

Abstract
The advent of Next-Generation sequencing technologies, which significantly increases the throughput and reduces the cost of large scale sequencing efforts, provides an unprecedented opportunity for discovery of novel gene mutations in human cancers. However, it remains a challenge to apply Next-Generation technologies to DNA extracted from formalin fixed paraffin embedded cancer specimens. We describe here the successful development of a custom DNA capture method using Next-Generation for detection of 140 driver genes in 5 formalin fixed paraffin embedded human colon cancer samples using an improved extraction process to produce high quality DNA. Isolated DNA was enriched for targeted exons and sequenced using the Illumina Next-Generation platform. An analytical pipeline using 3 software platforms to define single nucleotide variants was used to evaluate the data output. Approximately 250x average coverage was obtained with >96% of target bases having at least 30 sequence reads. Results were then compared to previously performed high throughput Sanger sequencing. Using an algorithm of needing a positive call from all 3 callers to give a positive result, 98% of the verified Sanger sequencing somatic driver gene mutations were identified by our method with a specificity of 90%. 13 insertions and deletions identified by Next-Generation were confirmed by Sanger sequencing. We also applied this technology to two components of a biphasic colon cancer which had strikingly differing histology. Remarkably, no new driver gene mutation accumulation was identified in the more undifferentiated component. Applying this method to profiling of formalin fixed paraffin embedded colon cancer tissue samples yields equivalent sensitivity and specificity for mutation detection as Sanger sequencing of matched cell lines derived from these cancers. This method directly enables high throughput comprehensive mutational profiling of colon cancer samples, and is easily extendable to enable targeted sequencing from formalin fixed paraffin embedded material for other tumor types.
Keywords: next generation sequencing, colon cancer, driver gene mutations
The advent of Next-Generation sequencing [NGS] technologies offers significant opportunity to perform broad mutational surveys on individual cancer specimens at considerably decreased costs per mutation detected compared to traditional Sanger sequencing. These technologies have been applied with increasing frequency to address fundamental questions in cancer pathogenesis and patient outcomes.(1-8) Moreover, with the increasing development of targeted therapeutic agents, there is increasing clinical importance to identifying somatic mutations that can impart either sensitivity or resistance to specific cancer therapies. (9, 10)
Most NGS studies report using DNA derived from fresh frozen material. This constraint significantly impedes use of NGS in clinical practice, since standard processing of clinically derived specimens entails fixation with formalin and embedding in paraffin wax. In addition, development of reliable methods to apply high throughput sequencing technology to formalin fixed paraffin embedded biospecimens would significantly improve the opportunity to perform large scale investigations of the relationship between somatic gene mutations and clinical cancer outcomes, by enabling the analysis of the extensive clinically annotated tumor samples held in pathology archives.
Colorectal cancers provide a particularly amenable system for NGS based analysis, as global sequencing of the RefSeq gene set in human colon cancers has identified a delimited set of 140 driver genes that are the targets for recurring mutations in human colon cancers. (11) The average microsatellite stable colon cancer bears somatic mutations in 15 of these driver genes, with the great majority of these driver genes being individually mutant in less than 10% of colon cancers and also showing no evidence for mutational hotspots. Thus, defining the driver gene mutational profile of an individual colon cancer essentially requires the sequencing of the full coding sequences of this complete 140 driver gene set.
In the current study, we investigated the feasibility of interrogating routinely processed formalin fixed paraffin embedded colon cancers for driver gene mutation status using a custom DNA capture method and Illumina high throughput sequencing. Our methods included tissue microdissection, prolonged DNA extractions to optimize DNA fragment size and yield, design and use of a customized DNA capture array that targeted the 140 colon cancer driver gene set, Illumina sequencing, and development and validation of an analysis pipeline for variant detection.
Materials and Methods
Pathology Processing
All specimens used were derived from standard colectomies from previously biopsy proven colon cancers. Specimens were received fresh into the Department of Pathology at Case Medical Center. After harvesting of fresh portions of cancer and normal mucosa, colectomy specimens were pinned onto a wax plate and immersed overnight in 10% neutral buffered formalin. The next day routine sections of cancer, normal mucosa and adjacent lymph nodes were submitted for routine paraffin embedding and H&E sections obtained. DNA for sequencing was isolated from these clinically derived specimens. Eight 5μm sections of formalin fixed paraffin embedded cancer sections were obtained per case. Cancers were microdissected to remove non-cancerous and necrotic tissue, resulting in specimens that contained approximately 80% viable cancer material.
