

Abstract
A series of heterocyclic quinones based on benzofuran, benzothiophene, indazole and benzisoxazole has been synthesized, and evaluated for their ability to function as substrates for recombinant human NAD(P)H:quinone oxidoreductase (NQO1), a two-electron reductase upregulated in tumor cells. Overall, the quinones are excellent substrates for NQO1, approaching the reduction rates observed for menadione
Keywords: Quinone, Benzofuran, Benzothiophene, Indazole, Quinone reductase, NQO1
1. Introduction
Quinones are widespread in nature,1 and participate in important biological redox processes. For example, the ubiquinones act as electron-transfer agents in the respiratory chain, and the heterocyclic pyrroloquinolinequinone (coenzyme PQQ) is a redox cofactor. Indeed heterocyclic derivatives form an important subset of quinones,2 often possessing potent biological activity, for example as phosphatase inhibitors.3 The best known heterocyclic quinone is the clinically used cancer therapeutic agent, the indolequinone natural product mitomycin C (MMC) 1,4–7 although other heterocyclic quinones such as streptonigrin 2 have also been widely studied.8
Given the biological importance of quinone reduction, our own studies have focused on the two-electron reduction of quinones by the enzyme NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase, QR1, EC 1.6.99.2).9–12 In particular, we have investigated a broad range of indolequinone based substrates and inhibitors,13–17 the quinoline-5,8-dione system found in streptonigrin,18 and both benzimidazole-4,7-diones 3 and benzothiazole-4,7-diones 4 (Figure 1).19 Others have also studied 5-undecyl-6-hydroxybenzothiazole-4,7-dione (UHDBT), an analogue of ubiquinone, as an inhibitor of electron transport by binding to cytochrome bc1.20, whilst the benzimidazole quinones have been widely investigated as analogues of MMC.21–27
Figure 1.

Some heterocyclic quinones.
Despite the aforementioned studies, we sought to extend the range of heterocyclic quinones studied as substrates for NQO1, focusing on benzofurans, benzothiophenes, and indazoles. Furoquinones are quite well described, and although most of their naturally occurring compounds are naphthofuranquinones,1 relatively simple derivatives such as acamelin 5 are known.28 Benzothiophene quinones are less common, although caldariellaquinone 6 appears to fulfil an important redox role in thermophilic and acidophlic archaea of the Sulfolobus genus that lack ubiquinones as electron-transfer agents.29,30 On the other hand, indazolequinones are somewhat rarer, although compounds such as 7 have been prepared as MMC analogues,31 whilst others have been investigated as substrates for carbonyl reductase.32 Therefore in an attempt to widen the group of NQO1 substrates/inhibitors, and to probe further the active site of the enzyme, we have explored a new series of heterocyclic quinones based on benzofuran, benzothiophene, indazole and benzisoxazole.
2. Results and Discussion
2.1. Chemistry
In order to make meaningful comparisons with the more widely studied indolequinones, we initially elected to investigate relatively simple 5-methoxy-heterocyclic quinones in the benzofuran and benzothiophene series. The synthesis of quinone 11 started with the known benzofuran 8, readily prepared from benzoquinone in a Nenitzescu type reaction.33 Methylation gave the known 5-methoxy derivative 9, nitration of which gave a mixture of the desired 4-nitro compound 10 along with its 6-nitro isomer in excellent overall yield, but in a 1:2 ratio. Although nitrobenzofuran 10 could be isolated, it was more convenient to reduce the mixture of nitro compounds to the corresponding amines, reduce the ketone with sodium borohydride and then oxidize the aniline with Fremy’s salt and purify the desired quinone 11 at the final stage (Scheme 1). The intermediate iminoquinone was not observed and was presumably readily hydrolyzed under the reaction conditions.
Scheme 1.

Reagents and conditions: a, KH, MeI, DMF, rt (42%); b, fuming HNO3, AcOH, rt (81% as 1:2 mixture of 4- and 6-nitro isomers); c, Sn, HCl, EtOH, reflux; d, NaBH4, MeOH, rt; e, Fremy’s salt, NaH2PO4, aq acetone, rt (20% over 3 steps).
The isomeric benzofuranquinone 13, with the alcohol group at the 2-position, was synthesized from the known benzofuran 12, readily available from 4-methoxyphenol by carbene O-H insertion, and cyclization.34 The sequence of nitration, reduction and oxidation to the quinone was carried out without purification of the intermediate compounds and delivered the pure benzofuranquinone 13 in 53% over the four steps (Scheme 2).
Scheme 2.

Reagents and conditions: a, HNO3, AcOH, rt; b, Sn, HCl, EtOH, rt; d, LiAlH4, THF, 0 °C; d, Fremy’s salt, NaH2PO4, aq acetone, rt (53% over 4 steps).
In the benzothiophene series, we started with the known 5-methoxy-2-methylbenzothiophene 14 (Scheme 3).35 Formylation under Vilsmeier conditions gave the desired 3-aldehyde 15, but in poor yield (20%), the major product being the unwanted 4-formyl compound. Nevertheless, subjecting aldehyde 15 to the usual sequence of nitration, reduction, and final oxidation to the quinone, provided the benzothiophenequinone 18 in reasonable overall yield (Scheme 3).
Scheme 3.

Reagents and conditions: a, POCl3, DMF, CH2Cl2, 0 to 50 °C (20%, plus 40% of 4-formyl isomer); b, HNO3, AcOH, rt (61%); c, Sn, HCl, EtOH, reflux (71%); d, NaBH4, MeOH, rt; e, Fremy’s salt, NaH2PO4, aq acetone, rt (41% over 2 steps).
By analogy with the conversion of benzofuran 12 into benzofuranquinone 13, the known starting material,36 ethyl 5-methoxy-3-methylbenzothiophene-2-carboxylate 19, was converted into benzothiophenequinone 23 by the now familiar sequence of nitration, reduction and final oxidation to the quinone (Scheme 4).
Scheme 4.

Reagents and conditions: a, HNO3, AcOH, rt (74%); b, Sn, HCl, EtOH, reflux (86%); d, LiAlH4, THF, rt (80%); d, Fremy’s salt, NaH2PO4, aq acetone, rt; then aq HCl (2 M), acetone. (67%).
A range of 3-unsubstituted benzothiophenequinones was also prepared as shown in Scheme 5. Reaction of 3-methoxycinnamic acid with thionyl chloride and methanol readily provided the known benzothiophene-2-carboxylate starting material 24 in 37% yield.37 Nitration, followed by hydrogenation over Pd/C resulted in reduction of the nitro group with concomitant hydrogenolysis of the chlorine, and gave the 4-aminobenzothiophene 26, which was either directly oxidized to quinone 27, or converted into quinone 29 by initial reduction of the ester (Scheme 5). Benzothiophenequinones 31–33 were also prepared by standard transformations as shown in Scheme 5.
Scheme 5.

Reagents and conditions: a, HNO3, AcOH, reflux (87%); b, H2, Pd/C, MeOH-THF (83%); c, Fremy’s salt, NaH2PO4, aq acetone, rt (75%); d, LiAlH4, THF, rt (78%); e, Fremy’s salt, NaH2PO4, aq acetone, rt (88%); f, MnO2, CH2Cl2, reflux (38%); g, Fremy’s salt, NaH2PO4, aq acetone, rt (79%); h, Ac2O, pyridine, rt (69%); i, MsCl, Et3N, CH2Cl2, rt; j, LiAlH4, THF, rt (5% over 2 steps).
The synthesis of indazolequinones started from the dimethoxyindazole 35, itself prepared by the literature method from 3,5-dimethoxyacetophenone 34 by electrophilic amination with bis(trichloroethyl) azodicarboxylate followed by zinc reduction and cyclization.38 Nitration, methylation, reduction and oxidation delivered the required indazolequinone 39 (Scheme 6). The methyl group was further functionalized by radical bromination and conversion into the 3-hydroxymethylindazolequinone 40 (Scheme 6).
Scheme 6.

Reagents and conditions: a, Cl3CCH2O2CN=NCO2CH2Cl3, BF3•OEt2, CH2Cl2, rt; b, Zn, AcOH, rt (35% over 2 steps); c, HNO3, AcOH, rt (81%); d, MeI, KOH, DMSO, rt (49%); e, Sn, HCl, EtOH, reflux (96%); f, Fremy’s salt, NaH2PO4, aq acetone, rt (78%); g, NBS, AIBN, CCl4; h, Ag-NO3, aq acetone (36% over 2 steps).
Finally an example of a benzisoxazolequinone was prepared. 2-Hydroxy-5-methoxyacetophenone 41 was converted into its oxime 42 that underwent facile intramolecular Mitsunobu reaction to provide the benzisoxazole 43 in quantitative yield. Thereafter, nitration, reduction and oxidation as before gave the required benzisoxazolequinone 45 (Scheme 7)
Scheme 7.

Reagents and conditions: a, NH2OH•HCl, EtOH, pyridine, rt (98%); b, DIAD, Ph3P, THF, rt (100%); c, HNO3, AcOH, rt (91%); d, Sn, HCl, EtOH, reflux; e, Fremy’s salt, NaH2PO4, aq acetone, rt (20% over 2 steps).
Reduction potentials were measured for benzofuran-, benzothiophene- and indazole-quinones 13, 18, 23, 39 and 40 using cyclic voltammetry in DMF as solvent with tetra-n-butylammonium tetrafluoroborate as supporting electrolyte as previously described.13 The Eredox values, with reference to ferrocene (Fc), are shown in Figure 2; values for the related indolequinones 46 and 47 are also shown. The data show that whilst the indazole quinone 39 has a similar redox potential to the indolequinones (Eredox v. Fc −1.20 to −1.40V), the other heterocyclic quinones are considerably easier to reduce. Consistent with this finding, quinone 39 had the lowest reduction rate by NQO1 (Table 1).
Figure 2.

Eredox values (v. Fc) for benzimidazole- and benzothiazole- quinones 11 and 25 compared to related indolequinones 46 and 47.13,14
Table 1.
