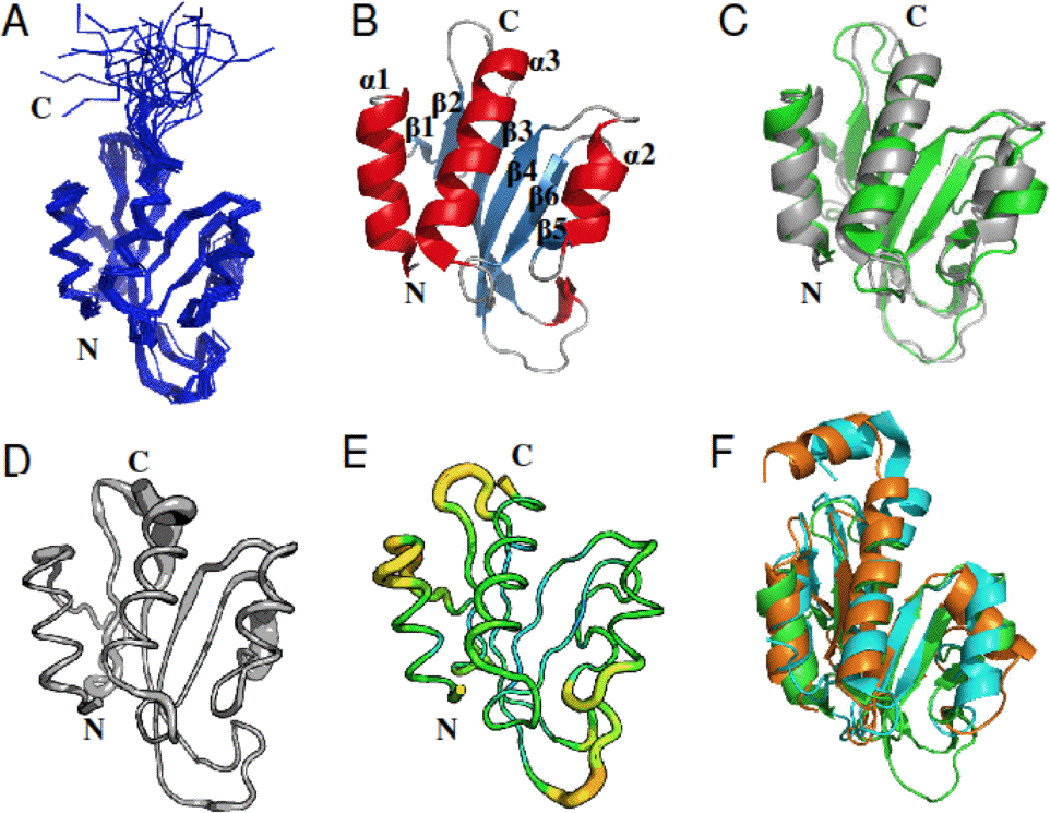
Three-dimensional structure of the Pspto_3016 protein from P. syringae (PDB IDs 2KFP and 3H9X). A) Backbone trace of the ensemble of 20 solution NMR conformers for residues 2–125 including the His6 affinity tag. B) Ribbon diagram of the NMR structure for the conformer with the lowest energy from CNS refinement. Secondary structure elements are denoted α1–3 and β1–6 for respective features. C) Alignment of the X-ray structure (green) with the lowest energy NMR conformer (gray). D) Sausage diagram of the NMR ensemble according to the RMSD in backbone Cα position represented as the thickness of the trace. E) Sausage diagram, of the X-ray structure where the thickness of the trace is reflected by increasing backbone B-factor (PyMOL script from PDBe). F) Alignment of the Pspto_3016 X-ray structure (green) with the X-ray structure of DR2400 (PDB ID 2A1V) from D. radiodurans (cyan) and NMR structure of YjbR (PDB ID 2FKI) from E. coli (orange). Unless otherwise noted, residues 2–117 are shown for Pspto_3016, residues 3–130 for DR2400, and residues 2–118 for YjbR. All structures were rendered withand alignments performed with PyMOL [27].
Table 1.