DNA Extraction from Formalin Fixed Paraffin Embedded Samples
Initially the tissues were de-paraffinized by dipping the slides in a series of solutions: Xylene – initially for 4 minutes and secondly for 2 minutes; 2 minutes each in 100%, 95%, and 70% ethanol followed by 2 rinses of 2 minutes each in 10mM of Tris solution. Tissues were then scraped into a polypropylene microcentrifuge tube. The QIAamp Micro DNA Kit was employed to extract the DNA. 15μl of buffer ATL and 10μl of proteinase K (20 mg/ml) were added per slide with up to sections from five slides in a polypropylene microcentrifuge tube. Tubes were then vortexed for 15 sec. The tubes were incubated at 60°C for 8 days – with the daily addition of 1.5μl of proteinase K (20 mg/ml) per tube.
After incubation, 25μl of buffer ATL is added per slide scraped into tube. (For example, if four slides were scraped then add 100 μl). Add 50 μl of buffer AL containing 1μl RNA carrier to the tube per slide scraped in step one. [1μl of RNA carrier is added to 50 μl of buffer AL previously: RNA stock is 1 μg/μl] then vortex solution for 15 sec then incubate at 70°C for 20 minutes. After a brief spin, add 50 μl of 100% ETOH per slide scraped from step one, vortex for 15 sec. incubate at room temperature for 5 minutes, spin briefly and add contents of tube to a micro spin column (provided with kit). (One may combine up to five digestions onto a single column.) Spin at 8,000 rpm for 1 minute, place spin column into a clean collection tube then add 500μl of AW1 wash buffer. Spin at 8,000 rpm for 1 minute. Place spin column into a clean collection tube then add 500μl of AW2 wash buffer to the column. Spin at 8,000 rpm for 1 minute. Place the spin column into a clean collection tube and spin for 3 minutes at 14,000 rpm to dry the column. Place the spin column into a clean collection tube then add 25μl of buffer AE to the center of the column and incubate for 5 minutes at room temperature. [Add 50μl of AE buffer if you have more than one digestion added to a single column]. Spin the column for 1 minute at 14,000 rpm to collect the DNA. Repeat with the same volume of buffer AE added to the center of the column and incubate for 5 minutes at room temperature without changing the collection tube. Spin the column for 1 minute at 14,000 rpm to collect the DNA. Transfer DNA to a closable tube for permanent storage.
Assessment of Sample Quality and Yield
To select samples for sequencing, each DNA sample isolated from formalin fixed paraffin embedded tissue was assessed for quality and yield. DNA concentration was determined using a Qubit fluorometer [Invitrogen Qubit High Sensitivity dsDNA Assay]. A minimum of 3 μg of DNA was required to initiate library preparation. Samples producing insufficient yield were removed for further analysis. DNA quality was assessed by the ability of the sample to produce a robust PCR amplicon of at least 420 bps. The formalin fixed paraffin embedded DNA was used as a template for a series of 5 PCR assays designed to produce PCR products ranging in size from 420–718 bps. The resulting amplicons were then examined on a 2% agarose gel. If a DNA sample was not able to robustly produce amplicons >420 bps in this PCR QC Assay, it was excluded from further analysis. The PCR primers used in the PCR QC Assay amplify different regions of the HLTF gene. These primer IDs and their sequences are depicted in Table 1.
Table 1.
Primers from the HLTF Gene.
| Primer ID | Primer Sequence |
|---|---|
| P-HLTF – 1755F | CCATGTCCTGGATGTTCAAGAGGTG |
| P-HLTF – 1931F | TGCATTGTCTTCGCTGGCGAGTAG |
| P-HLTF – 2350R | ACCCATTAGGTGTAACTGTTTCGGG |
| P-HLTF – 2425R | AACCACGCCACCGTGGGAAATTGG |
| P-HLTF – 2473R | ATCCATCACTGTAACTCCCTGCGAG |
Table 2 depicts the primer combinations used in the 5 different PCR assays designed to produce a range of different sized amplicons. Each PCR amplification was initiated in a 50μl reaction volume at 95°C for 9 minutes and a total of 35 PCR cycles were then carried out using AmpliTaq Gold as the polymerase. [PCR Cycle Conditions: 95°C for 30 sec; 64°C for 45 sec & 72°C for 45 sec].
Table 2.