Metabolism of heterocyclic quinones by recombinant human NQO1.
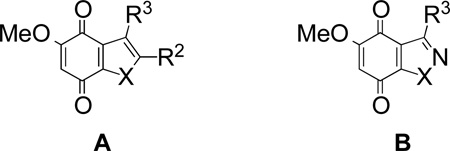 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ring | Cpd | X | R2 | R3 | NQO1 (ave)a µmol/min/mg |
NQO1 (init)a µmol/min/mg |
| 1 | A | 46 | NMe | Me | CH2OH | 1.25 ± 0.03b | ndc |
| 2 | A | 47 | NMe | CH2OH | Me | 2.49 ± 1.27d | ndc |
| 3 | A | 11 | O | Me | CH(OH)Me | 87.9±25.6 | 585±32 |
| 4 | A | 13 | O | CH2OH | Me | 80.8±6.7 | 1025±54 |
| 5 | A | 18 | S | Me | CH2OH | ndc | 236±35 |
| 6 | A | 23 | S | CH2OH | Me | ndc | 468±68 |
| 7 | A | 27 | S | CO2Me | H | 53.9±3.8 | 555±71 |
| 8 | A | 29 | S | CH2OH | H | 63.8±11.7 | 927±89 |
| 9 | A | 31 | S | CHO | H | 27.4±5.7 | 897±64 |
| 10 | A | 32 | S | CH2OAc | H | 24.5±7.9 | 900±86 |
| 11 | A | 33 | S | Me | H | 5.51±0.87 | 553±66 |
| 12 | B | 39 | NMe | - | Me | 19.6±5.3 | 55.3±5.1 |
| 13 | B | 40 | NMe | - | CH2OH | 38.1±7.1 | 247±14 |
| 14 | B | 45 | O | - | Me | 27.6±3.2 | 290±28 |
Ave and init refer to average and initial rates of metabolism as measured by the HPLC and spectrophotometric assays, respectively; average rates are determined from irreversible NADH oxidation whereas initial rates are determined from cytochrome c reduction. For reference, initial rate for menadione reduction was 1225 ± 15 µmol/min/mg;
Ref13;
nd = not determined;
Ref14.
2.2. Enzyme Studies
The new heterocyclic quinones were evaluated for their ability to act as substrates for NQO1. We used two assays for studying quinone metabolism by recombinant human NQO1 based on HPLC and spectrophotometry. The former HPLC system is capable of quantifying both NADH oxidation and quinone reduction, and gives average rates of reduction over a 30 – 40 minute period.39,13 Quinone reduction is reversible due to redox cycling of the hydroquinone, so results (Table 1) are reported as µmol NADH oxidized min−1 mg−1 NQO1. The alternative spectrophotometric method uses cytochrome c as the terminal electron acceptor and gives initial rates of reduction that are generally higher than the HPLC method.18 Nevertheless the relative order of metabolism is essentially the same with the two methods, and with the exception of Entries 1, 2, 5 and 6, both methods were used to enable reliable comparison between the new heterocyclic quinones. Interestingly, quinones 27 and 33 had similar initial reduction rates, but the average rate for 27 was 10-fold higher than for 33. This suggests that the 27 hydroquinone redox cycles more efficiently than the 33 hydroquinone, most likely due to the electron-withdrawing methyl carboxylate group present on 27.40 In contrast to our previous studies on indolequinones,13,39 electron-withdrawing groups did not appear to increase rates of reduction for the benzothiophene series.
The new quinones are all excellent substrates for rhNQO1. In the benzofuran and benzothiophene series, reduction rates were higher when the hydroxyalkyl substituent was at the C-2 position rather than C-3 (Table 1), possibly due to stabilizing hydrogen-bonding interactions with key amino acid residues in the NQO1 active site. As with the benzimidazole- and benzothiazole- quinones,19 all of the new quinones were much better substrates for NQO1 than the widely studied indolequinones,13,39,14 as seen by comparison with indolequinones 46 and 47 included in Table 1 for comparison. In fact, the reduction rates for benzofuran- and benzothiophene- quinones 13, 29, 31 and 32 approach the initial reduction rate observed for menadione (1225 ± 15 µmol/min/mg),19 a simple naphthoquinone that has been used to measure activity of the enzyme, making these compounds some of the best NQO1 substrates seen to date.
The results presented here complement our previous work on bioreductive activation of indolequinone antitumor agents by NQO1. Novel heterocyclic quinones have been synthesized, characterized and studied biologically as substrates for recombinant human NQO1. These data add to our understanding of the structural requirements for efficient metabolism by the quinone reductase enzyme.
3. Experimental Section
3.1. General Chemistry Experimental Details
Commercially available reagents were used throughout without purification unless otherwise stated. Light petroleum refers to the fraction with bp 40 – 60 °C and was distilled before use. Ether refers to diethyl ether. Reactions were routinely carried out under a nitrogen or argon atmosphere. Analytical thin layer chromatography was carried out on aluminum-backed plates coated with silica gel, and visualized under UV light at 254 and/or 360 nm. Flash chromatography was carried out on silica gel. Fully characterized compounds were chromatographically homogeneous.
Infrared spectra were recorded in the range 4000 – 600 cm−1 using FT spectrometers. NMR spectra were carried out at 300 and 400 MHz (1H frequencies, corresponding 13C frequencies are 75 and 100 MHz). Chemical shifts are quoted in ppm with TMS as internal standard. J values are recorded in Hz. In the 13C spectra, signals corresponding to CH, CH2 or CH3 groups, as assigned from DEPT, are noted; all others are C. High and low resolution mass spectra were recorded on a Micromass GCT TDF High Resolution mass spectrometer, or at the EPSRC Mass Spectrometry Service (Swansea).
3.2. Synthesis of Benzofuranquinones
3-Acetyl-5-methoxy-2-methylbenzofuran 9
3-Acetyl-5-hydroxy-2-methylbenzofuran 833 (1.62 g, 8.5 mmol) in DMF (30 mL) was added to a stirring suspension of potassium hydride (0.61 g, 15.2 mmol) in DMF (50 mL) at 0 °C. The mixture was stirred at room temperature for 45 min. Iodomethane (1.80 g, 12.7 mmol) was added dropwise at 0 °C and the mixture allowed to warm to room temperature. After 2 h saturated ammonium chloride solution was added and the mixture extracted with ethyl acetate. The ethyl acetate layer was washed thoroughly with hydrochloric acid (1 M), dried (MgSO4) and concentrated. The crude product was purified by chromatography eluting with dichloromethane to yield the title compound as a colorless solid (0.74 g, 42%), mp 69–70 °C (lit.,41 mp 72 °C); δH (300 MHz; CDCl3) 7.48 (1 H, d, J 2.6, 4-H), 7.33 (1 H, d, J 8.9, 7-H), 6.89 (1 H, dd, J 8.9, J 2.6, 6-H), 3.88 (3 H, s, OMe), 2.76 (3 H, s, Me), 2.62 (3 H, s, Me).
3-Acetyl-5-methoxy-2-methyl-4-nitrobenzofuran 10
To a solution of 3-acetyl-5-methoxy-2-methylbenzofuran 9 (0.217 g, 1.1 mmol) in acetic acid (4 mL) cooled to 0 °C was added a mixture of fuming nitric acid (0.5 mL) and acetic acid (2 mL). The mixture was stirred at room temperature for 2 h and then poured on to an ice water mixture and the precipitate obtained was filtered off and dried. NMR analysis of the mixture showed a 2:1 ratio of 6-nitro and 4-nitro products (0.21 g, 81%), used without further purification. Small quantities of each isomer were obtained by preparative TLC (dichloromethane elution) and were identified by 1H NMR spectroscopy: 3-acetyl-5-methoxy-2-methyl-4-nitrobenzofuran 10, δH (300 MHz; CDCl3) 7.50 (1 H, d, J 9.0, ArH), 7.03 (1 H, d, J 9.0 ArH), 3.93 (3 H, s, OMe), 2.74 (3 H, s, Me), 2.49 (3 H, s, Me); 3-acetyl-5-methoxy-2-methyl-6-nitrobenzofuran, δH (300 MHz; CDCl3) 7.97 (1 H, s, ArH), 7.76 (1 H, s ArH), 4.02 (3 H, s, OMe), 2.83 (3 H, s, Me), 2.62 (3 H, s, Me).
3-(1-Hydroxyethyl)-5-methoxy-2-methylbenzofuran-4,7-dione 11
To a mixture of 3-acetyl-5-methoxy-2-methyl-4-nitrobenzofuran 10 and its 6-nitro isomer (1:2 ratio) (0.80 g, 3.4 mmol) in ethanol (100 mL) were added tin powder (4.00 g, 34.0 mmol) and hydrochloric acid (3 M; 40 mL). The mixture was heated under reflux for 30 min. Upon cooling the solution was decanted from the tin and neutralized with saturated aqueous sodium hydrogen carbonate. The mixture was extracted with ethyl acetate and the organic layer dried (MgSO4) and concentrated, to yield a mixture of 4- and 6-amino derivatives (0.56 g) that was used directly. Sodium borohydride (0.10 g, 2.6 mmol) was added to a stirred solution of the above mixture (0.56 g, 2.6 mmol) in methanol (50 mL). After 20 min, acetone (10 mL) was added. The solvent was removed and the residue dissolved in ethyl acetate and washed with water. The organic layer was dried (MgSO4) and concentrated. To a solution of the crude product in acetone (40 mL) was added a solution of potassium nitrosodisulfonate (1.00 g, 3.7 mmol) in sodium dihydrogen phosphate buffer (0.3 M; 40 mL). The mixture was stirred at room temperature for 1 h. The acetone was removed in vacuo, and the resulting residue was extracted with dichloromethane and washed with water. The organic layer was dried (Na2SO4) and concentrated. The crude material was purified by chromatography eluting with dichloromethane/ethyl acetate (19:1) and recrystallized (dichloromethane/ether) to yield the title compound (0.052 g, 20%, over 3 steps based on the 4-nitro starting material), as an orange crystalline solid, mp 157–158 °C; λmax (MeOH)/nm 436 (log ε 3.18), 320 (3.66), 260 (3.98); νmax (KBr)/cm−1 3414, 3060, 2990, 2915, 1683, 1657, 1607, 1587; δH (300 MHz; CDCl3) 5.76 (1 H, s, 6-H), 4.81 (1 H, m, CHMe), 3.85 (3 H, s, OMe), 3.78 (1 H, d, J 10.7 Hz, OH), 2.38 (3 H, s, Me), 1.45 (3 H, d, J 6.7 Hz, CHMe); δC (100 MHz; CDCl3) 179.2 (C), 175.4 (C), 159.8 (C), 153.4 (C), 150.6 (C), 125.0 (C), 124.8 (C), 105.7 (CH), 62.4 (CH), 56.9 (Me), 24.4 (Me), 12.2 (Me).