Summary of NMR and structural statistics for Pspto_3016 (PDB ID 2KFP)
| Completeness of resonance assignmentsa | |||
| Backbone (%) | 93.3 | ||
| Side chain (%) | 91.8 | ||
| Aromatic (%) | 83.0 | ||
| Stereospecific methyl (%) | 100.0 | ||
| Conformationally-restricting constraintsb | |||
| Distance constraints | |||
| Total | 1822 | ||
| Intra-residue [i = j] | 418 | ||
| Sequential [| i - j | = 1] | 481 | ||
| Medium range [1 < | i - j | < 5] | 370 | ||
| Long range [| i - j | ≥ 5] | 553 | ||
| Dihedral angle constraints | 172 | ||
| Hydrogen bond constraints | 80 | ||
| Number of constraints per residue | 17.0 | ||
| Number of long range constraints per residue | 4.9 | ||
| Residual constraint violationsb | |||
| Average number of distance violations per structure | |||
| 0.1 - 0.2 Å | 11.9 | ||
| 0.2 – 0.5 Å | 3.5 | ||
| > 0.5 Å | 0.0 | ||
| Average number of distance violations per structure | |||
| 1 – 10° | 6.3 | ||
| > 10° | 0.0 | ||
| RMSD from average coordinates (Å) b,d | |||
| Backbone atoms | 0.7 | ||
| Heavy atoms | 1.1 | ||
| MolProbity Ramachandran statistics b,d | |||
| Most favored regions (%) | 98.5 | ||
| Allowed regions (%) | 1.2 | ||
| Disallowed regions (%) | 0.3 | ||
| Global quality scores (Raw/ Z-score) b | |||
| Verify3D | 0.4 | / | −1.1 |
| ProsaII | 0.6 | / | −0.1 |
| Procheck G-factor (phi-psi)d | 0.0 | / | 0.3 |
| Procheck G-factor (all)d | 0.0 | / | −0.2 |
| MolProbity Clashscore | 24.5 | / | −2.7 |
| RPF Scoresc | |||
| Recall / Precision | 0.90 | 0.80 | |
| F-measure / DP-score | 0.85 | 0.74 |
Refers to chemical shifts for residues 2–117.
Calculated for the ensemble of 20 structures using PSVS version 1.4 [4]. Average distance violations were calculated using the sum over r−6.
RPF scores [19] calculated for the ensemble of 20 structures reflecting the goodness-of-fit to the NOESY data and resonance assignments.
Ordered residue ranges: 2–36,40–53, 57–83, 90–99, 101–116, with the sum of φ and ψ order parameters > 1.8.
The X-ray crystal structure of Pspto_3016 (PDB ID 3H9X) is shown aligned to the NMR structure in Figure 1C, and the corresponding structural refinement statistics are shown in Table 2. The NMR and crystal structure are very similar to one another, with backbone RMSD of 1.39 ± 0.14 Å, for residues with ordered secondary structure (2–14, 19–20, 29–32, 41–47, 58–65, 70–77, 80–82, 92–95, and 101–116), between the ensemble of 20 solution NMR models and the X-ray model. The 1H-15N resonances for residues Tyr40, Lys88, and Leu96 had lower peak intensity than average compared to other amide 1H-15N cross peaks. Trp39 and Ala83-Asn87 amides, and Tyr40 side chain resonances could not be assigned. Trp39 and Tyr40 residues reside on the loop separating strands β2 and β3, and residues Ala83-Lys88 are located on the extended loop connecting strands β5 and β6. These loop regions are likely to have considerable flexibility in Pspto_3016 and are represented with sausage diagrams created for the X-ray structure according according to Cα RMSD for the NMR ensemble and according to B-factor, for the X-ray structure and NMR ensemble according to Cα RMSD for the NMR ensemble and according to B-factor for the X-ray structure (Figures 1D and 1E, respectively). The sausage diagram of the NMR model (Figure 1D) reflects the increase in backbone RMSD for the β2–3 and β5–6 loops based ondue to incomplete resonance assignments and therefore a lack of structural constraints in these regions of the structure. HoweverAnalysis of the sausage diagram for the crystal structure in the region of these loops indicated that, the B-factors for residues in the β2–3 loop were slightly higher than in structured regions and that the B-factors for residues in the β5–6 is loop region in the crystal structure (Figure 1E) arewere relatively lownot unusually high compared to structured regions in the protein. Analysis of the crystal packing indicated that Trp39 in the β2–3 loop was in close proximity (< 4 Å) from Glu69 in a symmetry related molecule, and that rResidues Tyr84-Met86 of the β5–6 loop are in close proximity (< 5 Å) to Pro68- Glu69 of thea symmetry mate are in close to proximity (less 5 Å) than to residues Tyr84-Met86 of the β5–6 loop, suggesting an effect ofindicating that crystal packing may limit structural disorder in both theon β2–3 and loop β5–6 loops. Additionally, the position of helix α2 is slightly different in the NMR ensemble fromcompared to the X-ray structure, again indicating a possible consequencelikely caused by conformational mobility due to packing of packing against the β5–6 loop, which would otherwise experience conformational variability.