Primer Combinations for PCR QC Assays.
| Forward Primer | Reverse Primer | Size of Amplicon |
|---|---|---|
| P-HLTF – 1931F | P-HLTF – 2350R | 420 bp Fragment |
| P-HLTF – 1931F | P-HLTF – 2425R | 494 bp Fragment |
| P-HLTF – 1931F | P-HLTF – 2473R | 542 bp Fragment |
| P-HLTF – 1755F | P-HLTF – 2425R | 670 bp Fragment |
| P-HLTF – 1755F | P-HLTF – 2473R | 718 bp Fragment |
Library Preparation
DNA samples successfully passing the screen for Sample Quality and Yield were then prepped for library production, followed by targeted hybridization-based capture and sequencing on the Illumina Genome Analyzer. Initially each 3 μg DNA sample was sheared to a peak distribution of 150-200 bp, a size range that is optimal for SureSelect target enrichment, using a Covaris S2. Following shearing and sample purification, an Experion 1K DNA LabChip was employed to ensure that the DNA was sheared to the appropriate size. The Agilent SureSelect Target Enrichment System for Illumina Multiplexed Sequencing was followed. The Adapter-Ligated Library was amplified for 4 cycles of PCR. Agencourt AMPure XP Beads were employed for all purifications steps in the Library Preparation. The captured, amplified DNA library was checked for both quantity and quality. The Qubit High Sensitivity dsDNA Assay was employed to assess library concentration, while analysis on the Experion DNA 1K Chip was used to ensure a peak size between 250 – 275 bp. To move forward the sample must have a 260/280 ratio of 1.8 to 2.0, a minimum yield of 500 ng [147 ng/μl], and a single peak between 250 – 275 bp. Although sufficient yield was required to move to capture, the Library Preparations for all samples were too dilute and required concentration using a speed-vac before proceeding to enrichment.
Enrichment of CAN Gene Exons
Agilent Technologies ‘SureSelect Target Enrichment System’ [or DNA capture], was used to enrich targeted regions of the genome for analysis with the Illumina sequencing platform. Through Agilent's Custom Design Portal, a customized SureSelect Kit was designed to target exons from the 140 CAN genes, comprising 2,934 target exon regions. Only 31 (~1%) additional exon regions did not meet criteria for bait design. The Custom SureSelect Kit designed for this project used custom oligonucleotides as capture probes.
Once size-selected libraries were prepared and confirmed as detailed above, they were incubated with the custom-designed SureSelect baits for 24 hours. RNA bait-DNA hybrids were then isolated from the complex mixture with streptavidin-labeled magnetic beads. After extensive washing the RNA bait was digested, leaving only the targeted DNA of interest. Following the capture, 14 cycles of DNA amplification were performed. The targeted sample was then analyzed using an Agilent 2100 Bioanalyzer High Sensitivity DNA Chip to ensure that the amplified prepped library DNA showed a single peak in the size range of 300 to 325bp. DNA concentration was determined using Agilent's QPCR NGS Library Quantification Kit [for Illumina]. This Real Time PCR assay, which employs SYBR Green as the fluorescent indicator was designed by Agilent to assess the quantity of index-tagged libraries. Samples successfully meeting the size and concentration criteria were then pooled at equimolar concentrations and subjected to Illumina Sequencing.
DNA Sequencing
Up to five samples, with unique index-tag adapter sequences were combined for multiplex sequencing in a single lane on the Illumina Genome Analyzer IIx [GA2x]. Paired-end 72-base reads were collected, with an additional seven bases collected for decoding the index tag sequence in each read. Sequence data quality was assessed using fastqc(http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Capture efficiency was determined based on the percentage of reads that map to or near CAN gene target regions using the HSmetrics component of the picard package (http://picard.sourceforge.net/).
Variant Detection
Three software systems were used to define single nucleotide variants in the Illumina sequence data: Atlas-SNP2(12), SOAPsnp(13), and the Unified Genotyper component of the Genome Analysis Toolkit (UGT) (14, 15). UGT was also used to predict small indel variants. Reads were aligned to the human genome (hg18) SOAP2 (for SOAPsnp) and BWA (for UGT and Atlas-SNP2). Default parameters were used for SOAP2 and BWA. For Atlas-SNP2, the minimum coverage was least three reads and the maximum coverage was set to approximately the maximum coverage of each sample based on SOAP2 alignment results. Variants that have been previously observed in other studies were identified by cross-reference with dbSNP version 130. As shown in Figure 1, nearly all variants present in dbSNP were predicted by all three callers. A significant majority of novel (non-dbSNP) variants were also predicted as present by all three callers (Figure 2).
Figure 1.

Overlap of SNP calls at positions of dbSNP variants
Figure 2.

Overlap of SNP calls at positions without dbSNP variants
Validation of Unique Variants
Novel variants could be germline polymorphisms, somatic mutations, or sequencing errors. To distinguish among these possibilities, candidate variants not previously reported in dbSNP were confirmed or validated by re-sequencing the appropriate exon in the FFP-derived DNA, DNA from a cell line derived from the cancer and the respective matched normal DNA. This sequencing was performed in the Genomics Core by traditional PCR amplification and Sanger sequencing on an Applied Biosystems 3730xl DNA sequencer.
