

Abstract
We previously reported that substituted 4-aminoquinolines with a phenylether substituent at the 7-position of the quinoline ring and the capability of intramolecular hydrogen bonding between the protonated amine on the side chain and a hydrogen bond acceptor on the amine’s alkyl substituents exhibited potent antimalarial activity against the multi-drug resistant strain P. falciparum W2. We employed a parallel synthetic method to generate diaryl ether, biaryl, and alkylaryl 4-aminoquinoline analogs, in the background of a limited number of side chain variations that had previously afforded potent 4-aminoquinolines. All subsets were evaluated for their antimalarial activity against the chloroquine-sensitive strain 3D7 and the chloroquine-resistant K1 and cytotoxicity mammalian cell lines. While all three arrays showed good antimalarial activity, only the biaryl-containing subset showed consistently good potency against the drug-resistant K1strain good selectivity with regard to mammalian cytotoxicity. Overall, our data indicate that the biaryl-containing series contains promising candidates for further study.
Introduction
Malaria, a devastating infectious disease caused by five species of Plasmodium, affects 200–500 million people and causes over 800,000 deaths annually.1 The most lethal form of malaria is caused by Plasmodium falciparum.2 Since the development of quinine, drugs containing the quinoline nucleus have been a mainstay of antimalarial therapy.3,4 However, the spread of chloroquine (CQ) and mefloquine resistant strains of P. falciparum has curbed the usefulness of this class of drugs.5,6 In the past two decades, a number of new potent quinolines that overcome chloroquine resistance have been reported.7–9 Most contain the 7-chloroquinoline nucleus of chloroquine and vary in the length and nature of their basic amine side chain.10–12
We have reported extensive studies on the role of side chains for the antimalarial activity and efficacy.13, 14 Additional work in our laboratory systematically examined modification of the quinoline ring and the basic side chain of 4-aminoquinolines (1) 13, 14,15, 16 demonstrating that intramolecular hydrogen bonding (2) between the protonated amine and the hydroxyl group on the tertiary basic amine is crucial for the potency against CQ-resistant strains. In a similar systematic survey we found that most modifications to the quinoline nucleus resulted in lowered potency and had little effect on relative potency in multi-drug resistant (MDR) strains, such as W2.15, 16 This finding was in line with the consensus model from the literature that requires an small, electron withdrawing group at this position (chlorine or trifluoromethyl). From these studies, one finding that did stand out was the high potency of the 7-PhO substitution in the context of a shorter amino containing side chain, which was generically more potent against MDR strains. In addition other studies showed that exchanging the quinoline ring system with an acridine ring system in the context of the same side chain gave improved antimalarial potency against both CQ-sensitive and CQ-resistant strains, suggesting that increased bulk and/or hydrophobicity might be advantageous.17 Given these previous SAR results, we designed a new array of 4-aminoquinoline analogs (3) to explore this hypothesis by using parallel medicinal chemistry. Here we report the design, synthesis, and biological evaluation of this array.
Our strategy for the design of this compound array is shown in Figure 1. We fixed the side chain portion using a propyl spacer (three carbons) between two amines because such shorter side chain variants have consistently given better antimalarial potency than longer chains (> four carbons).16 A fixed set of substitutions on the terminal alkyl amine was chosen to allow for combinatorial interactions with the quinoline nucleus within the highly potent “chemical space.” This diversity set was based on previously obtained SAR for chloroquine analogs. The 7-position of the quinoline ring system was varied through a range of substituents that were accessible by the Ullmann coupling,18, 19 Suzuki coupling,20, 21 and Negishi coupling reactions,22 which respectively generate diaryl ether, biaryl, and alkylaryl substitutions. These three sub-series were chosen to systematically vary the size, hydrogen bonding capabilities, electrostatics, and hydrophobicity at the 7-position. Overall this study was expected to firmly establish which types of substitutions are tolerated at the 7-position and how this position interacts with the side chain in defining potency in CQ-resistant Plasmodium species.
Figure 1. Rational design for 7-substituted 4-aminoquinolines.

The three carbon spacer between two amines and the terminal secondary amine was selected since this pair consistently gave high potency in previous work. To explore substitution on the quinoline ring at the 7-position metal assisted-coupling reactions including the Ullmann, Suzuki, and Negishi reactions were applied to generate diarylether, biaryl, and alkylaryl subsets, respectively.
Chemistry
The general approach used to produce this compound array followed our previously established route (Scheme 1). The main challenge in this work was the development of a robust, general route to 7-bromo-4-chloroquinoline 7a, which served as the key intermediate for the diversification. Since chloro and bromo substituted carbons in general have strongly differing reactivity, this intermediate allowed for the selective synthesis of all targeted compounds. The synthetic route to the key intermediate 7a followed a previously reported method.16 Condensation of 3-bromoaniline with Meldrum’s acid 4 and trimethyl orthoformate gave enamine 5. Cyclization with microwave acceleration gave regioisomers 6a and 6b, as a 2:1 mixture that was inseparable by both flash column chromatography and crystallization. Chlorination of the mixture with POCl3 allowed the successful separation of 7-bromo-4-chloroquinoline 7a, the key intermediate, from its isomer 5-bromo-4-chloroquinoline 7b, by column chromatography. Condensing 7b with 20 equivalents of 1,3-propyldiamine afforded compound 8, which was used in the next coupling reaction without purification.
Scheme 1. Synthesis of the compound array 3{x,y,z}.
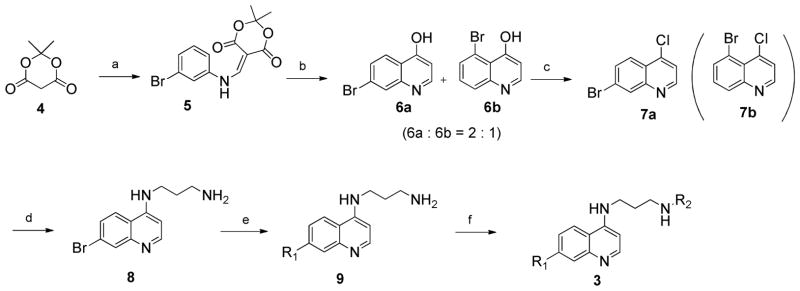
Reagents and conditions: (a) 3-bromoaniline, triethylorthoformate, reflux, 1h; (b) 225 °C, MW, Ph2O, 5 min; (c) POCl3, toluene, 100 °C, 1 h; (d) 1,3-aminopropane, K2CO3, TEA, NMP, 140 °C, 1 h; (e) for Ullmann reagent: phenols, CuI, Cs2CO3, Ligand, 1,4-dioxane, 4 Å MS, reflux, 24 h; for Suzuki reagents: boronic acids, PdCl2(dppf), Cs2CO3, toluene, 4 Å MS, 100 °C, 24 h; for Negishi reagents: zinc halides, Pd(PPh3)4, THF, reflux, 16 h.
Intermediate 8 was coupled with three sets of building blocks (diversity sets shown in Figure 2): phenols (x=1, y=1–8), boronic acids (x=2, y=1–8), and alkyl/benzyl zinc halides (x=3, y=1–12) in the presence of the corresponding metal catalyst. Each diversity set was designed to sample widely in sterics, electrostatics, and hydrophobicity. This parallel diversification step resulted in the production of the array of 7-substituted-4-aminoquinolines 9{x,y}. The final array was produced by an orthogonal diversification at the terminal amine. Each member of array 9{x,y} was treated in parallel with the four aldehydes defined from our previous studies (z=1–4) to give 4,7-disubstituted quinoline analogs 3{x,y,z}.
Figure 2.

Diversity Sets used in Generating the Compound Array.