2-Hydroxymethyl-5-methoxy-3-methylbenzofuran-4,7-dione 13
To a solution of methyl 5-methoxy-3-methylbenzofuran-2-carboxylate 1234 (0.20 g, 0.91 mmol) in acetic acid (6 mL), cooled to −10°C was added a mixture of concentrated nitric acid (1 mL) and acetic acid (4 mL). The mixture was stirred at room temperature for 1 h. The reaction mixture was poured in an ice/water mixture and the resulting precipitate filtered off and dried. The crude material was obtained as a 2.8:1 ratio of 4- and 6-nitro products and used directly in the next step. To a suspension of the above mixture in ethanol (30 mL) were added tin powder (0.48 g, 4.0 mmol) and hydrochloric acid (3 M; 7 mL). The mixture was stirred at room temperature for 2 h. The solution was decanted from the excess tin and neutralized with saturated aqueous sodium hydrogen carbonate. The suspension obtained was added to an equal volume of water. The mixture was extracted with ethyl acetate. The organic layer was dried (MgSO4) and concentrated. The crude material was used directly in the next step without purification.
To a suspension of lithium aluminum hydride (0.091 g, 2.4 mmol) in THF (10 mL) at 0°C was added a solution of the above mixture of benzofurans in THF (10 mL) and the reaction was stirred for 15 min. The reaction mixture was quenched by the addition of water (0.2 mL), sodium hydroxide (1 M; 0.2 mL) and silica gel (2 g). The granular precipitate was filtered off through a pad of Celite. The filtrate was dried (MgSO4) and concentrated in vacuo to give the alcohol that was used directly in the next step without purification, or characterization. To a solution of the benzofuran-2-methanol in acetone (50 mL) was added a solution of potassium nitrosodisulfonate (0.64 g, 2.4 mmol) in sodium dihydrogen phosphate buffer (0.3 M; 50 mL). The mixture was stirred at room temperature for 1 h. The excess acetone was removed in vacuo. The resulting residue was extracted with dichloromethane and washed with water. The organic layer was dried (Na2SO4) and concentrated. The crude product was purified by chromatography eluting with ethyl acetate/dichloromethane (1:4) and recrystallized (ethyl acetate) to yield the title compound as an orange solid (0.108 g, 53%, over 4 steps); mp 205–206 °C; (Found: C, 58.4; H, 4.5. C11H10O5 + 0.2H2O requires C, 58.5; H, 4.6%); (Found: M+, 222.0528. C11H10O5 requires 222.0528); νmax (KBr)/cm−1 3452, 3057, 2939, 1691, 1660, 1610, 1587; δH (300 MHz; CDCl3) 5.80 (1 H, s, 6-H), 4.69 (2 H, d, J 4.0 Hz, CH2OH), 3.86 (3 H, s, OMe), 2.31 (3 H, s, Me), 1.98 (1 H, br s, OH); δC (100 MHz; acetone) 177.8 (C), 175.3 (C), 160.3 (C), 156.6 (C), 149.2 (C), 125.4 (C), 116.7 (C), 105.7 (CH), 56.4 (Me), 54.0 (CH2), 7.6 (Me); m/z (EI) 222 (M+, 35%), 193 (13), 151 (23), 109 (25), 69 (100).
The intermediate nitro compound methyl 5-methoxy-3-methyl-4-nitrobenzofuran-2-carboxylate, can be isolated and characterized. To a solution of methyl 5-methoxy-3-methylbenzofuran-2-carboxylate 12 (0.130 g, 0.6 mmol) in acetic acid (5 mL), cooled to −10°C was added a mixture of concentrated nitric acid (0.5 mL) and acetic acid (3 mL). The mixture was stirred at room temperature for 1 h. The reaction mixture was poured in an ice/water mixture and the resulting precipitate filtered off and dried. The crude material was obtained as a 3:1 ratio of 4- and 6-nitro products (0.12 g, 80%). Purification by chromatography eluting with dichloromethane gave methyl 5-methoxy-3-methyl-4-nitrobenzofuran-2-carboxylate, mp 146–149 °C; νmax (KBr)/cm−1 3103, 2996, 2945, 1706, 1629, 1588; δH (300 MHz; CDCl3) 7.64 (1 H, d, J 9.1 Hz, ArH), 7.20 (1 H, d, J 9.1 Hz, ArH), 3.99 (3 H, s, OMe), 3.96 (3 H, s, OMe), 2.47 (3 H, s, Me); δC (75 MHz; CDCl3) 160.0 (C), 148.8 (C), 147.6 (C), 143.3 (C), 133.6 (C), 123.0 (C), 121.5 (C), 115.1 (CH), 113.7 (CH), 57.7 (Me), 52.4 (Me), 8.9 (Me).
3.3. Synthesis of Benzothiophenequinones
5-Methoxy-2-methylbenzothiophene-3-carboxaldehyde 15
To a mixture of dry DMF (19.6 mL, 169.40 mmol) and dry dichloromethane (9.1 mL) was added dropwise phosphorus oxychloride (16.4 mL, 169.40 mmol) at 0 °C. The ice bath was removed. The reaction mixture was stirred for an additional 30 min and cooled in an ice bath. 5-Methoxy-2-methylbenzothiophene 1435 (3.02 g, 16.94 mmol) was added portionwise to the solution over 5 min at 0 °C. The reaction mixture was stirred and heated to 50 °C overnight, poured into an ice-cold aqueous sodium hydroxide (1 M). The mixture was extracted with dichloromethane, dried over MgSO4, filtered, and concentrated. The residue obtained was purified by chromatography, eluting with dichloromethane, to yield the title compound (390 mg, 20%) as a yellow oil; (Found: MH+, 207.0477. C11H10O2S + H requires 207.0480); νmax (film)/cm−1 2957, 2930, 2834, 1735, 1670, 1597, 1559, 1455, 1413, 1351, 1270, 1247, 1224, 1151; δH (300 MHz; CDCl3) 10.31 (1 H, s, CHO), 8.14 (1 H, d, J 2.5, 4-H), 7.60 (1 H, d, J 9.0, 7-H), 7.00 (1 H, dd, J 9.0, 2.5, 6-H), 3.88 (3 H, s, OMe), 2.89 (3 H, s, Me); δC (75 MHz; CDCl3) 184.0 (CH), 159.6 (C), 159.3 (C), 138.9 (C), 130.5 (C), 129.3 (C), 122.6 (CH), 116.0 (CH), 106.4 (CH), 56.1 (Me), 14.8 (Me); m/z (CI) 207 (MH+, 100%), 179 (10); and the 4-formyl derivative (809 mg; 40 %) as a beige solid; mp 119–120 °C; (Found: MH+, 207.0477. C11H10SO2 + H requires 207.0480); νmax (KBr)/cm−1 3421, 2910, 2846, 1661, 1566, 1454, 1430, 1314, 1243, 1183, 1131, 1071; δH (300 MHz; CDCl3) 10.68 (1 H, s, CHO), 8.09 (1 H, br s, 3-H), 7.86 (1 H, d, J 8.8, ArH), 6.96 (1 H, d, J 8.8, ArH), 3.97 (3 H, s, OMe), 2.63 (3 H, s, Me); δC (100 MHz; CDCl3) 190.7 (CH), 162.3 (C), 148.0 (C), 139.9 (C), 134.1 (C), 129.7 (CH), 122.7 (CH), 118.1 (C), 108.6 (CH), 55.8 (Me), 17.0 (Me); m/z (CI) 207 (MH+, 100%), 179 (10).
5-Methoxy-2-methyl-4-nitrobenzothiophene-3-carboxaldehyde 16
To a solution of 5-methoxy-2-methylbenzothiophene-3-carboxaldehyde 15 (388 mg, 1.88 mmol) in acetic acid (3.2 mL), cooled to 0–5 °C was added a mixture of nitric acid (638 µl, 9.42 mmol) in acetic acid (4.7 mL). The mixture was stirred at room temperature overnight. The reaction mixture was poured into ice/water, neutralized with a saturated aqueous sodium hydrogen carbonate, extracted with dichloromethane, dried over MgSO4, filtered, and the filtrate evaporated under reduced pressure and azeotroped with toluene to remove the remaining acetic acid. The crude product obtained was purified by chromatography, eluting with dichloromethane, to yield the title compound (286 mg; 61 %) as a yellow solid; mp 205–207 °C; (Found: MH+, 252.0333. C11H9NO4S + H requires 252.0331); νmax (KBr)/cm−1 2975, 2941, 2845, 2771, 1679, 1598, 1536, 1459, 1425, 1375, 1271, 1117, 1082; δH (300 MHz; CDCl3) 10.05 (1 H, s, CHO), 7.80 (1 H, d, J 8.9, ArH), 7.66 (1 H, d, J 8.9, ArH), 3.97 (3 H, s, OMe), 2.90 (3 H, s, Me); δC (75 MHz; CDCl3) 183.7 (CHO), 160.4 (C), 150.4 (C), 131.0 (C), 129.4 (C), 128.8 (C), 125.1 (CH), 111.4 (CH), 57.7 (Me), 16.4 (Me), one C unobserved; m/z (CI) 252 (MH+, 100%), 222 (65), 206 (30), 194 (10).