Table 2.
Summary of crystals, X-ray diffraction data collection and refinement statistics for Pspto_3016 (PDB ID 3H9X).
| Data collection | |||
| Space group | P1 | ||
| X-ray wavelength (Å) | 0.979 | ||
| Unit-cell parameters | a =44.302 Å, b = 48.546 Å, c = 68.047 Å | ||
| α = 87.07°, β = 92.40°, γ = 93.57° | |||
| Resolution (Å) | 30-2.5 (2.5–2.59) | ||
| Temperature (K) | 100 | ||
| Completeness (%) | 96.9 (93.5) | ||
| Redundancy | 2.0 (1.8) | ||
| I/σ (I) | 12.0 (2.3) | ||
| Rmerge | 0.044 (0.255) | ||
| Refinement | |||
| Rcryst/Rfree* = | 0.210/0.245 | ||
| No. of subunits in ASU | 4 | ||
| No. of residues | 467 | ||
| No. of water molecules | 38 | ||
| RMSD in bond lengths (Å) | 0.010 | ||
| RMSD in bond angles (°) | 1.30 | ||
| Ramachandran statistics | |||
| Most favored regions (%) | 95.7 | ||
| Allowed regions (%) | 3.5 | ||
| Disallowed regions (%) | 0.9 | ||
| Global quality scores (Raw / Z-score) | |||
| Verify3D | 0.6 | / | 1.4 |
| ProsaII | 1.0 | / | 1.4 |
| Procheck G-factor (phi-psi) | −0.3 | / | −0.8 |
| Procheck G-factor (all) | −0.4 | / | −2.4 |
| MolProbity Clashscore | 30.1 | / | −3.6 |
Values in parantheses are representative of the highest resolution bin.
Rmerge = ∑khl∑i | Ii (hkl) - <I (hkl)> |/ ∑khl∑iIi (hkl), where I (hkl) is the intensity of reflection hkl, ∑khl, is the sum over all reflections and ∑i, is the sum over I measurements of reflection hkl.
R* = ∑khl | | Fobs | - | Fcalc | |/ ∑ khl Ii (hkl) | Fobs |; Rfree * is calculated using 10% of the reflection data omitted from refinement of resonance assignments and Rcryst is calculated for the remaining reflections.
Submission of the atomic coordinates for the NMR ensemble to the Dali [17] server identified two structures in the Protein Data Bank of orthologous bacterial proteins from Pfam PF04237 as structurally similar. The crystal structure of 132 residue hypothetical protein DR2400 from D. radiodurans (PDB ID 2A1V) was the most similar with a Z-score of 9.9 (seq ID = 32% and BLAST E-value = 4 ×10−4 obtained from the Kegg [20] database) followed by the NMR structure of 118-residue hypothetical protein YjbR (PDB ID 2FKI; NESG ID ER226) [30] from E. coli with a Z-score = 9.1 (seq ID = 33% and BLAST E-value = 4 ×10−4). The primary difference between these structures and Pspto_3016 is that DR2400 and YjbR lack the pronounced loop segment between strands β3 and β4, but also contain a short additional α-helix motif at the C-terminus (Figure 1F). Pspto_3016, like YjbR and DR2400, contains the highly conserved Asn-Lys-X-His-Trp motif [30] found on the loop connecting strands β5-β6. This sequence motif is characteristic of many members of Pfam PF04237. Further analysis of the Dali results identified the crystal structure of the 107-residue MotCF domain (PDB ID 1KAF) [21] of transcription factor MotA from bacteriophage T4, which has a lower Z-score of 4.9 but still exhibits many structural similarities despite a low sequence identity of 10%.