The crude products were purified by flash chromatography using a SP-1 (Biotage) purification system with dichloromethane and methanol mixture (1 -> 50% gradient condition). Each purified product was then dissolved in a saturated methanolic solution of hydrochloric acid and then evaporated to dryness to afford the HCl salts. Purity and identity were verified for all compounds by UPLC/MS and 1H-NMR. The yield, purity, and NMR data of final compounds are available in the Supporting Information. All purified products were dissolved in DMSO to a standard concentration of 10 mM for biological testing.
Biological and Pharmacological Testing
The compounds in array 3{x,y,z} were tested against CQ-sensitive (3D7) and CQ- resistant (K1) strains of P. falciparum in concentration-respons e experiments in a three-fold dilution schema spanning 15 μM to 0.7 nM, using a previously established method.23 All experiments were carried out in triplicate and each triplicate experiment being replicated on two separate days, for a total of 9 replicates. Data are reported as mean EC50 values with 95% confidence limits.
The compounds in array 3{x,y,z} were also tested for growth inhibition against four human cell lines: Raji, a Burkett’s lymphoma derived line; BJ, an immortalized foreskin fibroblast derived line; HEK 293, an embryonic kidney fibroblast derived line; and HepG2, a hepatocellular carcinoma derived line. The cytotoxicity was determined by detection of total cellular ATP levels (Cell Titer Glo). The same concentration-response plates were used for both antimalarial and growth proliferation experiments. All experiments were carried out in triplicate with one replicate for a total of 3 replicates. Data are reported as mean EC50 values with 95% confidence limits. Finally, the kinetic solubility and passive permeability of the molecules in array 3(x,y,z) were investigated to provide guidance in interpreting cell based assay results and to inform considerations about the development potential of the compounds. Solubility was determined using the pIon method by dilution from DMSO stocks into PBS buffer containing citric acid at neutral pH (7.4).24 Permeability was assessed using the pIon implementation of the parallel artificial membrane permeation assay (PAMPA) in PBS buffer containing citric acid at neutral pH (7.4), thus reflecting the conditions required for activity in cell-based assays.25 Both assays emulate the conditions used to determine cytotoxicity and antimalarial activity. The results of this testing schema are discussed below, broken out by subset of the analogs at the 7-position of the quinoline nucleus.
Diaryl Ether Series
Thirty two diaryl ethers derived from the Ullmann reaction were synthesized and evaluated. The data is summarized in the heat map in Table 1, which was produced by using the EC50 values determined by fitting curves to the concentration-response experiments described above. The tabulated experimental values and errors can be found in the supplemental materials. Of 32 diaryl ethers, 21 compounds showed reasonable potency according to the MMV lead criteria (EC50 < 50 nM) against the CQ-sensitive 3D7 strain. However, only two compounds (3{1,4,1} and 3{1,7,4}) exhibited highly potent activity (20 nM and 31 nM EC50, respectively) against the CQ-resistant strain K1. This is in stark contrast to the results with the analogous chloro compounds, which are all equivalent compounds are roughly equipotent against the two strains.13 One set of compounds stands out: 3{1,1,4}, 3{1,3,4}, and 3{1,5,4}, all carrying the 3-F-6-MeO-Bn alkyl amine substituent, showed enhanced potency in strain K1. Generally the furfuryl (z=1) and 3-F-6-MeO-Bn (z=4) substituted alkylamines in this series exhibited better potency against strain K1 than the 2-HO-3-MeO-Bn (z=2) and piperonyl (z=3) substituents. All of 2-HO-3-MeO-Bn (z=2) substituted alkylamines had poor antimalarial activity against strain K1, whereas they were highly potent against strain 3D7. Electronic effects were not strongly evident for the diaryl ethers, but electron-donating substituents showed slightly better activity against strain 3D7. The compounds were tested against a panel of four mammalian cell lines (HepG2, HEK293, Raji, and BJ). In general HepG2 cells were the most sensitive, while other lines were less sensitive against compounds (EC50 >5 uM). Table 1 shows only HepG2 data, while all cytotoxicity data is summarized in the Supporting Information. Most of the compounds active against strain 3D7 (< 50 nM EC50) showed moderate cytotoxicity (EC50 3–15 μM) in HepG2, resulting in a reasonable average selectivity for the parasite (> 100-fold). Actives against strain K1 (< 50 nM EC50) also showed at least 100-fold selectivity. All compounds in this series had reasonable solubility (14.5–46 μM) and good permeability (317–1309 × 10−6 cm/s) in pH 7.4 buffer (Table 1 in Supporting Information).
Table 1.
Biological and pharmacological evaluation of diaryl ethers: antimalarial activity and cytotoxicity.
| 3{x,y,z} | R1 | R2 | 3D7a EC50 (μM) | K1a EC50 (μM) | K1/3D7 | HepG2b EC50 (μM) | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|---|---|
| CQ | -- | -- | 0.03 | 1.0 | 33 | 25 | 833 | 33 |
| 3{1,4,1} | 4-MeO-PhO | Furfuryl | 0.004 | 0.020 | 5.6 | 5.3 | 1479 | 265 |
| 3{1,7,4} | 3-Me2N-PhO | 3-F-6-MeO-Bn | 0.007 | 0.031 | 4.7 | 3.3 | 514 | 110 |
| 3{1,1,3} | PhO | Piperonyl | 0.018 | 0.078 | 4.3 | 2.7 | 149 | 34 |
| 3{1,2,1} | 2-MeO-PhO | Furfuryl | 0.001 | 0.087 | 66.9 | 6.5 | 5014 | 75 |
| 3{1,5,1} | 4-F-PhO | Furfuryl | 0.026 | 0.096 | 3.6 | 16.0 | 611 | 167 |
| 3{1,6,4} | 4-Cl-PhO | 3-F-6-MeO-Bn | 0.031 | 0.109 | 3.5 | 4.5 | 143 | 41 |
| 3{1,5,4} | 4-F-PhO | 3-F-6-MeO-Bn | 0.297 | 0.135 | 0.5 | 6.6 | 22 | 49 |
| 3{1,1,4} | PhO | 3-F-6-MeO-Bn | 0.162 | 0.138 | 0.9 | 1.5 | 9 | 11 |
| 3{1,3,1} | 3-MeO-PhO | Furfuryl | 0.009 | 0.153 | 17.4 | 15.5 | 1763 | 102 |
| 3{1,7,1} | 3-Me2N-PhO | Furfuryl | 0.027 | 0.155 | 5.7 | 16.0 | 588 | 103 |