Results
Isolation of DNA from Formalin Fixed Paraffin Embedded Cancer Specimens
Our group had previously described successful extraction from formalin fixed paraffin embedded tumor material of DNAs of sufficient size to support PCR amplification and Sanger sequencing.(16) To further optimize the ability to recover DNA fragments of sufficient size to support construction of Illumina sequencing libraries, we tested the effect of prolonged extraction (8 days) of DNA from 5μM thick formalin fixed paraffin embedded sections. Using this approach (fully detailed in the methods section), DNA was isolated from 5 formalin fixed paraffin embedded colon cancer samples that were fixed in 2003 –2004. These samples were designated: 1 to 5. DNA samples from these patients’ matched normal tissues were also available for analysis. As a comparator, the full set of somatic mutations in these cases had also been previously identified by Sanger sequencing of the full RefSeq gene set, in a study that employed genomic DNA purified from the cell lines and xenografts that had been established from each of these cases.(11) The formalin fixed paraffin embedded derived tumor DNAs from these 5 cases were all of quality sufficient for construction of Illumina sequencing libraries, supporting PCR amplifications of fragments of 420bp or larger, and yielding at least 3μg of DNA (at 25ng/ul) from extraction of 5 formalin fixed paraffin embedded slides. These cases are however representative ones, as using our optimized methods for extracting DNA from formalin fixed paraffin embedded blocks, we found this threshold was met by 18 of 25 (72%) of formalin fixed paraffin embedded blocks we tested that spanned dates from between 1990 to 2005.
Sequence Level Analysis of CAN Gene Exons in DNA Isolated from 5 Archived Colon Cancers
We employed a custom SureSelect Enrichment Kit [Agilent Technologies] to perform region-specific DNA capture of all coding exons of the 140 colon cancer driver genes from libraries prepared from genomic DNA isolated from each of five different formalin fixed paraffin embedded colon cancer specimens selected for study. These 140 driver genes had previously been designated as the set of Colon Cancer CAN genes. (11, 17) While each library was individually captured, the five libraries were individually index tagged to allow pooling of the captured targets, sequencing of the pool, and then assignment of the sequence reads back to the corresponding case of origin. As shown in Table 3, 71-85% of reads obtained mapped to or near the regions targeted for capture. Approximately 250-fold average base coverage was obtained across the target exons, resulting in >96% of target bases being spanned by at least 30 sequence reads. This depth of coverage provided substantial power for detection of somatic mutations, despite an expected dilution of the cancer by up to 50% of stromal and other non-cancer cells present in the sequenced specimens.
Table 3.
Region-specific DNA capture and sequencing performance
| Cancer Designation | 1 | 2 | 3 | 4 | 5 | Biphasic Cancer: Low Grade Region | Biphasic Cancer: High Grade Region |
|---|---|---|---|---|---|---|---|
| Pass-Filter Read Pairs | 1751408 | 1552882 | 2834330 | 2059979 | 3459801 | 16908123 | 26247753 |
| Total reads aligned | 3391847 | 2995682 | 5468661 | 3970439 | 6567690 | 32671738 | 50742320 |
| % of reads aligned | 96.8 | 96.5 | 96.5 | 96.4 | 94.9 | 96.70% | 96.70% |
| % of Bases on/near targets* | 83.1% | 79.9% | 80.3% | 82.9% | 71.4% | 85.20% | 85.10% |
| Average target coverage | 209 | 177 | 318 | 243 | 331 | 1758 | 2680 |
| % Usable bases on target | 47.9% | 46.0% | 45.3% | 47.6% | 39.3% | 46.90% | 45.80% |
| Fold enrichment | 2379 | 2276 | 2247 | 2371 | 1967 | 2360 | 2316 |
| % zero-coverage targets | 1.0% | 1.1% | 0.8% | 1.1% | 0.9% | 0.80% | 0.80% |
| % of target bases with >=2x coverage | 99.3% | 99.3% | 99.1% | 99.2% | 99.5% | 99.50% | 99.50% |
| % of target bases with >=10x coverage | 98.3% | 98.5% | 98.4% | 98.5% | 98.9% | 99.10% | 99.30% |
| % of target bases with >=20x coverage | 97.6% | 97.5% | 97.9% | 97.7% | 98.4% | 98.80% | 99.00% |
| % of target bases with >=30x coverage | 96.6% | 96.4% | 97.5% | 96.7% | 98.0% | 98.60% | 98.80% |
Total target length = 546,336 bases; total bait length = 814,497 bases
Identification of Variants in Target Regions
Several software suites are available for detection of sequence variants from Illumina sequence data (18); however, these have chiefly been developed for detection of germline, not somatic variants, and there is little consensus approach on the best algorithm for detecting either germline or somatic variants in Illumina sequencing data. For example, the 1000 Genomics Pilot project used three algorithms and defined a working set of single nucleotide polymorphisms [SNPs] as those called by two programs (19). Detection of somatic mutations is further complicated by the fact that cancer cells carrying the mutation may easily be diluted by half or more by normal cellular elements, and thus there is no expectation that a somatic mutation in a cancer will be represented in 50% of the sequencing reads, in the manner that would support the confident identification of a heterozygous variant in the germline. Working in our favor though is that mutations in driver genes would be expected to be present in all of the cancer cells within the tumor specimen.