The X-ray crystal structure of MotCF (PDB ID 1KAF) revealed a novel DNA-binding α/β motif, composed of three α-helices and a six-stranded β-sheet [21], which together with the activation domain MotNF, function as the transcription factor MotA [11, 12, 22]. In MotCF, the putative DNA binding region of the double wing motif is composed of many electropositive and aromatic residues on the solvent exposed β-sheet, flanked by the two extended loop regions connecting strands β1-β2 and β5-β6, forming a binding pocket that resembles the shape of a saddle [21]. MotCF displays a similar fold to the aforementioned α/β sandwich present in Pspto_3016. The two extended loops separating strands β1-β2, and between β5-β6, are structurally conserved features in Pspto_3016 as well as in the structures for proteins DR2400 and YjbR. Analysis of the electrostatic surface potential of Pspto_3016 with APBS [3] reveals a highly positively charged surface corresponding to the loops and β-sheet of the saddle (Figure 2A), including the surface surrounding the strongly-conserved Asn-Lys-X-His-Trp motif (Figure 2B). In MotCF, the β5-β6 loop is predicted to interact with the DNA major groove [21]. The sequence Asn-Gly-Asn-Val-Tyr of MotCF, structurally aligns with the Asn-Lys-X-His-Trp motif on the β5-β6 loop of Pspto_3016, and includes Tyr191 on the β6 strand, a predicted DNA-binding residue [21]. In MotCF, additional residues, Tyr134 and Arg135 on the loop between strands β1-β2 and Lys166 on the C-terminal end of strand β4 (Figure 2C) are also predicted to interact with DNA [21]. These structurally align with Trp22, Lys24 and Lys65 in Pspto_3016 (Figure 2D).
Figure 2.
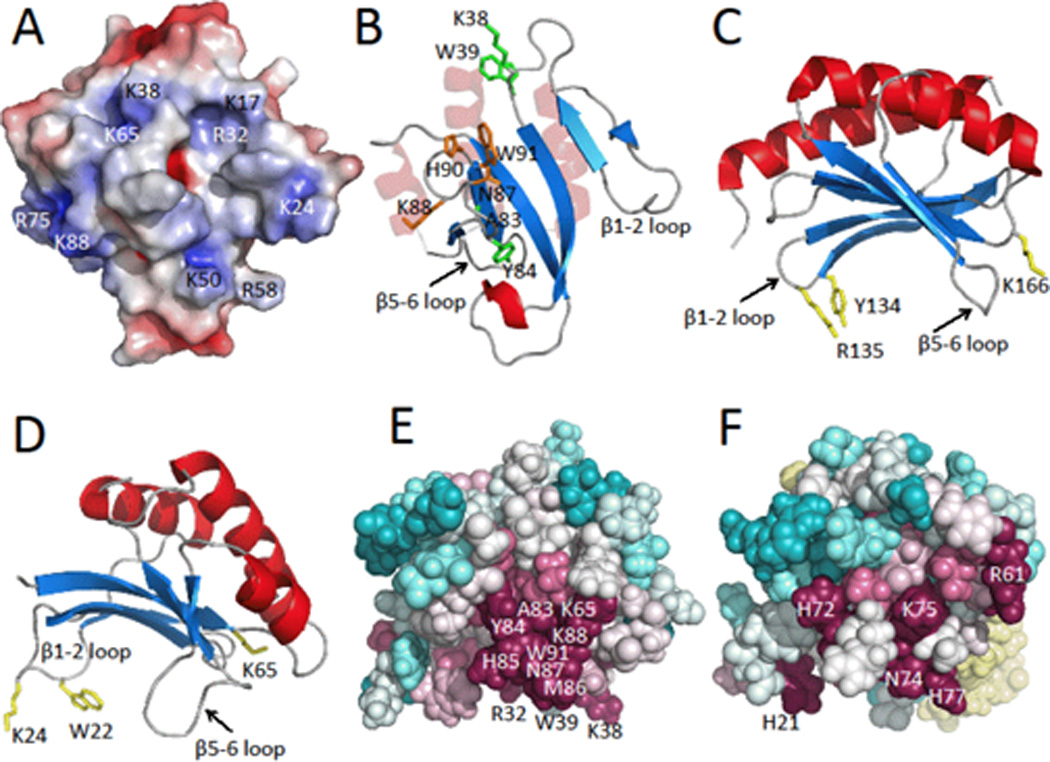
A) Electrostatic surface potential map of the lowest energy NMR conformer of Pspto_3016 generated with APBS [3] where negatively charged regions are represented in red, neutral regions in white, and positively charged regions in blue (± 5 kT/e). Locations of alkaline surface residues are indicated. B) Ribbon diagram with highlighted residue side chains represented as sticks. Residues Asn87, Lys88, His90, and Trp91 of the highly conserved Asn-Lys-X-His-Trp motif, are shown in orange, residues Lys38, Trp39, Ala83, and Tyr84 are shown in green. Pspto_3016 is shown in the same orientation as panel A. C) Ribbon diagram of MotCF (PDB ID 1KAF; residues 105–211 displayed) shown in the saddle