| 3{1,8,2} | 4-TERTBu-PhO | 2-HO-3-MeO-Bn | 0.054 | 0.249 | 4.6 | 1.5 | 28 | 6 |
| 3{1,3,4} | 3-MeO-PhO | 3-F-6-MeO-Bn | 2.360 | 0.296 | 0.1 | 7.1 | 3 | 24 |
| 3{1,6,3} | 4-Cl-PhO | Piperonyl | 0.038 | 0.320 | 8.4 | 7.2 | 189 | 22 |
| 3{1,4,2} | 4-MeO-PhO | 2-HO-3-MeO-Bn | 0.004 | 0.375 | 91.5 | 5.5 | 1347 | 15 |
| 3{1,2,3} | 2-MeO-PhO | Piperonyl | 0.014 | 0.517 | 37.7 | 5.9 | 427 | 11 |
| 3{1,3,3} | 3-MeO-PhO | Piperonyl | 0.012 | 0.600 | 50.0 | 14.1 | 1178 | 24 |
| 3{1,4,4} | 4-MeO-PhO | 3-F-6-MeO-Bn | 0.054 | 0.660 | 12.2 | 7.0 | 129 | 11 |
| 3{1,5,3} | 4-F-PhO | Piperonyl | 0.015 | 0.667 | 44.5 | 16.0 | 1067 | 24 |
| 3{1,6,2} | 4-Cl-PhO | Furfuryl | 0.011 | 0.785 | 70.7 | 5.7 | 514 | 7 |
| 3{1,6,1} | 4-Cl-PhO | Furfuryl | 0.040 | 0.793 | 19.8 | 7.8 | 194 | 10 |
| 3{1,2,4} | 2-MeO-PhO | 3-F-6-MeO-Bn | 0.022 | 0.960 | 44.0 | 6.4 | 293 | 7 |
| 3{1,4,3} | 4-MeO-PhO | Piperonyl | 0.046 | 1.424 | 30.9 | 7.9 | 172 | 6 |
| 3{1,1,2} | PhO | 2-HO-3-MeO-Bn | 0.015 | 1.843 | 122.0 | 6.8 | 450 | 4 |
| 3{1,7,2} | 3-Me2N-PhO | 2-HO-3-MeO-Bn | 0.010 | 2.044 | 215.1 | 7.3 | 765 | 4 |
| 3{1,8,3} | 4-TERTBu-PhO | Piperonyl | 0.137 | 2.451 | 17.9 | 0.9 | 7 | 0.4 |
| 3{1,8,4} | 4-TERTBu-PhO | 3-F-6-MeO-Bn | 0.106 | 2.780 | 26.2 | 7.2 | 68 | 3 |
| 3{1,3,2} | 3-MeO-PhO | 2-HO-3-MeO-Bn | 0.072 | 3.026 | 42.0 | 5.3 | 74 | 2 |
| 3{1,5,2} | 4-F-PhO | 2-HO-3-MeO-Bn | 0.011 | 3.132 | 282.2 | 11.0 | 990 | 4 |
| 3{1,7,3} | 3-Me2N-PhO | Piperonyl | 1.363 | 3.930 | 2.9 | 16.0 | 12 | 4 |
| 3{1,8,1} | 4-TERTBu-PhO | Furfuryl | 1.110 | 4.809 | 4.3 | 3.1 | 3 | 1 |
| 3{1,2,2} | 2-MeO-PhO | 2-HO-3-MeO-Bn | 0.009 | 5.701 | 626.4 | 11.2 | 1227 | 2 |
| 3{1,1,1} | PhO | Furfuryl | 0.314 | 5.971 | 19.0 | 15.2 | 48 | 3 |
| Heat map | 3D7 EC50 (μM) | K1 EC50 (μM) | K1/3D7 | HepG2 EC50 | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|
| <0.01 | <0.01 | >100 | >15 | >1000 | >1000 | |
| <0.05 | <0.05 | >50 | >10 | >500 | >500 | |
| <0.1 | <0.1 | >10 | >5 | >100 | >100 | |
| <0.5 | <0.5 | >1 | >1 | >50 | >50 | |
| >0.5 | >0.5 | <1 | <1 | <50 | <50 |
Activity summary of the compounds was sorted by EC50 value against K1 strain.
Values are the mean of three independent experiments in triplicate.
Values are means of one experiment in triplicate.
Biaryl Series
Next we evaluated the biological activity of the biaryl series derived from the Suzuki reaction. All data for this series is summarized in the Supporting Information and selected data are shown in Table 2. Most compounds in this series showed high potency against strain 3D7. Interestingly, many of them were also highly potent (<50 nM EC50) against strain K1. Thus there is a stark contrast between the diarylether and the biaryl series with respect to their activity against strain K1, which expresses the PfCRT transporter, the primary mediator of CQ resistance. However, compounds 3{2,1,2} and 3{2,5,2}, the most potent against 3D7 (both 9 nM EC50), only showed weak potency against K1 (850 nM and 1950 nM EC50 values, respectively) resulting in high selectivity for 3D7 (93 and 213-fold). As was the case with the diaryl ether series, all 2-HO-3-MeO-Bn substituted alkylamines showed very good activity in strain 3D7. A notable trend was observed: increasing hydrophobicity in this series was accompanied by decreasing antimalarial activity. For example, all tert-Bu-Ph (y=6) substituted, naphthyl substituted (y=3), and 3,5-CF3-Ph substituted (y=2) compounds showed poor activity in both 3D7 and K1. In contrast to the diaryl ether series, compounds with 2-HO-3-MeO-Bn (z=2) and piperonyl (z=3) substituents on the terminal alkylamine exhibited good activity against strain K1 (EC50 < 50 nM). Although this series displayed moderate overall cytotoxicity (0.7–15 μM EC50 in HepG2), all compounds with low nanomolar EC50’s against 3D7 and K1 had a good selectivity for the parasite relative to mammalian cells. All compounds in this series also have good solubility (32–42 μM) and good permeability (237–2101 × 10−6 cm/s) in pH 7.4 buffer (Table 2 in Supporting Information)
Table 2.
Biological and pharmacological evaluation for biaryls: antimalarial activity and cytotoxicity.
| No | R1 | R2 | 3D7a EC50 (μM) | K1a EC50 (μM) | K1/3D7 | HepG2b EC50 (μM) | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|---|---|
| CQ | -- | -- | 0.03 | 1.0 | 33 | 25 | 833 | 33 |
| 3{2,8,1} | 4-MeO-Ph | Furfuryl | 0.011 | 0.023 | 2.2 | 6.00 | 565.7 | 261.9 |
| 3{2,7,4} | Piperonyl | 3-F-6-MeO-Bn | 0.031 | 0.024 | 0.8 | 6.04 | 194.7 | 255.8 |
| 3{2,4,2} | 4-CF3-Ph | 2-HO-3-MeO-Bn | 0.013 | 0.024 | 1.8 | 2.21 | 166.2 | 91.0 |
| 3{2,8,3} | 4-MeO-Ph | Piperonyl | 0.029 | 0.026 | 0.9 | 5.63 | 192.9 | 218.3 |
| 3{2,7,3} | Piperonyl | Piperonyl | 0.026 | 0.027 | 1.0 | 4.79 | 185.0 | 178.8 |
| 3{2,8,4} | 4-MeO-Ph | 3-F-6-MeO-Bn | 0.096 | 0.029 | 0.3 | 5.96 | 62.1 | 205.5 |
| 3{2,5,3} | Ph | Piperonyl | 0.017 | 0.029 | 1.7 | 4.07 | 234.0 | 138.9 |
| 3{2,1,3} | 4-F-Ph | Piperonyl | 0.018 | 0.035 | 2.0 | 8.77 | 495.4 | 252.0 |
| 3{2,5,4} | Ph | 3-F-6-MeO-Bn | 0.011 | 0.045 | 4.2 | 2.96 | 276.4 | 66.3 |
| 3{2,7,1} | Piperonyl | Furfuryl | 0.019 | 0.046 | 2.4 | 6.50 | 340.2 | 142.2 |
| 3{2,7,2} | Piperonyl | 2-HO-3-MeO-Bn | 0.008 | 0.050 | 6.0 | 1.14 | 135.4 | 22.7 |
| 3{2,1,1} | 4-F-Ph | Furfuryl | 0.032 | 0.069 | 2.1 | 7.76 | 240.4 | 113.2 |
| 3{2,8,2} | 4-MeO-Ph | 2-HO-3-MeO-Bn | 0.013 | 0.078 | 6.0 | 1.60 | 122.9 | 20.6 |
| 3{2,4,4} | 4-CF3-Ph | 3-F-6-MeO-Bn | 0.054 | 0.089 | 1.7 | 5.19 | 96.8 | 58.4 |
| 3{2,1,4} | 4-F-Ph | 3-F-6-MeO-Bn | 0.018 | 0.097 | 5.5 | 0.77 | 44.0 | 7.9 |
| 3{2,4,3} | 4-CF3-Ph | Piperonyl | 0.025 | 0.098 | 3.9 | 1.89 | 74.5 | 19.2 |
| 3{2,5,1} | Ph | Furfuryl | 0.020 | 0.128 | 6.3 | 5.92 | 291.6 | 46.4 |
| 3{2,2,2} | 3,5-CF3-Ph | 2-HO-3-MeO-Bn | 0.029 | 0.209 | 7.2 | 0.94 | 32.4 | 4.5 |
| 3{2,4,1} | 4-CF3-Ph | Furfuryl | 0.020 | 0.223 | 11.0 | 3.47 | 171.0 | 15.5 |
| 3{2,6,1} | 4-TERT-Bu-Ph | Furfuryl | 0.704 | 0.325 | 0.5 | 3.15 | 4.5 | 9.7 |
| 3{2,6,2} | 4-TERT-Bu-Ph | 2-HO-3-MeO-Bn | 0.251 | 0.418 | 1.7 | 0.69 | 2.8 | 1.7 |