Three software programs for variant detection were evaluated: Atlas-SNP2 (12), SOAPsnp (20) and the Unified Genotyper (UGT) component of the Genome Analysis Toolkit (14, 15). For Atlas-SNP2 and UGT, the alignment program BWA (20) was used to map reads to the human genome; for SOAPsnp, SOAP2 (21) was used for alignment. We first considered the concordance of these three variant callers in recognizing the known SNP variants that were called by at least one of the variant callers and that corresponded to population polymorphisms already annotated in dbSNP (Figure 1). More than 95% of these single nucleotide variations [SNVs] identified in dbSNP, that were highly likely to represent germline variants, were predicted by all three variant callers. Examination of dbSNP variants called by only one or two callers showed that they were of marginal quality based on one or more metrics (data not shown).
We next applied the same standard – prediction by all three variant callers – to analysis of those SNVs called by at least one of the variant callers but that did not correspond to any known germline variant captured in dbSNP or in the 1000 genomes project. These calls would include private germline variants, somatic mutations, and potential sequence artifacts. Approximately one-third of these SNVs predictions were supported by all three variant callers (Figure 2). Another one-third of these SNV predictions were supported only by calls made by SOAPsnp (Figure 2).
Sensitivity of Somatic Mutation Detection
We evaluated the sensitivity of somatic mutation detection by our methods by comparison of SNVs predicted from Illumina sequencing by all three variant callers to the set somatic mutations previously identified in these cases through Sanger sequencing of the corresponding cell lines and xenografts established from these same set of tumors. (11) Of the 61 somatic mutations identified by Sanger sequencing of the cell lines derived from these cancers, 56 were successfully identified by all three variant callers in the Illumina sequences from the targeted CAN captures of the DNAs extracted from the 5 primary formalin fixed paraffin embedded tumor samples (Table 4). Thus, 92% of the previously identified somatic mutations detected by Sanger sequencing of purified cancer cell lines were identified by targeted capture and Illumina sequencing of DNA extracted from formalin fixed paraffin embedded crude tumor samples. One additional somatic mutation detected in our Sanger based analysis was also detected in the Illumina sequencing, but was filtered out because this somatic mutation corresponded to a variant also annotated in dbSNP130. There are two possible explanations for why the other four variants were not predicted: 1) there were insufficient Illumina reads for recognition of the variant; and 2) adequate Illumina sequence data was obtained, but did not support a variant call. In the latter instance, the variant could have been a false positive call in the prior high throughput Sanger sequencing data, or the variant could represent a bona fide difference between the cell line used for Sanger sequencing and the original formalin fixed paraffin embedded cancer studied by Illumina sequencing. To distinguish among these possibilities we repeated PCR amplification and Sanger sequencing of these 4 variants in DNA from patient's normal colon tissue, their colon cancer tumor, and their tumor derived cell line. In three of the four cases, repeat targeted Sanger sequencing failed to detect the putative mutations in any of the patients’ normal colon, colon cancer, or colon cancer cell line (Table 5), suggesting that these mutations were false positive results in the previous high throughput Sanger sequencing study, and were not failures of detection by the targeted capture and Illumina sequencing approach.(11) The remaining prior identified somatic mutation was indeed validated as a somatic mutation in repeat Sanger sequencing and so is a legitimate false negative (one of 61) of our approach. Nevertheless, this mutation was in fact correctly called in the Illumina data by two of the three SNP callers (Table 5), but failed to be called by Atlas-SNP2.
Table 4.
Concordance with known somatic mutations in sequenced cancer DNA
| Cancer Designation | Novel variants predicted by NGS* | Novel variants predicted by NGS not in Wood et al. set | Somatic mutations defined by Wood et al. | Wood et al. somatic mutations [i.e. novel SNVs] detected by NGS | % of Wood et al. somatic mutations detected by NGS | Revised % of Wood et al. somatic mutations detected by NGS† |
|---|---|---|---|---|---|---|
| 1 | 25 | 14 | 13 | 11 | 85 | 100 |
| 2 | 29 | 15 | 16 | 14 | 88 | 94a |
| 3 | 19 | 13 | 7 | 6 | 86 | 100 |
| 4 | 28 | 20 | 8 | 8 | 100 | 100 |
| 5 | 33 | 16 | 17 | 17 | 100 | 100 |
| TOTAL | 134 | 78 | 61 | 56 |
Called by all three variant detection programs: SOAPsnp, Atlas-SNP2, and UGT
After reclassification of three SNVs as absent from the cancer specimen used for this study by Sanger sequencing
One somatic mutation was called by UGT and SOAPsnp; in Atlas-SNP2, the variant was suppressed due to the strand bias score. [NB: chr1:29491107 in 2]
Table 5.