4-Amino-5-methoxy-2-methylbenzothiophene-carboxaldehyde 17
To a suspension of 5-methoxy-2-methyl-4-nitrobenzo-thiophenecarboxaldehyde 16 (280 mg, 1.12 mmol) in ethanol (23 mL) was added tin powder (601 mg, 5.02 mmol) and hydrochloric acid (3 M; 8.1 mL). The mixture was stirred and heated under reflux for 1 h. Upon cooling, the reaction mixture was decanted from the excess of tin and neutralized with a saturated aqueous solution of NaHCO3. The suspension obtained was filtered through Celite and extracted with ethyl acetate. The combined organic layer was dried over MgSO4 and evaporated under reduced pressure to yield the title compound (175 mg, 71%) as a yellow solid; mp 88–90 °C; (Found: MH+, 222.0587. C11H11NO2S + H requires 222.0589); νmax (KBr)/cm−1 3437, 2960, 2921, 2852, 1655, 1602, 1559, 1459, 1421, 1340, 1224, 1201; δH (300 MHz; CDCl3) 10.08 (1 H, s, CHO), 6.96 (1 H, d, J 8.5, ArH), 6.91 (1 H, d, J 8.5, ArH), 6.02 (2 H, br s, NH2), 3.89 (3 H, s, OMe), 2.85 (3 H, s, Me); δC (100 MHz; CDCl3) 184.7 (CH), 160.8 (C), 144.2 (C), 134.3 (C), 132.1 (C), 130.9 (C), 123.1 (C), 110.5 (CH), 108.6 (CH), 56.5 (Me), 14.9 (Me); m/z (CI) 222 (MH+, 100%), 206 (5).
3-Hydroxymethyl-5-methoxy-2-methylbenzothiophene-4,7-dione 18
Sodium borohydride (44 mg, 2.31 mmol) was added in one portion to a solution of 4-amino-5-methoxy-2-methylbenzothiophene-3-carboxaldehyde 17 (170 mg, 0.77 mmol) in dry methanol (21 mL) cooled at 0 °C. The reaction mixture was stirred at room temperature for 1 h. The reaction mixture was evaporated off and the crude product obtained was purified by chromatography, eluting with ethyl acetate-dichloromethane 1:1, to yield the unstable amino alcohol intermediate as a yellow solid that was used in the next step with no further purification.
To a solution of the amino alcohol intermediate dissolved in acetone (50 mL) was added a solution of potassium nitrosodisulfonate (907 mg, 3.32 mmol) in sodium dihydrogen phosphate buffer (0.3 M; 40 mL). The reaction was stirred at room temperature for 1 h. The excess acetone was removed in vacuo. The resulting residue was extracted with dichloromethane and the combined organic layer evaporated off. The residue obtained was purified by chromatography, eluting with dichloromethane, to yield the title compound (75 mg, 41%) as a yellow crystalline solid; mp 196–198 °C; (Found: MH+, 239.0372. C11H10O4S + H requires 239.0378); λmax (acetonitrile)/nm 200 (log ε 4.14), 232 (4.12), 288 (4.18), 348 (3.47), 416 (3.21); νmax (KBr)/cm−1 3425, 2925, 2852, 1663, 1636, 1601, 1447, 1344, 1324, 1255, 1220, 1120, 1086, 1070; δH (300 MHz; CDCl3) 5.96 (1 H, s, 6-H), 4.68 (2 H, s, CH2OH), 3.88 (3 H, s, OMe), 2.53 (3 H, s, Me); δC (100 MHz; CDCl3) 179.5 (C), 178.1 (C), 160.1 (C), 144.4 (C), 142.9 (C), 138.4 (C), 137.0 (C), 108.1 (CH), 56.9 (Me), 56.5 (CH2), 13.7 (Me); m/z (CI) 239 (MH+, 85%), 223 (50), 221 (100).
Ethyl 5-methoxy-3-methyl-4-nitrobenzothiophene-2-carboxylate 20
To a solution of ethyl 5-methoxy-3-methylbenzothiophene-2-carboxylate 1936 (480 mg, 1.92 mmol) in acetic acid (3.2 mL), cooled to 0–5 °C was added a mixture of nitric acid (1.3 mL, 19.20 mmol) in acetic acid (4.8 mL). The mixture was stirred at room temperature overnight. The reaction mixture was dropped in ice/water, extracted with dichloromethane, dried over MgSO4, filtered, evaporated and azeotroped with toluene to remove the acetic acid. The crude product obtained was purified by chromatography, eluting with dichloromethane, to yield the title compound (417 mg, 74%) as a light yellow solid; mp 158–160 °C; (Found: MH+, 296.0589. C13H13NO5S + H requires 296.0592); νmax (KBr)/cm−1 3422, 2988, 2939, 2850, 1711, 1608, 1527, 1446, 1370, 1297, 1247, 1193, 1174, 1151, 1051; δH (300 MHz; CDCl3) 7.85 (1 H, d, J 8.9, ArH), 7.26 (1 H, d, J 8.9, ArH), 4.40 (2 H, q, J 7.1, OCH2Me), 3.98 (3 H, s, OMe), 2.64 (3 H, s, Me), 1.41 (3 H, t, J 7.1, CH2Me); δC (100 MHz; CDCl3) 162.6 (C), 148.6 (C), 137.1 (C), 136.7 (C), 133.8 (C), 131.5 (C), 130.6 (C), 125.1 (CH), 113.3 (CH), 61.7 (CH2), 57.3 (Me), 14.2 (Me), 12.1 (Me); m/z (CI) 296 (MH+, 100%), 279 (8), 266 (15).
Ethyl 4-amino-5-methoxy-3-methylbenzothiophene-2-carboxylate 21
To a suspension of ethyl 5-methoxy-3-methyl-4-nitrobenzothiophene-2-carboxylate 20 (370 mg, 1.25 mmol) in ethanol (27 mL) was added tin powder (676 mg, 5.64 mmol) and hydrochloric acid (3 M; 9 mL). The mixture was stirred and heated under reflux for 1 h. Upon cooling, the reaction mixture was decanted from the excess of tin and neutralized with a saturated aqueous solution of NaHCO3. The suspension obtained was filtered through Celite and extracted with ethyl acetate. The combined organic layer was dried over MgSO4, filtered and evaporated under reduced pressure to yield the title compound (285 mg, 86%) as a yellow solid; mp 120–123 °C; (Found: MH+, 266.0844. C13H15NO3S + H requires 266.0851); νmax (KBr)/cm−1 3460, 3368, 2968, 2925, 1705, 1621, 1536, 1471, 1440, 1332, 1259, 1209, 1178, 1144, 1094, 1055, 1009; δH (300 MHz; CDCl3) 7.11 (1 H, d, J 8.6, ArH), 7.03 (1 H, d, J 8.6, ArH), 4.35 (2 H, q, J 7.0, OCH2Me), 3.89 (3 H, s, OMe), 3.06 (3 H, s, Me), 1.40 (3 H, t, J 7.0, CH2Me); NH2 not observed; δC (100 MHz; CDCl3) 165.7 (C), 145.5 (C), 143.3 (C), 136.7 (C), 136.5 (C), 130.5 (C), 127.3 (C), 115.3 (CH), 113.4 (CH), 63.1 (CH2), 59.0 (Me), 18.1 (Me), 16.4 (Me); m/z (CI) 266 (MH+, 100%), 265 (40), 220 (10).
4-Amino-5-methoxy-3-methylbenzothiophene-2-methanol 22
To a suspension of lithium aluminum hydride (162 mg, 4.23 mmol) in dry THF (6 mL) at 0 °C was added a solution of ethyl 4-amino-5-methoxy-3-methylbenzothiophene-2-carboxylate 21 (280 mg, 1.06 mmol) in dry THF (3 mL). The mixture was allowed to warm up to room temperature and stirred for 2 h. The mixture was cooled to 0 °C and quenched by the addition of water (0.5 mL), aqueous sodium hydroxide (1 M; 0.5 mL) and silica gel. The granular precipitate was filtered off through a pad of Celite. The filtrate was dried over MgSO4, filtered and concentrated in vacuo to yield the title compound (188 mg, 80%) as an orange solid; mp 114–115 °C; (Found: MH+, 224.0753. C11H11NO2S + H requires 224.0745); νmax (KBr)/cm−1 3436, 3352, 3273, 2923, 2851, 1599, 1462, 1265, 1203, 1157, 1136, 1044, 1019; δH (300 MHz; CDCl3) 7.12 (1 H, d, J 8.6, ArH), 6.90 (1 H, d, J 8.6, ArH), 4.75 (2 H, s, CH2OH), 3.88 (3 H, s, OMe), 2.54 (3 H, s, Me); OH, NH2 not observed; δC (100 MHz; CDCl3) 142.3 (C), 134.8 (C), 131.7 (C), 131.1 (C), 127.5 (C), 126.8 (C), 110.6 (CH), 109.1 (CH), 56.9 (CH2), 55.5 (Me), 13.4 (Me); m/z (CI) 224 (MH+, 60%), 223 (M+, 80), 206 (100), 194 (15).