| 3{2,6,3} | 4-TERT-Bu-Ph | Piperonyl | 0.223 | 0.490 | 2.2 | 1.37 | 6.2 | 2.8 |
| 3{2,3,3} | 1-Naphtyl | Piperonyl | 0.598 | 0.538 | 0.9 | 6.57 | 11.0 | 12.2 |
| 3{2,2,3} | 3,5-CF3-Ph | Piperonyl | 0.284 | 0.604 | 2.1 | 1.16 | 4.1 | 1.9 |
| 3{2,2,4} | 3,5-CF3-Ph | 3-F-6-MeO-Bn | 0.092 | 0.812 | 8.8 | 0.87 | 9.4 | 1.1 |
| 3{2,1,2} | 4-F-Ph | 2-HO-3-MeO-Bn | 0.009 | 0.854 | 92.8 | 6.84 | 742.9 | 8.0 |
| 3{2,2,1} | 3,5-CF3-Ph | Furfuryl | 0.285 | 0.972 | 3.4 | 2.41 | 8.5 | 2.5 |
| 3{2,3,4} | 1-Naphtyl | 3-F-6-MeO-Bn | 1.154 | 1.034 | 0.9 | 16.00 | 13.9 | 15.5 |
| 3{2,3,2} | 1-Naphtyl | 2-HO-3-MeO-Bn | 0.033 | 1.119 | 33.9 | 2.52 | 76.3 | 2.3 |
| 3{2,3,1} | 1-Naphtyl | Furfuryl | 0.031 | 1.295 | 41.2 | 5.60 | 178.2 | 4.3 |
| 3{2,6,4} | 4-TERT-Bu-Ph | 3-F-6-MeO-Bn | 0.144 | 1.594 | 11.0 | 0.62 | 4.3 | 0.4 |
| 3{2,5,2} | Ph | 2-HO-3-MeO-Bn | 0.009 | 1.936 | 212.7 | 3.21 | 353.0 | 1.7 |
| Heat map | 3D7 EC50 (μM) | K1 EC50 (μM) | K1/3D7 | HepG2 EC50 (μM) | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|
| <0.01 | <0.01 | >100 | >15 | >1000 | >1000 | |
| <0.05 | <0.05 | >50 | >10 | >500 | >500 | |
| <0.1 | <0.1 | >10 | >5 | >100 | >100 | |
| <0.5 | <0.5 | >1 | >1 | >50 | >50 | |
| >0.5 | >0.5 | <1 | <1 | <50 | <50 |
Activity summary of the compounds was sorted by EC50 value against K1 strain.
Values are the mean of three independent experiments in triplicate.
Values are means of one experiment in triplicate.
Alkylaryl Series
The alkylaryl series, derived from Negishi couplings, was evaluated in same test cascade. This set of compounds also showed good antimalarial activity against strain 3D7. However, their activity against strain K1 was very weak (EC50 > 200 nM). Most of the active compounds in this series showed moderate to strong cytotoxicity (0.4–16 μM) in HepG2, resulting in a reasonable selectivity relative to mammalian cells (>100). However, given the poor potency on K1, there was almost no selectivity relative to mammalian cells that strain. Interestingly, the most potent compound 3 with 4 nM EC50 against 3D7 was quite toxic (403 nM EC50) to HepG2 cell, but it still gave good selectivity (>100-fold) between parasite and mammalian cells. Overall this series is the least promising with respect to potential for development. In general the compounds in this series tended to be less soluble and less permeable than the other series studied. They had values ranging from modest to good solubility (4.2–41 μM) and modest to good permeability (23–2110 × 10−6 cm/s) in pH 7.4 buffer (Table 3 in Supporting Information)
Discussion
In earlier work we identified a previously unappreciated aspect to the structure-activity relationships of the 4-aminoquinoline antimalarials in which the 7-chloro substituent of the quinolone A-ring could be replaced by a hydrophobic substituent with retention of potency. In order to more fully explore this observation, we applied three different coupling chemistries to a common synthetic intermediate to give access to fairly widely ranging substituents at this position (R1 group). This array of compounds provided a wide range of physico-chemical properties for the quinoline including variant pKa of the quinoline nitrogen; overall electrostatics and sterics, based on the Topliss and Hammett approaches; and overall lipophilicity.
It is known that the pKa of the ring and the basic side chain affect the activity against both CQ-sensitive and CQ-resistant strains of Plasmodium.12, 26,27 We calculated overall lipophilicity and pKa of quinoline nitrogens (data summarized in Supporting Information).28 Surprisingly, the biaryl series (x=2) and alkylaryl series (x=3) showed identical pKa values (9.47) on the nitrogen of the quinoline ring, whereas the diaryl ether series (x=1) showed a slightly lower pKa value (9.28). Overall this did not appear to correlate with activity trends. Interestingly, no strong stereo-electronic effects were observed for substituents on the R1 group. Unfortunately, we did not find a good correlation between antimalarial activity and pKa value.
Kaschula et al. reported that a more lipophilic group at the 7-position of 4-aminoquinoline enhanced antimalarial activity by increasing hematin affinity.27 Therefore, correlations with ALogP were explored to elucidate the relationship between activity and lipophilicity. (Supporting Information, Figure 2) Lipophilic compounds in our tests exhibited slightly weaker antimalarial activity in both the 3D7 and K1 strains when all series were combined (Figure 3A). The same trend was observed in the diarylether series alone (Figure 3B) and in the biaryl series (Figure 3C). However, the trend was reversed in the alkylaryl series (Figure 3D). We noted further more that in the diarylether series the antimalarial activity in strain 3D7 was more sensitive to lipophilicity than in strain K1. However, none of these trends was strong enough to have any real interest or to warrant further mechanistic studies with hematin.
Figure 3.

Relationship between lipophilicity and antimalarial activity (3D7 strain (○) and K1 strain (▲)). (A) ALogP vs LogEC50 - complete compound array; (B) ALogP vs LogEC50 – Diarylethers; (C) ALogP vs LogEC50 - Biaryls; (D) ALogP vs LogEC50 - Alkylaryls.
Overall, there were no strong trends in activity that could be easily explained using any conventional models for chloroquine mechanism of action.
Another possible explanation for variations in activity would be variant cellular bioavailability of the compounds. The quinolines are well known to accumulate in the acidic food vacuole of the parasite, so permeability is usually a poor predictor of potency, beyond a requirement that the compounds be able to cross a membrane. As expected we observed no strong correlations between permeability or solubility and potency. The most striking finding was that the alkyl aryl series had fairly extreme cross-resistance with CQ-resistant strains. Presumably, this substitution at the 7-position increases transport by PfCRT.