Resolution of discordant variant calls between Sanger Sequencing of fresh frozen cancers and Illumina sequencing of matching FFPE cancer.
| Cancer Designation | Gene Name | Chr:Position | Ref. | Var | FFPE | Cancer | Normal | Status [SNP, Mutant, or No Variant] | Notes |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NAV3 | Chr12_77117387 | G | A | No | No | No | No Variant | |
| 2 | PTPRU | chr1:29491107 | C | T | Yes | Yes | No | Mutant | Called by SOAPsnp and UGT but not Atlas-SNP2 |
| 2 | PLCG2 | Chr16_80460330 | C | T | No | No | No | No variant | |
| 3 | SCN3B | Chr11_123018543 | T | A | No | No | No | No Variant |
Specificity of Somatic Mutation Detection
In addition to detecting 56 of 61 somatic mutations previously identified by high throughput Sanger sequencing, targeted capture and Illumina sequencing of the 140 CAN gene set yielded 78 novel SNVs predicted by all three variant callers, all of which were found to be coding variants. 31 of these 78 novel SNVs were selected for further analysis by PCR amplification and Sanger sequencing in DNA extracted from formalin fixed paraffin embedded primary colon tumors, as well as in DNA from corresponding cancer cell lines and from matched normal patient tissues. 28 of these 31 novel SNVs predicted by Illumina sequencing were confirmed by Sanger analysis. Four were identified as somatic mutations that had been missed in the previous high throughput Sanger analysis, and 22 of these 31 were confirmed as private germline polymorphisms. In two cancers, Illumina detected novel SNVs were confirmed by Sanger sequencing but sequencing of the patients’ germline DNA gave indeterminate results. Three of the predictions from the Illumina sequencing could not be confirmed by directed re-sequencing by Sanger methods. Thus, requiring that a variant be supported by all 3 of the variant callers yields a very high 90% specificity (28/31) for the predictions made from the Illumina sequencing of targeted captured DNAs extracted from crude formalin fixed paraffin embedded colon cancer tumors (Table 6). Quality scores (consensus quality score, best base quality score and 2nd base quality score) and coverage for 31 tested variants were similar to the remaining 47 SNVs not tested.
Table 6.
Validation of novel single nucleotide variants by Sanger sequencing
| Validation Status | Number |
|---|---|
| SNVs with validation attempted | 31 |
| Sanger data confirmed Illumina data | 28 (90%) |
| Somatic mutation | 4 |
| Germline polymorphism | 22 |
| Somatic status unknown* | 2 |
Variant as detected in Illumina data was confirmed by Sanger sequencing, but the genotype in the normal DNA could not be determined with accuracy.
Additionally, our analysis of the Illumina sequencing data also identified 13 potential insertions and deletion variants (indel variants) not annotated as known polymorphisms in dbSNP. Indels were called using the default parameters of Atlas and UGT after passing criteria for quality scores and absence of strand bias. Sanger sequencing to interrogate these indel variants confirmed all 13 of them as being real, with 1 indel proving to be a somatic mutation that had escaped detection in the high throughput Sanger sequencing effort. The remaining novel 12 indel variants were all confirmed as private germline variants (Table 7).
Table 7.
Validation of novel indel variants by Sanger sequencing
| Validation Status | Number |
|---|---|
| Indels with validation attempted | 13 |
| Sanger data confirmed Illumina data | 13 (100%) |
| Somatic mutation | 1 |
| Germline polymorphism | 12 |
In summary, of 61 previously detected somatic mutations in these 5 colon cancer cases, 57 were validated upon repeat analysis, and 56 of these were detected by targeted capture and Illumina sequencing of formalin fixed paraffin embedded extracted DNAs. Moreover, the Illumina analysis also identified 5 new somatic variants (4 single base variants and one indel) that had escaped detection in the high throughput Sanger sequencing approach. Thus Illumina analysis correctly detected 61 somatic variants present in these 5 colon cancers. Illumina analysis incorrectly predicted only 3 variants that could not be confirmed by Sanger analysis. Thus, setting aside detection of private germline variants, the prediction accuracy for somatic mutations of the targeted capture and Illumina sequencing approach equals 95% (61/64).