2-Hydroxymethyl-5-methoxy-3-methylbenzothiophene-4,7-dione 23
To a solution of 4-amino-5-methoxy-3-methylbenzothiophene-2-methanol 22 (180 mg, 0.81 mmol) in acetone (48 mL) was added a solution of potassium nitrosodisulfonate (889 mg, 3.26 mmol) in sodium dihydrogen phosphate buffer (0.3 M; 39 mL). The reaction was stirred at room temperature for 1 h. The excess acetone was removed in vacuo. The resulting residue was extracted with dichloromethane and the combined organic layer concentrated. The residue obtained was stirred at room temperature in a 1:1 mixture of hydrochloric acid (2 M)-acetone for 1 h. The acetone was removed in vacuo. The resulting residue was extracted with dichloromethane. The organic layer was washed with water, dried over MgSO4, filtered and evaporated under reduced pressure to yield the title compound (130 mg, 67%) as an orange crystalline solid; mp 215–216 °C; (Found: MH+, 239.0379. C11H10O4S + H requires 239.0378); λmax (acetonitrile)/nm 200 (log ε 3.86), 232 (3.88), 284 (3.90), 352 (3.24), 412 (3.09); νmax (KBr)/cm−1 3385, 3054, 2920, 2843, 1677, 1638, 1600, 1538, 1461, 1442, 1342, 1327, 1253, 1227, 1134; δH (300 MHz; CDCl3) 5.96 (1 H, s, 6-H), 4.87 (2 H, s, CH2OH), 3.87 (3 H, s, OMe), 2.47 (3 H, s, Me), 1.71 (1 H, br s, OH); δC (100 MHz; CDCl3) 180.0 (C), 176.9 (C), 160.4 (C), 146.6 (C), 143.2 (C), 136.5 (C), 135.7 (C), 107.7 (CH), 58.2 (CH2), 56.7 (Me), 13.2 (Me); m/z (CI) 239 (MH+, 100%), 221 (30).
Methyl 3-chloro-5-methoxy-4-nitrobenzothiophene-2-carboxylate 25
Methyl 3-chloro-5-methoxybenzothiophene-2-carboxylate 2437 (1.00 g, 3.90 mmol) was dissolved in acetic acid (20 mL). Fuming nitric acid (0.25 mL) was added carefully, and the mixture was stirred at room temperature before being heated under reflux overnight. After cooling, the mixture was poured into water (125 mL), and the precipitate collected, washed with water and dried to give the title compound (1.03 g, 87%) used without further purification. A sample was recrystallized from methanol to give yellow crystals, mp 170–173 °C; (Found: M+, 300.9821. C11H835ClNO5S requires 300.9812); νmax (KBr)/cm−1 1721, 1607, 1534, 1514, 1438, 1374, 1293, 1241; δH (300 MHz; CDCl3) 7.86 (1 H, d, J 9.0, ArH), 7.33 (1 H, d, J 9.0, ArH), 3.99 (3 H, s, OMe), 3.97 (3 H, s, OMe); δC (100 MHz; CDCl3) 160.8 (C), 149.4 (C), 132.1 (C), 130.1 (C), 127.4 (C), 125.4 (CH), 122.8 (C), 114.6 (CH), 57.4 (Me), 52.9 (Me); 1 C unobserved; m/z (EI) 303/301 (MH+, 100%).
Methyl 4-amino-5-methoxybenzothiophene-2-carboxylate 26
The 3-chloro-4-nitrobenzothiophene 25 (1.00 g, 3.3 mmol) and sodium acetate (0.60 g, 7.3 mmol) were dissolved in methanol (200 mL) and THF (55 mL). Palladium-on-carbon (5%; 0.5 g) was added, and the mixture was shaken under a hydrogen atmosphere for 5 h. The mixture was filtered through Celite, the filtrate evaporated, and the residue purified by chromatography eluting with light petroleum / ethyl acetate (4:1) to give the title compound (0.69 g, 88%) as a pale yellow solid, mp 149–151 °C; (Found: C, 55.6; H, 4.7; N, 5.7. C11H11NO3S requires C, 55.7; H, 4.7; N, 5.9%); νmax (KBr)/cm−1 3445, 3359, 1702, 1625, 1528, 1470, 1206; δH (300 MHz; CDCl3) 8.01 (1 H, d, J 0.8, 3-H), 7.20 (1 H, dd, J 8.6, 0.8, ArH), 7.09 (1 H, d, J 8.6, ArH), 4.27 (2 H, br s, NH2), 3.93 (3 H, s, OMe), 3.91 (3 H, s, OMe); δC (100 MHz; CDCl3) 163.3 (C), 142.9 (C), 136.0 (C), 132.4 (C), 131.8 (C), 127.8 (C), 126.5 (CH), 113.7 (CH), 111.4 (CH), 56.8 (Me), 52.3 (Me); m/z (EI) 237 (M+, 70%), 222 (100), 194 (35), 83 (68).
Methyl 5-methoxy-4,7-dioxobenzothiophene-2-carboxylate 27
A mixture of 4-aminobenzothiophene 26 (50 mg, 0.21 mmol) and potassium nitrosodisulfonate (230 mg, 0.84 mmol) in acetone (7.5 mL) and sodium dihydrogen phosphate buffer (0.3 M; 7.5 mL) was stirred at room temperature overnight. The acetone was evaporated, water (70 mL)was added, and the mixture was extracted with dichloromethane (3 × 25 mL). The combined extracts were dried (Na2SO4), evaporated and the residue purified by chromatography eluting with dichloromethane to give the title compound (40 mg, 75%) as a dark yellow solid, mp 234 °C; (Found: C, 52.8; H, 3.3. C11H8O5S requires C, 52.4; H, 3.2%); νmax (KBr) 3088, 2960, 1720, 1689, 1633, 1597, 1534, 1439, 1244, 1268, 1085; δH (300 MHz; CDCl3) 8.15 (1 H, s, 3-H), 6.08 (1 H, s, 6-H), 3.94 (3 H, s, OMe), 3.90 (3 H, s, OMe); δC (75 MHz; CDCl3) 179.6 (C), 175.1 (C), 161.4 (C), 160.8 (C), 148.0 (C), 139.3 (C), 138.5 (C), 130.7 (CH), 109.0 (CH), 57.0 (Me), 53.1 (Me); m/z (EI) 252 (M+, 100%), 237 (34).
4-Amino-5-methoxybenzothiophene-2-methanol 28
A solution of the benzothiophene 26 (2.00 g, 8.43 mmol) in dry THF (28 mL) was added dropwise to lithium aluminum hydride (1.07 g, 28.1 mmol) in THF (55 mL). The mixture was stirred at room temperature for 16 h, and then water was carefully added. The mixture was filtered through Celite, and extracted with ethyl acetate (3 × 30 mL). The combined extracts were washed with water (30 mL), dried (MgSO4), and evaporated to a solid that was recrystallized from methanol to give the title compound (1.38 g, 78%), mp 130–132 °C; (Found: C, 57.2; H, 5.3; N, 6.6. C10H11NO2 requires C, 57.4; H, 5.3; N, 6.7%); νmax (KBr)/cm−1 3401, 3310, 3016, 1602, 1480, 1463; δH (300 MHz; CDCl3) 7.19 (1 H, d, J 8.6, ArH), 7.09 (1 H, s, 3-H), 6.95 (1 H, d, J 8.6, ArH), 4.87 (2 H, s, CH2), 3.90 (3 H, s, OMe); OH, NH2 not observed; δC (75 MHz; CDCl3) 144.0 (C), 143.3 (C), 133.7 (C), 130.4 (C), 128.6 (C), 117.1 (CH), 111.9 (CH), 110.7 (CH), 61.0 (CH2), 56.8 (Me); m/z (EI) 209 (M+, 62%), 194 (100), 166 (25).
2-Hydroxymethyl-5-methoxybenzothiophene-4,7-dione 29
A mixture of the 4-aminobenzothiophene 28 (0.32 g, 1.5 mmol) and potassium nitrosodisulfonate (1.61 g, 6.0 mmol) in acetone (47 mL) and sodium dihydrogen phosphate buffer (0.3 M; 47 mL) was stirred at room temperature for 1 h. Work-up as described above gave the title compound (0.29 g, 88%) as a dark orange solid, mp 213 °C (from methanol); (Found: C, 53.3; H, 3.3. C10H8O4S requires C, 53.6; H. 3.60%); νmax (KBr)/cm−1 3413, 3062, 2986, 2947, 1680, 1628, 1598, 1572, 1326, 1246, 1140, 1084, 1043, 864, 792; δH (300 MHz; CDCl3) 7.43 (1 H, s, 3-H), 5.99 (1 H, s, 6-H), 4.91 (2 H, d, J 6.0, CH2OH), 3.88 (3 H, s, OMe), 2.05 (1 H, t, J 6.0, CH2OH); δC (100 MHz; CDCl3) 180.2 (C), 175.9 (C), 160.6 (C), 156.3 (C), 142.3 (C), 139.2 (C), 121.5 (CH), 108.7 (CH), 58.8 (CH2), 57.3 (Me); m/z (EI) 224 (M+, 100%) 209 (28), 194 (35), 125 (26).
4-Amino-5-methoxybenzothiophene-2-carboxaldehyde 30
A solution of the benzothiophene-2-methanol 28 (0.90 g, 4.30 mmol) in dichloromethane (100 mL) was stirred with manganese(IV) oxide (3.78 g, 43 mmol) under reflux for 24 h. The solution was filtered through Celite, washed through with dichloromethane, and the combined filtrate and washings concentrated under vacuum. The residue was purified by chromatography eluting with light petroleum/ethyl acetate (4:1) to give the title compound (0.29 g, 33%) as a pale yellow solid, mp 128–130 °C; (Found: C, 57.9; H, 4.3; N, 6.5. C10H9NO2S requires C, 57.9; H, 4.4; N, 6.8%); νmax (KBr)/cm−1 3474, 3373, 2826, 1667, 1520, 1483; δH (300 MHz; CDCl3) 10.00 (1 H, s, CHO), 7.97 (1 H, s, H-3), 7.19 (1 H, d, J 8.6, ArH), 7.09 (1 H, d, J 8.6, ArH), 4.42 (2 H, br s, NH2), 3.90 (3 H, s, OMe); δC (75 MHz; CDCl3) 184.4 (CH), 143.0 (C), 142.2 (C), 136.2 (C), 133.2 (C), 130.8 (CH), 127.7 (C), 114.6 (CH), 111.9 (CH), 56.7 (Me); m/z (EI) 207 (M+, 70%), 192 (100), 164 (40), 77 (35).