orientation with highlighted putative DNA binding residues Tyr134, Arg135, and Lys166 in yellow. D) Ribbon diagram of Pspto_3016 in the saddle orientation with residues Trp22, Lys24, and Lys65 highlighted in yellow. E) Pspto_3016 ConSurf [2] representation shown with spheres, using the aligned Pfam PF04237 sequences clustered from the CLANS [13] sub-family. Conserved surface residues Arg32, Lys38, Trp39, Lys65, Ala83, Tyr84, His85, Met86, Asn87, Lys88, and Trp91 are indicated. EF) ConSurf representation of YjbR from Escherichia coli using the corresponding CLANS sub-family sequences. YjbR was aligned to Pspto_3016 in the same orientation as panels D and E. Residues N74, K75, and H77 of the Asn-Lys-X-His-Trp motif are indicated on the surface and Trp78 is buried. Additional conserved residues His21, Arg61, and His72 are indicated.
A CLANS (13) cluster analysis of the 1,845 Uniprot sequences from Pfam PF04237 revealed two major groupings of closely related sequences in the family (Figure 3). Using a CLANS p-value cutoff of 1 × 10−10 Pspto_3016 was found in a distinct sub-family of 374 sequences, whereas YjbR was found in a separate sub-family with 1,073 other sequences (Figure 3), including the 33% identical protein DR2400. The sequences of each sub-family were extracted to generate multiple alignment files using ClustalW [32], and submitted to the ConSurf server [2] to map conserved surface residues from each sub-family onto the corresponding structures of Pspto_3016 and YjbR (Figures 2E and 2F). The location of the conserved surface patches for each sub-family was similarly located, surrounding the surface of the extended β5-β6 loop, presumably the site of DNA major groove interactions. Uniquely conserved in the Pspto_3016-containing sub-family was an insertion of residues 50–59 corresponding to the loop between strands β3-β4 (Alignment in Supplementary Figure S3). Additionally present was a Lys-Trp motif (Lys38 and Trp39) in the short loop connecting strands β2-β3 and an Ala-Tyr motif (Ala83 and Tyr84) of the β5-β6 loop (Figure 2B and 2E), which was not conserved in the YjbR sub-family. However, YjbR residues His21, Arg61, and His72 were conserved surface residues among its corresponding sub-family (Figure 2F). The conserved residues in each sub-family may be important for sequence specificity to the respective DNA substrate for the proteins from Pfam PF04237.
Figure 3.