Mutational Comparison of Different Components of a Biphasic Colon Cancer
The ability to identify mutations from formalin fixed paraffin embedded archived tumors enables investigation of many basic questions in cancer pathogenesis. To illustrate this, we examined the case of an unusual colon cancer that demonstrated two different and distinct histologies within the tumor. One region was composed of standard type colon adenocarcinoma [Figure 3A] and another region composed of poorly differentiated [non-gland forming] adenocarcinoma [Figure 3B]. These two morphologically distinct regions were purified by microdissection and the corresponding extracted DNAs were then compared by CAN gene capture and sequencing. Despite their markedly different histologies, the mutational profiles of these two tumor regions were virtually identical [Table 3]. 354 SNP variants were identified by all three SNP callers that mapped to known germline variants annotated in dbSNP. As expected, nearly all, 351, of these variants were detected in both samples. The remaining 3 variants that corresponded to known dbSNP annotations were called by two of the SNP callers in the Low Grade sample samples, and by three of the SNP callers in the High Grade sample [Figure 4A].
Figure 3. Colon cancer with two distinct morphologies in one cancer mass.
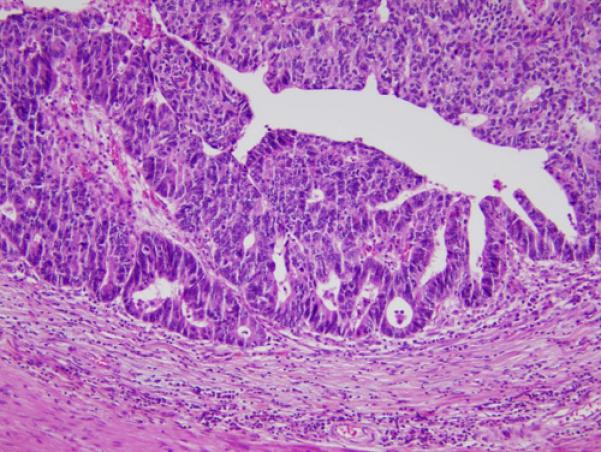
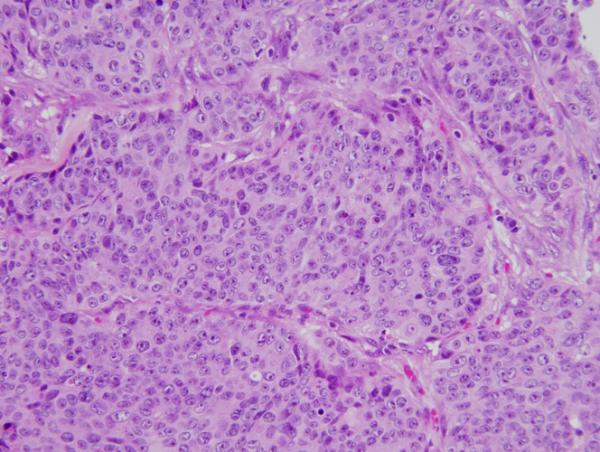
Each component was microdissected and Illumina sequenced separately. A: Low grade [standard type] component adenocarcinoma; B: High grade adenocarcinoma component [note lack of glandular formation compared to 3A]
Figure 4. Comparison of SNP calls for each component of Biphasic Colon Cancer.

Figure 4A: Overlap of SNP calls at positions of dbSNP variants in 2 distinct components of a colon adenocarcinoma
Figure 4B: Overlap of SNP calls at positions of nondbSNP variants in 2 distinct components of a colon adenocarcinoma
An additional 29 variants were also called by all 3 SNP callers, and these corresponded to novel variants not annotated in dbSNP. 28 of these 29 SNPs were identified as present in both samples. The one discordant SNP was called by two of the variant callers in the Low Grade sample but was called by all 3 SNP variant callers in the High Grade sample [Figure 4B]. To test whether these findings were derived from germline line polymorphisms or somatic mutations in the tumors we designed primers for amplifying these SNPs from the patient's normal colon and from both regions of his tumor. Single amplicons were obtainable for 24 of these 28 variants, and 23 of the 24 variants identified by Illumina sequencing proved confirmable by direct Sanger sequencing. 16 of the 23 were identified as germline SNPs and 7 as somatic mutations that were all present in both portions of the biphasic cancer. No somatic mutations were identified as present in only one of the tumor components. The 7 somatic mutations common to both tumor histologies were in APC, KRAS, ACAN, PTPRS, KIAA1409, MYO5C and MYO18B. Thus, in spite of the significant morphological differences between the two regions of this colon cancer, NGS CAN gene profiles revealed essential genetic identity, arguing for a common origin of both tumor histologies.