Conclusions
In conclusion, a parallel synthetic strategy was employed to generate a rationally designed array of 7-substituted 4-aminoquinolines. Utilizing 7-bromo-4-chloroquinoline as a key intermediate enabled the exploration of two-dimensional diversity, using a fixed propyl spacer between the quinoline and the distal basic center and a small set of amine substituents previously shown to be active in CQ- resistant Plasmodium strains when used with the chloroquine nucleus. The diversity explored at the 7- position included diarylethers, biaryls, and alkylaryls and was produced using the Ullmann, Suzuki, and Negishi reactions, respectively. The compounds were screened against CQ-sensitive and CQ-resistant strains of P. falciparum and a panel of mammalian cell line to establish antimalarial potency and selectivity for mammalian cells.
Physico-chemical properties of 4-aminoquinolines such as pKa and lipophilicity can be influenced by the substitution at the 7-position, resulting in changes of the drug-sensitivity against antimalarial. However in the series explored, the calculated pKa of the compounds was not significantly changed by substitution at 7-position, and no correlation was observed between pKa and antimalarial activity. However, lipophilicity did weakly correlate with antimalarial activity. Strikingly, less lipophilic compounds were generally more potent against both the CQ-sensitive (3D7) and CQ-resistant strain (K1) an effect that was especially strong for the biaryl compounds. Taken as a whole, this study highlights the potential for developing novel 4-aminoquinolines including a biaryl at the 7-position of the quinoline. Further exploration in preclinical studies is warranted.
Experimental Section
General
All materials were obtained from commercial suppliers and used without further purification. All solvents were dried using an aluminum oxide column. Thin-layer chromatography was performed on pre-coated silica gel 60 F254 plates. Purification of synthesized compounds was carried out by normal phase column chromatography (SP1 [Biotage], Silica gel 230–400 mesh) followed by evaporation (HT-4X evaporator [Genevac]. Initiator [Biotage] was used for microwave reaction. 1H NMR spectra were recorded using a Bruker 400-MHz spectrometer, in CDCl3 or DMSO-d6 solvent. Chemical shifts were reported as parts per million (ppm) downfield from solvent reference. Coupling constants (J values) were measured in Hertz (Hz). NMR peaks were assigned by MestReNova (5.2.2) Identity of all compounds was confirmed by proton and carbon NMR and by mass spectrometry. The purity of all compounds was assessed using LC/MS/UV/ELSD/CLND with the purity being assigned as the average determined by UV/ELSD/CLND. All compounds used for subsequent studies had a minimum purity of 95%.
Chemistry
5-((3-Bromophenylamino)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione (5)
A solution of 2,2-dimethyl-1,3-dioxane-4,6-dione (4) (21.6 g, 150 mmol) in triethyl orthoformate (100 mL) was refluxed for 1h. After cooling down to room temperature 3-bromoaniline (17.2 g, 100 mmol) was added to the solution. The mixture was refluxed for 2 h, then cooled down to room temperature. The precipitates were collected and washed with ether to give compound 5 (32.8 g, quant. yield) as pale yellow crystals. 1H NMR (CDCl3) δ ppm 11.51–10.89 (m, 1H), 8.60 (d, J = 14.1 Hz, 1H), 7.47–7.37 (m, 2H), 7.31 (t, J = 8.0 Hz, 1H), 7.21–7.15 (m, 1H), 1.76 (s, 6H).
7-Bromoquinolin-4-ol (6a) and 5-bromoquinolin-4-ol (6b)
Compound 5 (1.0 g, 3.1 mmol) and diphenyl ether (10 mL) were irradiated by microwave at 225 °C for 5 minutes. Ether (10 mL) was added to the mixture, and the resulting brown precipitates were collected and washed with ether to give a mixture of compounds 6a and 6b. Replicating this process 16 times in parallel gave 9.4 g (85% yield) of the product mixture. Caution: The reaction vessel develops high pressure. 1H NMR (DMSO-d6) δ ppm 11.79 (brs, 1H), 7.99 (d, J = 8.6 Hz, 2/3H), 7.92 (d, J = 7.4 Hz, 2/3H), 7.82 (d, J = 7.4 Hz, 1/3H), 7.75 (d, J = 1.9 Hz, 2/3H), 7.52 (dd, J = 1.2, 8.0 Hz, 1/3H), 7.48 (dd, J = 1.4, 8.0 Hz, 1/3H), 7.45 (dd, J = 1.8, 8.6 Hz, 2/3H), 7.44 (dd, J = 8.0, 8.4 Hz, 1/3H), 6.05 (d, J = 7.5 Hz, 2/3H), 6.01 (d, J = 7.5 Hz, 1/3H).
7-Bromo-4-chloroquinoline (7a)
To a suspension of a mixture of compounds 6a and 6b (9.4 g, 42 mmol) in toluene (70 mL) was added phosphorus oxychloride (13 g, 84 mmol). The mixture was stirred for 1 h at 100 °C, then cooled to room temperature. Excess phosphorus oxychloride was quenched by adding a small portion of ice water and then stirred for 1h. Additional water (100 mL) was added and the resulting mixture extracted with dichloromethane. The organic phase was dried over MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (EtOAc in hexane = 5 -> 25%) to give compound 7a (6.2 g, 61% yield) as white crystals and compound 7b (3.0 g, 30% yield) as white crystals. 7a: 1H NMR (CDCl3) δ ppm 8.79 (d, J = 4.8 Hz, 1H), 8.32 (d, J = 1.9 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.74 (dd, J = 9.0, 1.9 Hz, 1H), 7.51 (d, J = 4.7 Hz, 1H). 7b: 1H NMR (CDCl3) δ ppm 8.73 (d, J = 4.6 Hz, 1H), 8.12 (dd, J = 8.4, 1.2 Hz, 1H), 7.96 (dd, J = 7.6, 1.2 Hz, 1H), 7.56 (d, J = 4.4 Hz, 1H), 7.52 (t, J = 8.4, 7.6 Hz, 1H).
N1-(7-bromoquinolin-4-yl)propane-1,3-diamine (8)
To a mixture of compound 7a (6.18 g, 25.6 mmol) and 1,3-diaminopropane (37.9 g, 512 mmol) in N-methylpyrrolidinone (60 mL) was added triethylamine (777 mg, 7.68 mmol) and K2CO3 (1.17 g, 8.45 mmol). The resulting mixture was stirred at 140 °C for 1h and then cooled to room temperature. Water (200 mL) was added to the solution and the product was precipitated by cooling the mixture to 4 °C. The resulting white precipitates were collected, washed with water, and dried to give compound 8 (5.69 g, 80% yield) as white crystals. 1H NMR (DMSO-d6) δ ppm 8.38 (d, J = 5.4 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.93 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 9.0, 2.1 Hz, 1H), 7.52 (brs, 1H), 6.48 (d, J = 5.5 Hz, 1H), 3.32 (brs, 2H), 2.68 (t, J = 6.5 Hz, 2H), 1.68–1.77 (m, 2H).
Procedure for parallel synthesis of Compounds 9{x=1}:Ullmann coupling
To a mixture of compound 8 (279 mg, 1 mmol) in 1,4-dioxane (6 mL) was added the phenol reagent (4 mmol), Cs2CO3 (1.3 g, 4 mmol), MS 4 Å powder (ca. 280 mg, dried in oven), salicylaldoxime (55 mg, 0.4 mmol) and copper iodide (I) (38 mg, 0.2 mmol). The mixture was heated to reflux and maintained at that temperature for 24 h, and then cooled to room temperature. A solution of dichloromethane (10 mL) and methanol (1 mL) was added to the mixture. After filtration the solvents were removed in vacuo. The crude product was purified by flash chromatography (MeOH/TEA in CH2Cl2 = 10%/1% ~ 40%/4% gradient) to give the corresponding compound 9{x=1}.