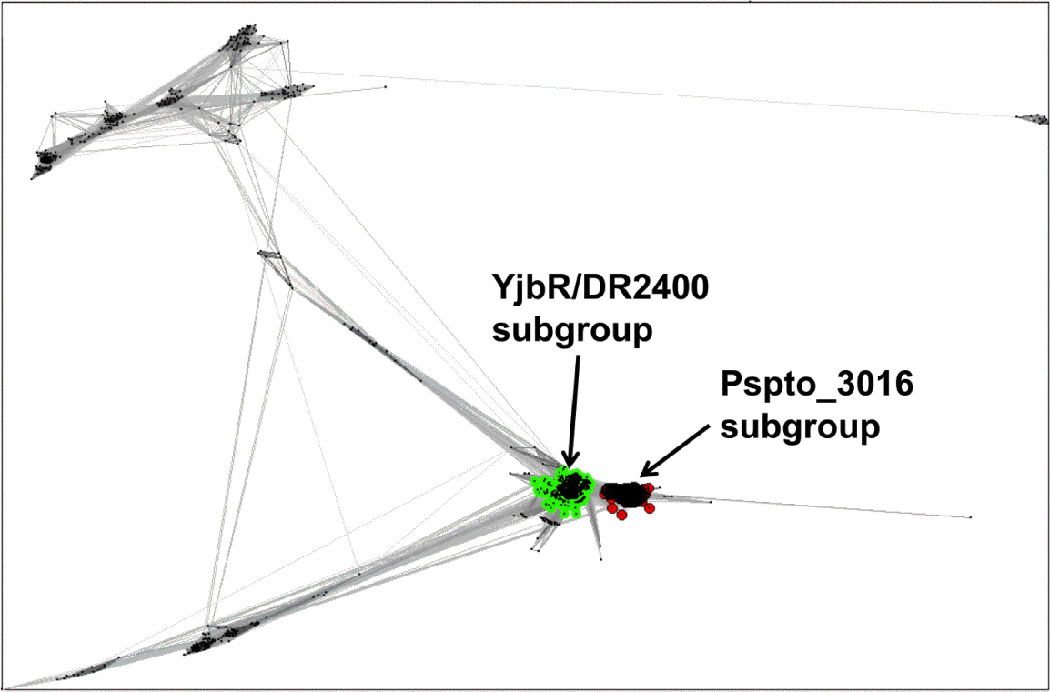
Cluster map of the Pfam PF04237 protein family using CLANS [13]. Clusters were determined using a BLAST p-value cutoff of 1 × 10−10 to run the CLANS all-to-all pairwise comparison of the 1,845 sequences for PF04237 from the Uniprot database. The cluster containing Pspto_3016 is indicated on the right, and the cluster containing YjbR and DR2400 is on the left.
In summary, we have presented the solution NMR and X-ray crystal structures of Pspto_3016 from P. syringae, a member of the uncharacterized protein family PF04237 (DUF419). Sequence homology and structural similarities led to the identification of the MotCF domain of transcription factor MotA from T4 bacteriophage as a potential functional homolog and the putative function as a DNA-binding protein. In addition, other double wing proteins from Pfam family PF04237 with known three-dimensional structures: , YjbR from E. coli and DR4200 from D. radiodurans, are structurally similar and a have a high degree of conservation for several exposed electropositive and aromatic residues in the potential DNA-binding regions, including the highly conserved Asn-Lys-X-His-Trp motif [30]., These observations as well as the overall structural similarity with the known DNA-binding protein MotCF, strongly suggest that Pspto_3016 is a sequence-specific DNA binding protein, and that this molecular function is common to members of the PF04237 domain family. However, since MotCF is the putative DNA-binding domain of the larger MotA protein that utilizes the activation domain MotNF for associating with other protein complexes, the independent gene product Pspto_3016 may require other yet unknown proteins in order to form complexes with DNA to carry out its physiological function.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank John A. Schwanof and Randy Abramowitz for their support at the X4 beam line, National Synchrotron Light Source, Brookhaven National Lab.
This work was supported by grants from the National Institute of General Medical Sciences Protein Structure Initiative (PSI) of the National Institutes of Health, PSI-2 (P50 GM 074958) and PSI:Biology (U54-GM094597). NMR data collection was conducted at the Ohio Center of Excellence in Biomedicine in Structural Biology and Metabonomics at Miami University.