2-Formyl-5-methoxybenzothiophene-4,7-dione 31
A mixture of the 4-aminobenzothiophene 30 (50 mg, 0.24 mmol) and potassium nitrosodisulfonate (260 mg, 0.96 mmol) in acetone (7.5 mL) and sodium dihydrogen phosphate buffer (0.3 M; 7.5 mL) was stirred at room temperature overnight. Work-up as described above gave the title compound (42 mg, 79%) as a dark orange solid, mp 218 °C; (Found: C, 53.9; H, 2.8. C10H6O4S requires C, 54.0; H, 2.7%); νmax (KBr)/cm−1 3070, 1688, 1649, 1600, 1522, 1325, 1249, 1148, 1079, 867; δH (300 MHz; CDCl3) 10.03 (1 H, s, CHO), 8.16 (1 H, s, 3-H), 6.12 (1 H, s, 6-H), 3.92 (3 H, s, OMe); δC (75 MHz; CDCl3) 183.0 (CH), 179.4 (C), 174.9 (C), 161.0 (C), 149.6 (C), 147.6 (C), 138.7 (C), 132.6 (CH) 109.2 (CH), 57.1 (Me); m/z (EI) 222 (M+, 100%), 207 (32), 192 (37).
2-Acetoxymethyl-5-methoxybenzothiophene-4,7-dione 32
Acetic anhydride (2.7 mL) was added to a solution of the alcohol 29 (75 mg, 0.33 mmol) in pyridine (16 mL), and the mixture was stirred overnight at room temperature. The mixture was diluted with water (27 mL), and extracted with dichloromethane (3 × 50 mL). The combined extracts were washed with water (50 mL) and brine (50 mL), dried (MgSO4) and evaporated. The residue was purified by chromatography eluting with dichloromethane/ethyl acetate (19:1) to give the title compound as a yellow solid (58 mg, 66%), mp 153–155 °C; (Found: C, 54.0; H, 3.7. C12H10O5S requires C, 54.1; H, 3.8%); νmax (KBr)/cm−1 3091, 2983, 2963, 1725, 1677, 1642, 1602, 1534, 1467, 1438, 1333, 1232, 1141, 1085, 1030, 962, 854, 798; δH (300 MHz; CDCl3) 7.49 (1 H, s, 3-H), 5.99 (1 H, s, 6-H), 5.26 (2 H, s, CH2), 3.88 (3 H, s, OMe), 2.13 (3 H, s, Me); δC (75 MHz; CDCl3) 179.7 (C), 175.6 (C), 170.3 (C), 160.3 (C), 146.0 (C), 144.6 (C), 138.7 (C), 125.5 (CH), 108.5 (CH), 60.2 (CH2), 56.8 (Me), 20.7 (Me); m/z (EI) 266 (M+, 32%), 251 (8), 224 (100), 207 (36), 178 (20).
5-Methoxy-2-methylbenzothiophene-4,7-dione 33
(a) A stirred solution of the alcohol 29 (200 mg, 0.89 mmol) in dichloromethane (11 mL) at −10 °C was treated with triethylamine (140 mg, 0.19 mL, 1.34 mmol). After 10 min, methanesulfonyl chloride (110 mg, 0.08 mL, 0.98 mmol) was added, and the mixture allowed to stir at room temperature overnight. Water (11 mL) and dichloromethane (30 mL) were added, the organic layer separated, washed with water (50 mL), brine (50 mL), dried (MgSO4) and evaporated to give the mesylate as a yellow solid, used without any purification.
(b) The above product was dissolved in THF (11 mL) and added to lithium aluminum hydride (330 mg, 8.63 mmol) in THF (9 mL). The mixture was stirred overnight at room temperature, before the careful addition of water. The mixture was filtered through Celite, and the filtrate extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with saturated aqueous sodium hydrogen carbonate (10 mL), water (10 mL), dried (MgSO4) and evaporated. The residue was purified by chromatography to give the title compound (9 mg, 5%) as a yellow solid, mp 217–220 °C; (Found: M+, 208.0194. C10H8O3S requires 208.0194); νmax (KBr)/cm−1 3066, 2923, 1680, 1640, 1596, 1535, 1468, 1329, 1246, 1083, 968, 858, 792; δH (300 MHz; CDCl3) 7.22 (1 H, s, 3-H), 5.94 (1 H, s, 6-H), 3.86 (3 H, s, OMe), 2.56 (3 H, s, Me); δC (75 MHz; CDC13) 179.9 (C), 175.9 (C), 160.0 (C), 148.5 (C), 142.4 (C), 139.4 (C), 124.1 (CH), 108.3 (CH), 56.7 (Me), 15.9 (Me); m/z (EI) 208 (M+, 64%), 193 (21), 178 (30), 149 (47), 71 (44), 69 (100).
3.4. Synthesis of Indazolequinones
5,7-Dimethoxy-3-methylindazole 35
To a solution of 3,5-dimethoxyacetophenone 34 (1.58 g, 8.81 mmol) in dry dichloromethane (44 mL) were added bis(trichloroethyl) azodicarboxylate (3.35 g, 8.81 mmol) and BF3•EtO (539 µl, 4.40 mmol). The mixture was stirred overnight at room temperature, quenched with aqueous ammonium acetate solution (25%; 70 mL) and extracted with ethyl acetate (4 × 70 mL). The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. The crude product obtained was purified by flash chromatography. A mixture of hydrazine intermediate and starting material (20:1) was obtained. To a solution of the above product in glacial acetic acid (49 mL) was added zinc dust (4.94 g, 75.55 mmol). The mixture was stirred at room temperature for 1 h and water (50 mL) followed by aqueous sodium hydroxide (1 M) were added until pH = 10. The mixture was extracted with ethyl acetate. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography, eluting with ethyl acetate-light petroleum 1:1 to yield the title compound (590 mg; 35%) as a beige solid; mp 154–156 °C (lit.,38 mp 155–156 °C); δH (300 MHz; CDCl3) 11.72 (1 H, br s, NH), 6.64 (1 H, d, J 2.3, ArH), 6.44 (1 H, t, J 2.3, ArH), 3.95 (3 H, s, OMe), 3.80 (3 H, s, OMe), 2.45 (3 H, s, Me).
5,7-Dimethoxy-3-methyl-4-nitroindazole 36
To a solution of 5,7-dimethoxy-3-methylindazole 35 (200 mg, 1.04 mmol) in acetic acid (8 mL), cooled to 0–5 °C, was added a mixture of nitric acid (69 µl, 1.04 mmol) in acetic acid (1 mL). The mixture was stirred at room temperature for 1 h. The reaction was quenched by addition of brine, extracted with ethyl acetate, dried over MgSO4, filtered, evaporated and azeotroped with toluene to remove the acetic acid. The crude product obtained was purified by flash chromatography, eluting with ethyl acetate/light petroleum (1:1), to yield the title compound (201 mg, 81%) as a bright yellow crystalline solid, recrystallized from dichloromethane-pentane; mp 219–220 °C; (Found: C, 50.7; H, 4.5; N, 17.6. C10H11N3O4 requires C, 50.6; H, 4.7; N, 17.7%); (Found: MH+, 238.0829. C10H11N3O4 + H requires 238.0828); νmax (KBr)/cm−1 3398, 3149, 2917, 1597, 1517, 1308, 1223, 1115, 1059; δH (300 MHz; CDCl3) 13.43 (1 H, s, NH), 6.53 (1 H, s, ArH), 4.05 (3 H, s, OMe), 4.00 (3 H, s, OMe), 2.50 (3 H, s, Me); δC (100 MHz; d6-DMSO) 148.9 (C), 148.2 (C), 138.6 (C), 129.0 (C), 124.3 (C), 114.7 (C), 94.8 (CH), 58.3 (Me), 56.6 (Me), 13.2 (Me); m/z (CI) 238 (MH+, 95%), 221 (55), 207 (100), 192 (80).
5,7-Dimethoxy-1,3-dimethyl-4-nitroindazole 37
To a stirred solution of 5,7-dimethoxy-3-methyl-4-nitroindazole 36 (116 mg, 0.60 mmol) in DMSO (2 mL) was added potassium hydroxide (135 mg, 2.42 mmol). The mixture was stirred at room temperature for 30 min and iodomethane (150 µl, 2.42 mmol) was added dropwise to the solution. The reaction mixture was then stirred at room temperature for a further 4 h. The crude mixture was diluted with ethyl acetate, washed thoroughly with hydrochloric acid (2 M), dried over MgSO4, filtered and evaporated. The crude product obtained was purified by flash chromatography, eluting with ethyl acetate/light petroleum (1:1), to yield the title compound (71 mg, 49%) as a bright yellow crystalline solid, recrystallized from dichloromethane-pentane; mp 168–169 °C; (Found: C, 52.8; H, 5.1; N, 16.9. C11H13N3O4 requires C, 52.6; H, 5.2; N, 16.7%); (Found: M+, 251.0910. C11H13N3O4 requires 251.0906); νmax (KBr)/cm−1 3435, 2929, 2841, 1577, 1522, 1324, 1280, 1220, 1050; δH (300 MHz; CDCl3) 6.45 (1 H, s, ArH), 4.19 (3 H, s, NMe), 4.02 (3 H, s, OMe), 3.97 (3 H, s, OMe), 2.40 (3 H, s, Me); δC (75 MHz; CDCl3) 149.0 (C), 148.6 (C), 138.9 (C), 128.9 (C), 126.3 (C), 117.1 (C), 94.6 (CH), 58.9 (Me), 56.4 (Me), 39.0 (Me), 13.4 (Me); m/z (EI) 251 (M+, 100%), 234 (28), 221 (29), 206 (72), 175 (40). No nOe enhancement observed after pre-irradiation of the Me group at 2.39 ppm. 4.9% nOe enhancement observed on the aromatic proton at 6.45 ppm after pre-irradiation at 4.02 ppm (5-OMe). 2.3% nOe enhancement observed at 3.97 ppm (7-OMe) after pre-irradiation of NMe at 4.19 ppm. 3.5% nOe enhancements observed on the aromatic proton at 6.45 ppm and 1.3% nOe enhancement observed at 4.02 ppm (5-OMe) after pre-irradiation at 3.97 ppm (7-OMe).