Procedure for parallel synthesis of Compounds 9{x=2}: Suzuki coupling
To a mixture of compound 8 (279 mg, 1 mmol) in toluene (6 mL) was added the boronic acid reagent (4 mmol), Cs2CO3 (1.3 g, 4 mmol), MS 4 Å powder (ca. 280 mg) and PdCl2(dppf) (41 mg, 0.05 mmol). The mixture was heated to 100 °C for 24 hrs and then cooled to room temperature. Water (50 mL) was added and the resulting mixture extracted with dichloromethane and 10% methanol. The organic phase was dried over MgSO4, filtered, and dried in vacuo. The crude product was purified by flash chromatography (MeOH/TEA in CH2Cl2 = 10%/1% ~ 80%/8% gradient) to give the corresponding compounds 9{x=2}.
Procedure for parallel synthesis of Compounds 9{x=3}: Negishi coupling
To a solution of compound 8 (279 mg, 1 mmol) and Pd(PPh3)4 (58 mg, 0.05 mmol) was added the alkyl zinc bromide reagent (8 mL, 0.5 M in THF, 4 mmol). The mixture was refluxed for 16 hrs, then cooled to room temperature (one hour is sufficient for benzyl zinc halide). 1N HCl (10 mL) was added to the mixture and stirred. Additional water (40 mL) was added to the mixture, and the solution then neutralized by adding solid K2CO3. The mixture was extracted with 10% methanol in dichloromethane, the organic phase dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash chromatography (MeOH/TEA in CH2Cl2 = 10%/1% ~ 80%/8% gradient) to give the corresponding compounds 9{x=3}. Benzyl zinc bromide solution was prepared as follows: To a suspension of Zn powder (261 mg, 4 mmol) in THF (6 mL) was added benzyl bromide (4 mmol). The solution was heated to 50 °C for 30 min. The solution was used immediately for Negishi coupling.
Procedure for parallel synthesis of Compounds 3{x,y,z}: Reductive amination
To a solution of compound 9{x} (0.1~0.2 mmol) in methanol (1~2 mL) was added 1M aldehyde stock solution in methanol (1 eq.) After stirring for 1 h, sodium borohydride (0.5 eq.) was added to the reaction mixture and the resulting mixture stirred for 30 min. The reaction was quenched by addition of water (2 mL), extracted with CH2Cl2 and concentrated in vacuo. The crude product was purified by flash chromotography (1% methanol in CH2Cl2 (12 mL) and 30% methanol in CH2Cl2 (16 mL)). The solvent was removed in vacuo, and the product was suspended in 1.25 M HCl in methanol solution (1 mL). The solvent and excess of HCl were evaporated in vacuo to give the corresponding HCl salt.
Antimalarial Activity
Two P. falciparum strains were used in this study and were provided by the MR4 Unit of the American Type Culture Collection (ATCC, Manassas, VA). Those two strains were the chloroquine sensitive strain 3D7 and the chloroquine resistant strain K1. Asynchronous parasites were maintained in culture based on the method of Trager29. Parasites were grown in presence of fresh group O-positive erythrocytes (Lifeblood Memphis, TN) in Petri dishes at a hematocrit of 4–6% in RPMI based media(RPMI 1640 supplemented with 0.5% AlbuMAX II, 25 mM HEPES, 25 mM NaHCO3 (pH 7.3), 100ug/mL hypoxanthine, and 5 Pg/mL gentamycin). Cultures were incubated at 37 °C in a gas mixture of 90% N2, 5% O2, 5% CO2. For IC50 determinations, 20 μL of RPMI 1640 with 5 μg/ml gentamycin were dispensed per well in an assay plate (Corning 384-well microtiter plate, clear bottom, tissue culture treated, cat. 8807BC). 40 nL of compound, previously serial diluted in a separate 384-well white polypropylene plate (Corning, cat 8748BC), were dispensed to the assay plate by hydrodynamic pin transfer (FP1S50H, V&P Scientific Pin Head) and then 20 μl of a synchronized culture suspension (1% rings, 4% hematocrit) were added per well thus making a final hematocrit and parasitemia of 2% and 1%, respectively. Assay plates were incubated for 72 h and the parasitemia was determined by a method previously described 30: Briefly, 10 μl of the following solution in RPMI (10 X Sybr Green I, 0.5% v/v triton, 0.5 mg/ml saponin) were added per well, assay plates were shaken for 30 s, incubated in the dark for 90 min, then read with the Envision spectrophotometer at Ex/Em 485 nm/535 nm. IC50’s were calculated with the robust investigation of screening experiments (RISE) with four parameter logistic equation.
Cytotoxicity in mammalian cells
BJ, HEK293, Hep G2, and Raji cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured according to recommendations. Cell culture media were purchased from ATCC. Cells were routinely tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza). Exponentially growing cells were plated in Corning 384 well white custom assay plates, and incubated overnight at 37° C in a humidified, 5% CO2 incubator. DMSO compound solutions were added the following day to a top final concentration of 25 μM and then diluted 1/3 for a total of ten testing concentrations. Cytotoxicity was determined following a 72-hour incubation using Promega Cell Titer Glo Reagent according to the manufacturer’s recommendation. Luminescence was measured on an Envision plate reader (Perkin Elmer)
Solubility
The solubility assay was carried out on Biomek FX lab automation workstation (Beckman Coulter, Inc.). Ten μL of compound stock was added to 190 μL 1-propanol to make a reference stock plate. Five μL from this reference stock plate was mixed with 70 μL 1-propanol and 75 μL phosphate buffered saline (PBS, pH 7.4) to make the reference plate and the UV spectrum (250 – 500 nm) of the reference plate was measured using a SPECTRAmax PLUS plate reader (Molecular Devices). Six μL of 10 mM test compound stock was added to 600 μL PBS in a 96-well storage plate and mixed. The storage plate was sealed and incubated at room temperature for 18 h. The suspension was then filtered through a 96-well filter plate (pION Inc.). Seventy-five μL of filtrate was mixed with 75 μL 1-propanol to make the sample plate for UV spectroscopic analysis. A single experiment was performed in triplicate for each compound. Solubility was calculated using PSOL Evolution software based on the AUC (area under the curve) of the UV spectrum of the sample plate and the reference plate.
Permeability assay
The Parallel Artificial Membrane Permeability Assay (PAMPA) was carried out on a Biomek FX lab automation workstation (Beckman Coulter, Inc.). Three μL of test compound stock (10 mM in DMSO) was mixed with 600 μL of SSB (system solution buffer, pH 7.4 or 4, pION Inc.) to dilute the test compound. One hundred fifty μL of diluted test compound in SSB was transferred to a UV plate (pION Inc.) and the UV spectrum was measured on a SPECTRAmax PLUS plate reader (Molecular Devices) to establish a reference plate. The membrane on a pre-loaded PAMPA sandwich (pION Inc.) was painted with 4 μL GIT lipid (pION Inc.). The acceptor chamber was then filled with 200 μL ASB (acceptor solution buffer, pION Inc.) and the donor chamber was filled with 180 μL test compound diluted in SSB. The PAMPA sandwich (donor and acceptor chamber) was assembled, placed on the Gut-box (pION Inc.) and stirred for 30 minutes. The Aqueous Boundary Layer was set to 40 μM for stirring and the UV spectrum (250–500 nm) of the donor and the acceptor chambers were read. A single experiment was performed in triplicate for each compound. The permeability coefficient was calculated using PAMPA Evolution 96 Command software (pION Inc.) based on the AUC of the reference, donor, and acceptor plates.
Supplementary Material
Table 3.