Discussion
We demonstrate the efficacy of genomic profiling of human colon cancer samples using Next Generation sequencing from formalin fixed paraffin embedded archived materials to globally type all somatic mutations in the previously defined 140 set of colon cancer driver genes (CAN genes), that are the recurrent targets of somatic mutations in human colon cancers. (11) The method has high sensitivity and specificity for identification of somatic mutations in tumors. Specifically, targeted capture and Illumina sequencing from formalin fixed paraffin embedded tumor samples provided essentially the same power for mutation detection as did high throughput Sanger sequencing of cell lines derived from the same tumors, with the failure of the Illumina sequencing approach to detect one somatic mutation previously identified by Sanger sequencing offset by the Illumina approach identifying 5 variants that had been missed by the high throughput Sanger approach, and with the false prediction of 3 variants by the Illumina approach, that could not be confirmed on Sanger re-sequencing, offset by the finding of 3 somatic mutations that had been called in the original high throughput Sanger approach but that could also not be confirmed on further re-sequencing.
An example of the capabilities of our methods was the finding that two morphologically distinct regions from one cancer have identical variant profiles. This virtual complete concordance of somatic mutation profiles between a well differentiated- and a poorly differentiated- component of the same colon cancer is in keeping with the hypothesis that both tumor histologies are descended from a common transformed cell of origin.(16, 22)
The Illumina method provides a straightforward and inexpensive approach for comprehensive mutation typing of human tumors. This approach provides much greater ability to identify all the mutations likely to impact on tumor phenotype than do methods that test for only a compendium of previously know somatic base changes. The method is thus directly applicable for use in identifying mutations that may guide decisions regarding use of targeted therapies in the clinic. The methods also extends the utility of mutation typing by Next Generation sequencing to archived formalin fixed paraffin embedded samples that represent by far the most common tissue processing method for Pathology Departments worldwide. The ability to interrogate these biospecimens on NGS-based platforms should be of significant utility for clinical studies that compare tumor genotype with clinical outcomes. They could also be useful in confirming mutational differences of cancers at different sites along the colon.(23) The method presented here for colon cancer should be directly applicable to other human tumors, whose defined sets of driver genes are also being rapidly elucidated by individual laboratories and by the TCGA. (1, 3, 6, 7, 18, 24) In summary, we present an inexpensive, high-throughput approach for comprehensive mutation profiling of driver gene mutations in human tumors. The method is applicable to fresh clinical samples, and also to formalin fixed paraffin embedded archived material, and should be of value for both clinical and research applications.
Supplementary Material
Acknowledgements
We gratefully acknowledge the DNA extraction techniques used in these experiments from Dr Bert Vogelstein, Director, Ludwig Center at Johns Hopkins University.
Grant Support: This project was supported by the following:
NIH1P50CA150964-01A1: Case GI Specialized Program of Research Excellence (SPORE)
NIH P30 CA043703-21: Case Comprehensive Cancer Center Support Grant
1R21CA149349-01A1: Significant Race Associated Colon Cancer Driver Gene Mutations
1U01 CA152756: EDRN Validation of Serum Biomarkers
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Cancer Genome Atlas Research N Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117:915–9. doi: 10.1182/blood-2010-08-303305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holbrook JD, Parker JS, Gallagher KT, et al. Deep sequencing of gastric carcinoma reveals somatic mutations relevant to personalized medicine. J Transl Med. 2011;9:119. doi: 10.1186/1479-5876-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nature Gen. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, White TA, MacKenzie AP, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci USA. 2011;108:17087–92. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:18032–7. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PloS One. 5:e15661. doi: 10.1371/journal.pone.0015661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lievre A, Laurent-Puig P. Genetics: Predictive value of KRAS mutations in chemoresistant CRC. Nat Rev Clin Oncol. 2009;6:306–7. doi: 10.1038/nrclinonc.2009.69. [DOI] [PubMed] [Google Scholar]
- 10.Bonanno L, Schiavon M, Nardo G, et al. Prognostic and predictive implications of EGFR mutations, EGFR copy number and KRAS mutations in advanced stage lung adenocarcinoma. Anticancer Res. 2010;30:5121–8. [PubMed] [Google Scholar]
- 11.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 12.Shen Y, Wan Z, Coarfa C, Drabek R, et al. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res. 2010;20:273–80. doi: 10.1101/gr.096388.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–32. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. J Genet Genomics. 2011;38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genomes Project C A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 22.Siegmund KD, Marjoram P, Tavare S, Shibata D. High DNA methylation pattern intratumoral diversity implies weak selection in many human colorectal cancers. PloS One. 2011;6:e21657. doi: 10.1371/journal.pone.0021657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamauchi M, Morikawa T, Kuchiba A, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61:847–54. doi: 10.1136/gutjnl-2011-300865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–61. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.