REFERENCES
- 1.Acton TB, Xiao R, Anderson S, Aramini J, Buchwald W, Ciccosanti C, Conover K, Everett JK, Hamilton K, Huang Y, Janjua H, Kornhaber G, Lau J, Lee D, Liu G, Maglaqui M, Ma L, Mao L, Patel D, Rossi P, Sahdev S, Shastry R, Swapna GVT, Tang Y, Tong S, Wang D, Wang H, Zhao L, Montelione GT. Preparation of protein samples for NMR structure, function, and small-molecule screening studies. Methods in Enzymology. 2010;493:21–60. doi: 10.1016/B978-0-12-381274-2.00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Battacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consotria. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 5.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 6.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 7.Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, Deboy RT, Durkin AS, Kolonay JF, Madupu R, Daugherty S, Brinkac L, Beanan MJ, Haft DH, Nelson WC, Davidsen T, Zafar N, Zhou L, Liu J, Yuan Q, Khouri H, Fedorova N, Tran B, Russell D, Berry K, Utterback T, Van Aken SE, Feldblyum TV, D'Ascenzo M, Deng WL, Ramos AR, Alfano JR, Cartinhour S, Chatterjee AK, Delaney TP, Lazarowitz SG, Martin GB, Schneider DJ, Tang X, Bender CL, White O, Fraser CM, Collmer A. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci U S A. 2003;100:10181–10186. doi: 10.1073/pnas.1731982100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 9.Dessailly BH, Nair R, Jaroszewski L, Fajardo JE, Kouranov A, Lee D, Fiser A, Godzik A, Rost B, Orengo C. PSI-2: structural genomics to cover protein domain family space. Structure. 2009;17:869–881. doi: 10.1016/j.str.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 11.Finnin MS, Cicero MP, Davies C, Porter SJ, White SW, Kreuzer KN. The activation domain of the MotA transcription factor from bacteriophage T4. EMBO J. 1997;16:1992–2003. doi: 10.1093/emboj/16.8.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finnin MS, Hoffman DW, White SW. The DNA-binding domain of the MotA transcription factor from bacteriophage T4 shows structural similarity to the TATA-binding protein. Proc Natl Acad Sci U S A. 1994;91:10972–10976. doi: 10.1073/pnas.91.23.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- 14.Gabanyi MJ, Adams PD, Arnold K, Bordoli L, Carter LG, Flippen-Andersen J, Gifford L, Haas J, Kouranov A, McLaughlin WA, Micallef DI, Minor W, Shah R, Schwede T, Tao YP, Westbrook JD, Zimmerman M, Berman HM. The Structural Biology Knowledgebase: a portal to protein structures, sequences, functions, and methods. J Struct Funct Genomics. 2011;12:45–54. doi: 10.1007/s10969-011-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 16.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang YJ, Moseley HN, Baran MC, Arrowsmith C, Powers R, Tejero R, Szyperski T, Montelione GT. An integrated platform for automated analysis of protein NMR structures. Methods Enzymol. 2005a;394:111–141. doi: 10.1016/S0076-6879(05)94005-6. [DOI] [PubMed] [Google Scholar]
- 19.Huang YJ, Powers R, Montelione GT. Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J Am Chem Soc. 2005b;127:1665–1674. doi: 10.1021/ja047109h. [DOI] [PubMed] [Google Scholar]
- 20.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li N, Sickmier EA, Zhang R, Joachimiak A, White SW. The MotA transcription factor from bacteriophage T4 contains a novel DNA-binding domain: the 'double wing' motif. Mol Microbiol. 2002;43:1079–1088. doi: 10.1046/j.1365-2958.2002.02809.x. [DOI] [PubMed] [Google Scholar]
- 22.Li N, Zhang W, White SW, Kriwacki RW. Solution structure of the transcriptional activation domain of the bacteriophage T4 protein, MotA. Biochemistry. 2001;40:4293–4302. doi: 10.1021/bi0028284. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Montelione GT, Rost B. Novel leverage of structural genomics. Nat Biotechnol. 2007;25:849–851. doi: 10.1038/nbt0807-849. [DOI] [PubMed] [Google Scholar]
- 24.Moseley HN, Monleon D, Montelione GT. Automatic determination of protein backbone resonance assignments from triple resonance nuclear magnetic resonance data. Methods Enzymol. 2001;339:91–108. doi: 10.1016/s0076-6879(01)39311-4. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 27.Schrödinger L. The PyMOL Molecular Graphics System, Version 0.99rc6. [Google Scholar]
- 28.Schwieters C, Kuszewski J, Clore G. Using Xplor-NIH for NMR molecular structure determination. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;48:47–62. [Google Scholar]
- 29.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 30.Singarapu KK, Liu G, Xiao R, Bertonati C, Honig B, Montelione GT, Szyperski T. NMR structure of protein yjbR from Escherichia coli reveals 'double-wing' DNA binding motif. Proteins. 2007;67:501–504. doi: 10.1002/prot.21297. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.