4-Amino-5,7-dimethoxy-1,3-dimethylindazole 38
To a suspension of 5,7-dimethoxy-1,3-dimethyl-4-nitroindazole 37 (55.3 mg, 0.22 mmol) in ethanol (3.7 mL) were added tin powder (120 mg, 0.99 mmol) and hydrochloric acid (3 M; 1.5 mL). The mixture was heated under reflux for 1 h. Upon cooling, the solution was decanted from the excess tin and neutralized with a saturated aqueous solution of sodium hydrogen carbonate. The precipitate obtained was extracted with ethyl acetate. The organic layer was filtered through a pad of Celite, dried over MgSO4, filtered and evaporated under reduced pressure to yield the title compound (47 mg; 96%) as a colorless solid; mp 79–80 °C; (Found: M+, 221.1163. C11H15N3O2 requires 221.1164); νmax (KBr)/cm−1 3434, 3343, 2927, 2836, 1590, 1527, 1453, 1351, 1271, 1197, 1111; δH (300 MHz; CDCl3) 6.48 (1 H, s, 6-H), 4.11 (3 H, s, NMe), 3.84 (3 H, s, OMe), 3.82 (3 H, s, OMe), 2.66 (3 H, s, Me); δC (75 MHz; CDCl3) 139.9 (C), 138.4 (C), 138.3 (C), 130.5 (C), 123.2 (C), 116.3 (C), 99.3 (CH), 59.2 (Me), 56.4 (Me), 38.3 (Me), 14.5 (Me); m/z (EI) 221 (M+, 27%), 206 (100), 191 (14), 163 (8).
5-Methoxy-1,3-dimethylindazole-4,7-dione 39
To a solution of 4-amino-5,7-dimethoxy-1,3-dimethylindazole 38 (0.250 g, 1.15 mmol) in acetone (70 mL) was added a solution of potassium nitrosodisulfonate (1.250 g, 4.59 mmol) in sodium dihydrogen phosphate buffer (0.3 M, 58 mL). The reaction was stirred at room temperature for 1h. The excess acetone was removed in vacuo. The resulting residue was extracted with dichloromethane. The organic layer was washed with water, dried over MgSO4, filtered, evaporated under reduced pressure to yield the title compound (208 mg, 78%) as a bright yellow crystalline solid, recrystallized from dichloromethane-pentane; mp 185–186 °C; (Found: C, 58.2; H, 4.8; N, 13.6. C10H10N2O3 requires C, 58.2; H, 4.9; N, 13.6%); (Found: M+, 206.0683. C10H10N2O3 requires 206.0691); λmax (acetonitrile)/nm 272 (log ε 3.91), 320 (3.84); νmax (KBr)/cm−1 1680, 1657, 1590, 1529, 1510, 1340, 1216, 1018; δH (300 MHz; CDCl3) 5.79 (1 H, s, 6-H), 4.16 (3 H, s, NMe), 3.85 (3 H, s, OMe), 2.49 (3 H, s, Me); δC (75 MHz; CDCl3) 177.8 (C), 176.1 (C), 162.0 (C), 149.0 (C), 137.4 (C), 117.1 (C), 107.3 (CH), 57.0 (Me), 38.4 (Me), 12.9 (Me); m/z (EI) 206 (M+, 100%), 191 (85), 177 (62), 123 (30).
3-Hydroxymethyl-5-methoxy-1-methylindazole-4,7-dione 40
A solution of 5-methoxy-1,3-dimethylindazole-4,7-dione 39 (51 mg, 0.25 mmol), AIBN (12 mg, 0.07 mmol) and N-bromosuccinimide (86 mg, 0.49 mmol) in CCl4 (3 mL) was purged five times with vacuum followed with nitrogen. The reaction mixture was heated at reflux under a nitrogen atmosphere overnight. The mixture was concentrated under reduced pressure and purified by flash chromatography, eluting with dichloromethane, to yield the bromide intermediate, which was used in the next step with no further purification. The bromide intermediate dissolved in acetone (9 mL) and water (4 mL) was added to a suspension of silver nitrate (92 mg, 0.54 mmol) in an aqueous solution of acetone (50%; 13 mL). The reaction mixture was heated under reflux overnight and extracted with ethyl acetate. The combined organic layer was dried over MgSO4, filtered and evaporated under reduced pressure. The residue obtained was purified by flash chromatography, eluting with light petroleum-ethyl acetate 1:2, to yield the title compound (29 mg; 36%) as a yellow solid; mp 177–179 °C; (Found: MH+, 223.0721. C10H10N2O4 + H requires 223.0719); λmax (acetonitrile)/nm 276 (log ε 4.23), 308 (3.74); νmax (KBr)/cm−1 3368, 2928, 1689, 1653, 1593, 1525, 1505, 1240, 1204, 1015; δH (300 MHz; CDCl3) 5.87 (1 H, s, 6-H), 4.86 (2 H, s, CH2OH), 4.17 (3 H, s, NMe), 3.90 (3 H, s, OMe); δC (75 MHz; CDCl3) 177.5 (C), 177.0 (C), 161.8 (C), 152.6 (C), 137.8 (C), 117.4 (C), 107.7 (CH), 58.2 (CH2), 57.2 (Me), 38.7 (Me); m/z (ES) 223 (MH+, 95%), 164 (35), 149 (30), 90 (100).
3.5. Synthesis of Benzisoxazolequinones
(E)-2-Hydroxy-5-methoxyacetophenone oxime 42
To a solution of 2-hydroxy-5-methoxyacetophenone 41 (2.50 g, 15.1 mmol) and hydroxylamine hydrochloride (2.10 g, 30.1 mmol) in ethanol (50 mL) was added dropwise dry pyridine (2.68 mL, 30.1 mmol). The reaction mixture was stirred at room temperature overnight and evaporated under reduced pressure. The crude product obtained was purified by flash chromatography, eluting with ethyl acetate-light petroleum 1:2, to yield the title compound (1.68 g, 98%) as a colorless solid; mp 112–113 °C (lit.,42 mp 121 °C); (Found: MH+, 182.0813. C9H11NO3 + H requires 182.0817); νmax (KBr)/cm−1 3348, 2964, 2920, 2831, 1641, 1497, 1404, 1368, 1284, 1232, 1208, 1176, 1052, 1011; δH (300 MHz; CDCl3) 10.70 (1 H, br s, OH), 7.53 (1 H, br s, OH), 7.17 (1 H, d, J 3.0, 6-H), 7.11 (1 H, dd, J 9.0, 3.0, 4-H), 6.93 (1 H, d, J 9.0, 3-H), 3.80 (3 H, s, OMe), 2.62 (3 H, s, Me); δC (100 MHz; CDCl3) 159.3 (C), 152.3 (C), 151.6 (C), 118.7 (C), 117.7 (CH), 116.5 (CH), 113.1 (CH), 56.0 (Me), 10.9 (Me); m/z (CI) 182 (MH+, 63%), 166 (100), 151 (15), 125 (5).
5-Methoxy-3-methylbenzisoxazole 43
Diisopropyl azodicarboxylate (8.5 mL, 43.5 mmol) was added dropwise to a stirred solution of the oxime 42 (5.24 g, 29.0 mmol) and triphenylphosphine (11.35 g, 43.5 mmol) in dry THF (192 mL) under nitrogen. The reaction mixture was stirred at room temperature overnight, and evaporated under reduced pressure. The crude product was purified by chromatography, eluting with ethyl acetate/light petroleum (1:3), to yield the title compound (4.72 g, 100%) as a yellow solid; mp 28–30 °C (lit.,43 mp not given); (Found: MH+, 164.0708. C9H9NO2 + H requires 164.0712); νmax (KBr)/cm−1 2972, 2935, 1730, 1528, 1484, 1458, 1439, 1252, 1219, 1127, 1079, 1028; δH (300 MHz; CDCl3) 7.44 (1 H, d, J 9.0, 7-H), 7.16 (1 H, dd, J 9.0, 2.3, 6-H), 6.96 (1 H, d, J 2.3, 4-H), 3.87 (3 H, s, OMe), 2.56 (3 H, s, Me); δC (75 MHz; CDCl3) 158.4 (C), 156.2 (C), 154.9 (C), 122.5 (C), 120.2 (CH), 110.5 (CH), 101.3 (CH), 55.9 (Me), 10.1 (Me); m/z (CI) 164 (MH+, 100%), 148 (5).
5-Methoxy-3-methyl-4-nitrobenzisoxazole 44
To a solution of nitric acid/sulfuric acid 9:1 (22 mL), cooled in a salt and ice bath, was added 5-methoxy-3-methylbenzisoxazole 43 (2.22 g, 13.6 mmol) portionwise. The mixture was stirred at room temperature overnight. The mixture was poured into ice/water, basified with a saturated aqueous sodium hydrogen carbonate, extracted with dichloromethane, dried over MgSO4, and evaporated under reduced pressure to yield the title compound (2.58 g, 91%) as a yellow solid; mp 103–105 °C; (Found: MH+, 209.0551. C9H8N2O4 + H requires 209.0562); νmax (KBr)/cm−1 3092, 2954, 2850, 1525, 1513, 1475, 1371, 1325, 1267, 1060; δH (300 MHz; CDCl3) 7.69 (1 H, d, J 9.2, ArH), 7.36 (1 H, d, J 9.2, ArH), 4.00 (3 H, s, OMe), 2.50 (3 H, s, Me); δC (75 MHz; CDCl3) 158.2 (C), 153.0 (C), 149.1 (C), 131.7 (C), 116.9 (CH), 115.3 (C), 113.6 (CH), 58.1 (Me), 10.9 (Me); m/z (CI) 209 (MH+, 100%), 192 (5).