Biological and pharmacological evaluation for alkylaryls: antimalarial activity and cytotoxicity.
| No | R1 | R2 | 3D7a EC50 (μM) | K1a EC50 (μM) | K1/3D7 | HepG2b EC50 (μM) | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|---|---|
| CQ | -- | -- | 0.03 | 1.0 | 33 | 25 | 833 | 33 |
| 3 | 1-Et-Pr | 2-HO-3-MeO-Bn | 0.004 | 0.273 | 68.3 | 0.41 | 103.3 | 1.5 |
| 3{3,5,2} | 3,5-Me-Bn | 2-HO-3-MeO-Bn | 0.009 | 1.360 | 160.0 | 2.68 | 315.8 | 2.0 |
| 3{3,5,1} | 3,5-Me-Bn | Furfuryl | 0.009 | 0.678 | 77.0 | 6.27 | 712.1 | 9.2 |
| 3{3,3,2} | 4-CN-Bn | 2-HO-3-MeO-Bn | 0.011 | 5.443 | 513.5 | 7.15 | 674.8 | 1.3 |
| 3{3,4,4} | Bn | 3-F-6-MeO-Bn | 0.011 | 1.424 | 129.5 | 7.99 | 726.7 | 5.6 |
| 3{3,6,2} | 2-Cl-4-F-Bn | 2-HO-3-MeO-Bn | 0.013 | 5.136 | 404.4 | 2.35 | 185.4 | 0.5 |
| 3{3,9,3} | PhEt | Piperonyl | 0.013 | 1.533 | 117.1 | 4.98 | 380.3 | 3.2 |
| 3{3,1,2} | 4-F-Bn | 2-HO-3-MeO-Bn | 0.015 | 7.150 | 473.5 | 4.78 | 316.6 | 0.7 |
| 3{3,9,2} | PhEt | 2-HO-3-MeO-Bn | 0.017 | 5.517 | 324.5 | 6.80 | 400.0 | 1.2 |
| 3{3,4,2} | Bn | 2-HO-3-MeO-Bn | 0.017 | 6.971 | 400.6 | 5.72 | 328.6 | 0.8 |
| 3{3,11,2} | cHex | 2-HO-3-MeO-Bn | 0.019 | 16.000 | 864.9 | 1.08 | 58.1 | 0.1 |
| 3{3,6,1} | 2-Cl-4-F-Bn | Furfuryl | 0.020 | 1.223 | 62.1 | 5.73 | 290.8 | 4.7 |
| 3{3,10,2} | iso-butyl | 2-HO-3-MeO-Bn | 0.020 | 8.130 | 402.5 | 2.92 | 144.5 | 0.4 |
| 3{3,7,2} | iso-pentyl | 2-HO-3-MeO-Bn | 0.022 | 8.000 | 365.3 | 5.39 | 245.9 | 0.7 |
| 3{3,3,3} | 4-CN-Bn | Piperonyl | 0.023 | 3.069 | 136.4 | 6.08 | 270.2 | 2.0 |
| 3{3,2,2} | 3-CF3-Bn | 2-HO-3-MeO-Bn | 0.025 | 2.189 | 87.9 | 1.72 | 69.0 | 0.8 |
| 3{3,6,3} | 2-Cl-4-F-Bn | Piperonyl | 0.027 | 0.953 | 35.9 | 16.00 | 603.8 | 16.8 |
| 3{3,12,3} | 1-Et-Pr | Piperonyl | 0.027 | 1.065 | 39.5 | 6.35 | 235.1 | 6.0 |
| 3{3,7,3} | iso-pentyl | Piperonyl | 0.027 | 0.206 | 7.6 | 2.80 | 103.1 | 13.5 |
| 3{3,3,4} | 4-CN-Bn | 3-F-6-MeO-Bn | 0.028 | 0.770 | 27.5 | 6.05 | 216.2 | 7.9 |
| 3{3,5,4} | 3,5-Me-Bn | 3-F-6-MeO-Bn | 0.034 | 0.794 | 23.1 | 0.94 | 27.4 | 1.2 |
| 3{3,3,1} | 4-CN-Bn | Furfuryl | 0.039 | 0.382 | 9.7 | 7.91 | 201.9 | 20.7 |
| 3{3,2,3} | 3-CF3-Bn | Piperonyl | 0.039 | 1.314 | 33.5 | 2.20 | 56.1 | 1.7 |
| 3{3,4,1} | Bn | Furfuryl | 0.040 | 3.550 | 89.9 | 16.00 | 405.1 | 4.5 |
| 3{3,2,4} | 3-CF3-Bn | 3-F-6-MeO-Bn | 0.040 | 0.323 | 8.1 | 0.44 | 11.0 | 1.3 |
| 3 | 1-Et-Pr | 3-F-6-MeO-Bn | 0.040 | 1.933 | 48.0 | 7.86 | 194.9 | 4.1 |
| 3{3,8,3} | cHexmethyl | Piperonyl | 0.041 | 0.701 | 17.3 | 6.29 | 155.2 | 9.0 |
| 3{3,5,3} | 3,5-Me-Bn | Piperonyl | 0.041 | 1.040 | 25.6 | 2.25 | 55.5 | 2.2 |
| 3{3,9,1} | PhEt | Furfuryl | 0.046 | 2.930 | 63.7 | 16.00 | 347.8 | 5.5 |
| 3{3,8,2} | cHexmethyl | 2-HO-3-MeO-Bn | 0.050 | 2.610 | 52.7 | 0.97 | 19.6 | 0.4 |
| 3{3,8,1} | cHexmethyl | Furfuryl | 0.055 | 0.621 | 11.2 | 1.70 | 30.7 | 2.7 |
| 3{3,2,1} | 3-CF3-Bn | Furfuryl | 0.056 | 0.743 | 13.2 | 3.19 | 56.7 | 4.3 |
| 3{3,1,1} | 4-F-Bn | Furfuryl | 0.057 | 1.659 | 28.9 | 5.60 | 97.5 | 3.4 |
| 3{3,10,3} | iso-butyl | Piperonyl | 0.066 | 1.814 | 27.7 | 2.38 | 36.3 | 1.3 |
| 3{3,1,4} | 4-F-Bn | 3-F-6-MeO-Bn | 0.075 | 0.580 | 7.8 | 3.53 | 47.1 | 6.1 |
| 3{3,12,1} | 1-Et-Pr | Furfuryl | 0.086 | 4.300 | 50.1 | 6.76 | 78.7 | 1.6 |
| 3{3,10,4} | iso-butyl | 3-F-6-MeO-Bn | 0.089 | 2.119 | 23.9 | 2.90 | 32.7 | 1.4 |
| 3{3,11,4} | cHex | 3-F-6-MeO-Bn | 0.090 | 1.596 | 17.7 | 2.29 | 25.4 | 1.4 |
| 3{3,7,4} | iso-pentyl | 3-F-6-MeO-Bn | 0.116 | 0.360 | 3.1 | 0.89 | 7.7 | 2.5 |
| 3{3,11,1} | cHex | Furfuryl | 0.118 | 3.900 | 33.1 | 4.76 | 40.4 | 1.2 |
| 3{3,1,3} | 4-F-Bn | Piperonyl | 0.127 | 0.256 | 2.0 | 5.55 | 43.7 | 21.7 |
| 3{3,11,3} | cHex | Piperonyl | 0.128 | 4.709 | 36.7 | 3.02 | 23.6 | 0.6 |
| 3{3,9,4} | PhEt | 3-F-6-MeO-Bn | 0.132 | 2.143 | 16.2 | 4.24 | 32.1 | 2.0 |
| 3{3,8,4} | cHexmethyl | 3-F-6-MeO-Bn | 0.138 | 0.687 | 5.0 | 1.83 | 13.3 | 2.7 |
| 3{3,7,1} | iso-pentyl | Furfuryl | 0.176 | 4.605 | 26.1 | 11.24 | 63.7 | 2.4 |
| 3{3,6,4} | 2-Cl-4-F-Bn | 3-F-6-MeO-Bn | 0.208 | 1.112 | 5.4 | 2.46 | 11.8 | 2.2 |
| 3{3,10,1} | iso-butyl | Furfuryl | 0.581 | 4.500 | 7.7 | 16.00 | 27.5 | 3.6 |
| 3{3,4,3} | Bn | Piperonyl | 0.720 | 1.237 | 1.7 | 6.28 | 8.7 | 5.1 |
| Heat map | 3D7 EC50 (3M) | K1 EC50 (3M) | K1/3D7 | HepG2 EC50 (3M) | HepG2/3D7 | HepG2/K1 |
|---|---|---|---|---|---|---|
| <0.01 | <0.01 | >100 | >15 | >1000 | >1000 | |
| <0.05 | <0.05 | >50 | >10 | >500 | >500 | |
| <0.1 | <0.1 | >10 | >5 | >100 | >100 | |
| <0.5 | <0.5 | >1 | >1 | >50 | >50 | |
| >0.5 | >0.5 | <1 | <1 | <50 | <50 |
Activity summary of the compounds was sorted by EC50 value against 3D7 strain.
Values are the mean of three independent experiments in triplicate.
Values are means of one experiment in triplicate.
Acknowledgments
This work was supported by NIH/NIAID (Al075517), the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude Children’s Research Hospital.
Abbreviations
- CQ
Chloroquine
- MDR
multi-drug resistant
- PAMPA
parallel artificial membrane permeation assay
- WHO
World Health Organization
- MMV
Medicines for Malaria Venture
- HTS
High Throughput Screening
- SAR
Structure Activity Relationship
- Clint’
Intrinsic Clearance
Footnotes
All biological data (antimalarial, cytotoxicity in mammalian cells, solubility, and permeability). 1H NMR spectra and purity of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO. World Malaria Report 2010. 2010. [Google Scholar]
- 2.Kappe SH, Vaughan AM, Boddey JA, Cowman AF. That was then but this is now: malaria research in the time of an eradication agenda. Science. 2010;328:862–826. doi: 10.1126/science.1184785. [DOI] [PubMed] [Google Scholar]
- 3.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew Chem Int Ed Engl. 2003;42:5274–5293. doi: 10.1002/anie.200200569. [DOI] [PubMed] [Google Scholar]
- 4.Baird JK. Effectiveness of antimalarial drugs. N Engl J Med. 2005;352:1565–1577. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 5.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rieckmann KH, Trenholme GM, Williams RL, Carson PE, Frischer H, Desjardins RE. Prophylactic activity of mefloquine hydrochloride (WR 142490) in drug-resistant malaria. Bull World Health Organ. 1974;51:375–377. [PMC free article] [PubMed] [Google Scholar]
- 7.O’Neill PM, Ward SA, Berry NG, Jeyadevan JP, Biagini GA, Asadollaly E, Park BK, Bray PG. A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs. Curr Top Med Chem. 2006;6:479–507. doi: 10.2174/156802606776743147. [DOI] [PubMed] [Google Scholar]
- 8.Wipf P, Mo T, Geib SJ, Caridha D, Dow GS, Gerena L, Roncal N, Milner EE. Synthesis and biological evaluation of the first pentafluorosulfanyl analogs of mefloquine. Org Biomol Chem. 2009;7:4163–4165. doi: 10.1039/b911483a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellot F, Cosledan F, Vendier L, Brocard J, Meunier B, Robert A. Trioxaferroquines as new hybrid antimalarial drugs. J Med Chem. 2010;53:4103–4109. doi: 10.1021/jm100117e. [DOI] [PubMed] [Google Scholar]
- 10.Kaur K, Jain M, Reddy RP, Jain R. Quinolines and structurally related heterocycles as antimalarials. Eur J Med Chem. 45:3245–3264. doi: 10.1016/j.ejmech.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Yearick K, Ekoue-Kovi K, Iwaniuk DP, Natarajan JK, Alumasa J, de Dios AC, Roepe PD, Wolf C. Overcoming drug resistance to heme-targeted antimalarials by systematic side chain variation of 7-chloro-4-aminoquinolines. J Med Chem. 2008;51:1995–1998. doi: 10.1021/jm800106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheruku SR, Maiti S, Dorn A, Scorneaux B, Bhattacharjee AK, Ellis WY, Vennerstrom JL. Carbon isosteres of the 4-aminopyridine substructure of chloroquine: effects on pK(a), hematin binding, inhibition of hemozoin formation, and parasite growth. J Med Chem. 2003;46:3166–3169. doi: 10.1021/jm030038x. [DOI] [PubMed] [Google Scholar]
- 13.Madrid PB, Liou AP, DeRisi JL, Guy RK. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug-resistant P. falciparum. J Med Chem. 2006;49:4535–4543. doi: 10.1021/jm0600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray S, Madrid PB, Catz P, LeValley SE, Furniss MJ, Rausch LL, Guy RK, DeRisi JL, Iyer LV, Green CE, Mirsalis JC. Development of a new generation of 4-aminoquinoline antimalarial compounds using predictive pharmacokinetic and toxicology models. J Med Chem. 2010;53:3685–3695. doi: 10.1021/jm100057h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parallel synthesis and antimalarial screening of a 4-aminoquinoline library. J Comb Chem. 2004;6:437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madrid PB, Sherrill J, Liou AP, Weisman JL, Derisi JL, Guy RK. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg Med Chem Lett. 2005;15:1015–1018. doi: 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 17.May BC, Witkop J, Sherrill J, Anderson MO, Madrid PB, Zorn JA, Prusiner SB, Cohen FE, Guy RK. Structure-activity relationship study of 9-aminoacridine compounds in scrapie-infected neuroblastoma cells. Bioorg Med Chem Lett. 2006;16:4913–4916. doi: 10.1016/j.bmcl.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 18.Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 19.Cristau HJ, Cellier PP, Hamada S, Spindler JF, Taillefer M. A general and mild Ullmann-type synthesis of diaryl ethers. Org Lett. 2004;6:913–916. doi: 10.1021/ol036290g. [DOI] [PubMed] [Google Scholar]
- 20.Miyaura N, Yamada K, Suzuki A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Letters. 1979;20:3437–3440. [Google Scholar]
- 21.Barder TE, Walker SD, Martinelli JR, Buchwald SL. Catalysts for Suzuki-Miyaura coupling processes: scope and studies of the effect of ligand structure. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 22.King A, Okukado N, Negishi E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. Journal of the Chemcial Society Chemical Communications. 1977:683–684. [Google Scholar]
- 23.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avdeef A, Bendels S, Tsinman O, Tsinman K, Kansy M. Solubility-excipient classification gradient maps. Pharm Res. 2007;24:530–545. doi: 10.1007/s11095-006-9169-0. [DOI] [PubMed] [Google Scholar]
- 25.Avdeef A, Bendels S, Di L, Faller B, Kansy M, Sugano K, Yamauchi Y. PAMPA--critical factors for better predictions of absorption. J Pharm Sci. 2007;96:2893–2909. doi: 10.1002/jps.21068. [DOI] [PubMed] [Google Scholar]
- 26.Natarajan JK, Alumasa JN, Yearick K, Ekoue-Kovi KA, Casabianca LB, de Dios AC, Wolf C, Roepe PD. 4-N-, 4-S-, and 4-O-chloroquine analogues: influence of side chain length and quinolyl nitrogen pKa on activity vs chloroquine resistant malaria. J Med Chem. 2008;51:3466–3479. doi: 10.1021/jm701478a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaschula CH, Egan TJ, Hunter R, Basilico N, Parapini S, Taramelli D, Pasini E, Monti D. Structure-activity relationships in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. J Med Chem. 2002;45:3531–3539. doi: 10.1021/jm020858u. [DOI] [PubMed] [Google Scholar]
- 28.Avdeef A, Kansy M, Bendels S, Tsinman K. Absorption-excipient-pH classification gradient maps: sparingly soluble drugs and the pH partition hypothesis. Eur J Pharm Sci. 2008;33:29–41. doi: 10.1016/j.ejps.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 30.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.