5-Methoxy-3-methylbenzisoxazole-4,7-dione 45
To a suspension of 5-methoxy-3-methyl-4-nitrobenzisoxazole 44 (2.5 g, 12.0 mmol) in ethanol (200 mL) was added tin powder (3.6 g, 120.2 mmol) and hydrochloric acid (3 M; 83 mL). The mixture was stirred and heated under reflux for 1 h. Upon cooling, the reaction mixture was decanted from the excess of tin and neutralized with a saturated sodium hydrogen carbonate. The suspension obtained was added to an equal volume of water. The precipitate and aqueous layer were stirred overnight with dichloromethane, filtered through Celite and the layers separated. The organic layer was dried over Na2SO4 and concentrated to yield the 4-amino compound, used in the next step with no further purification.
To a solution of the 4-amino compound in acetone (490 mL) was added a solution of potassium nitrosodisulfonate (8.9 g, 33.2 mmol) in sodium dihydrogen phosphate buffer (0.3 M; 400 mL). The reaction was stirred at room temperature for 1 h, and concentrated under reduced pressure. The residue was extracted with dichloromethane. The organic layer was washed with water, dried over MgSO4, filtered, evaporated under reduced pressure to yield the title compound (250 mg, 20%) as a dark orange crystalline solid; mp 126–129 °C; (Found: MH+, 194.0451. C9H7NO4 + H requires 194.0453); λmax (acetonitrile)/nm 220 (log ε 3.92), 256 (3.89), 380 (3.30); νmax (KBr)/cm−1 3437, 3060, 2917, 1698, 1667, 1578, 1486, 1467, 1440, 1355, 1336, 1251, 1205, 1186, 1032, 1013; δH (300 MHz; CDCl3) 5.98 (1 H, s, 6-H), 3.93 (3 H, s, OMe), 2.59 (3 H, s, Me); δC (75 MHz; CDCl3) 176.0 (C), 174.5 (C), 165.2 (C), 161.8 (C), 158.1 (C), 117.7 (C), 107.1 (CH), 57.9 (Me), 11.0 (Me); m/z (CI) 194 (MH+, 100%), 166 (5).
3.6. Biology
HPLC analysis
Reduction of the quinones was followed by HPLC using an Alltech C18 (5 µm, 250 mm × 4.6 mm) column with a Waters HPLC system (2487 Dual λ Absorbance detector, two 515 HPLC pumps, 717plus Autosampler, Millennium32 Chromatography Manager). The solvent program used a linear gradient of 5% to 80% B over 10 min, 80% B for 5 min, then 80% B to 5% B over 5 min (solution A, 10 mM potassium phosphate buffer, pH 6.0; solution B, methanol). Reactions were run in 25 mM Tris-HCl (pH 7.4) containing 200 µM NADH (Sigma), 50 µM quinone, and recombinant human NQO1 (gift from David Ross, University of Colorado, Denver, CO). NADH oxidation was quantified at 340 nm following 30–40 min incubations at 22 °C.
Spectrophotometric method
Quinone reduction by recombinant human NQO1 was also quantified using a modification of an assay that uses cytochrome c as the terminal electron acceptor.44 Reaction mixtures contained 1 mM NADH (Sigma), 25 µM quinone, 70 µM cytochrome c (Sigma) and 0.1–3.0 µg/ml rhNQO1 in 25 mM Tris-HCl (pH 7.4) with 0.07% BSA and 0.1% Tween-20. Reactions were run at least in triplicate at 22 °C in a Beckman DU 7500 spectrophotometer at 550 nm (molar absorptivity 21.1 mM−1 cm−1 for cytochrome c). Initial reduction rates (µmol cytochrome c reduced/min/mg NQO1) were calculated from the linear portion (0–30 s) of the reaction curves.
Acknowledgments
This work was supported by the FORCE Cancer Charity, the Cancer Research Campaign (as was) [CRUK], and NIH Grant P20RR017670 (H.D.B.). We also thank the EPSRC Mass Spectrometry Centre at Swansea for mass spectra, Dr Stephen Green for help with the electrochemical experiments, and Professor David Ross (University of Colorado, Denver) for generous gifts of enzyme.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Thomson RH. Naturally Occurring Quinones IV. Recent advances. 4th ed. London: Blackie; 1997. [Google Scholar]
- 2.Tisler M. Adv. Heterocycl. Chem. 1989;45:37–150. [Google Scholar]
- 3.Boutros R, Lobjois V, Ducommun B. Nat. Rev. Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 4.Carter SK, Crooke ST. Mitomycin C; Current Status and New Developments. New York: Academic Press; 1979. [Google Scholar]
- 5.Remers WA, Dorr RT. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. New York: Wiley; 1988. pp. 1–74. [Google Scholar]
- 6.Franck RW, Tomasz M. In. In: Chemistry of Antitumor Agents. Wilman DEV, editor. Glasgow: Blackie and Son Ltd.; 1990. pp. 379–393. [Google Scholar]
- 7.Danishefsky SJ, Schkeryantz JM. Synlett. 1995:475–490. [Google Scholar]
- 8.Bringmann G, Reichert Y, Kane VV. Tetrahedron. 2004;60:3539–3574. [Google Scholar]
- 9.Ernster L. Chem. Scripta. 1987;27A:1–13. [Google Scholar]
- 10.Ross D, Siegel D, Beall H, Prakash AS, Mulcahy RT, Gibson NW. Cancer Metast. Rev. 1993;12:83–101. doi: 10.1007/BF00689803. [DOI] [PubMed] [Google Scholar]
- 11.Beall HD, Winski SL. Frontiers in Bioscience. 2000;5:D639–D648. doi: 10.2741/beall. [DOI] [PubMed] [Google Scholar]
- 12.Colucci MA, Couch GD, Moody CJ. Org. Biomol. Chem. 2008;6:637–656. doi: 10.1039/b715270a. [DOI] [PubMed] [Google Scholar]
- 13.Beall HD, Winski S, Swann E, Hudnott AR, Cotterill AS, O'Sullivan N, Green SJ, Bien R, Siegel D, Ross D, Moody CJ. J. Med. Chem. 1998;41:4755–4766. doi: 10.1021/jm980328r. [DOI] [PubMed] [Google Scholar]
- 14.Newsome JJ, Swann E, Hassani M, Bray KC, Slawin AMZ, Beall HD, Moody CJ. Org. Biomol. Chem. 2007;5:1629–1640. doi: 10.1039/b703370b. [DOI] [PubMed] [Google Scholar]
- 15.Faig M, Bianchet MA, Winski S, Hargreaves R, Moody CJ, Hudnott AR, Ross D, Amzel LM. Structure. 2001;9:659–667. doi: 10.1016/s0969-2126(01)00636-0. [DOI] [PubMed] [Google Scholar]
- 16.Winski SL, Swann E, Hargreaves RHJ, Dehn DL, Butler J, Moody CJ, Ross D. Biochem. Pharmacol. 2001;61:1509–1516. doi: 10.1016/s0006-2952(01)00631-1. [DOI] [PubMed] [Google Scholar]
- 17.Winski SL, Faig M, Bianchet MA, Siegel D, Swann E, Fung K, Duncan MW, Moody CJ, Amzel M, Ross D. Biochemistry. 2001;40:15135–15142. doi: 10.1021/bi011324i. [DOI] [PubMed] [Google Scholar]
- 18.Fryatt T, Pettersson HI, Gardipee WT, Bray KC, Green SJ, Slawin AMZ, Beall HD, Moody CJ. Bioorg. Med. Chem. 2004;12:1667–1687. doi: 10.1016/j.bmc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 19.Newsome JJ, Colucci MA, Hassani M, Beall HD, Moody CJ. Org. Biomol. Chem. 2007;5:3665–3673. doi: 10.1039/b713044a. [DOI] [PubMed] [Google Scholar]
- 20.Esser L, Quinn B, Li YF, Zhang MQ, Elberry M, Yu L, Yu CA, Xia D. J. Mol. Biol. 2004;341:281–302. doi: 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 21.Skibo EB, Islam I, Schulz WG, Zhou R, Bess L, Boruah R. Synlett. 1996:297–309. [Google Scholar]
- 22.Flader C, Liu JW, Borch RF. J. Med. Chem. 2000;43:3157–3167. doi: 10.1021/jm000179o. [DOI] [PubMed] [Google Scholar]
- 23.Garuti L, Roberti M, Malagoli M, Rossi T, Castelli M. Bioorg. Med. Chem. Lett. 2000;10:2193–2195. doi: 10.1016/s0960-894x(00)00429-7. [DOI] [PubMed] [Google Scholar]
- 24.Lynch M, Hehir S, Kavanagh P, Leech D, O'Shaughnessy J, Carty MP, Aldabbagh F. Chem. Eur. J. 2007;13:3218–3226. doi: 10.1002/chem.200601450. [DOI] [PubMed] [Google Scholar]
- 25.Gellis A, Kovacic H, Boufatah N, Vanelle P. Eur. J. Med. Chem. 2008;43:1858–1864. doi: 10.1016/j.ejmech.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 26.Bonham S, O'Donovan L, Carty MP, Aldabbagh F. Org. Biomol. Chem. 2011;9:6700–6706. doi: 10.1039/c1ob05694h. [DOI] [PubMed] [Google Scholar]
- 27.Fagan V, Bonham S, Carty MP, Saenz-Mendez P, Eriksson LA, Aldabbagh F. Bioorg. Med. Chem. 2012;20:3223–3232. doi: 10.1016/j.bmc.2012.03.063. [DOI] [PubMed] [Google Scholar]
- 28.Scannell RT, Stevenson R. J. Org. Chem. 1983;48:127–129. [Google Scholar]
- 29.De Rosa M, De Rosa S, Gambacorta A, Minale L, Thomson RH, Worthington RD. J. Chem. Soc. Perkin Trans. 1. 1977:653–657. doi: 10.1039/p19770000653. [DOI] [PubMed] [Google Scholar]
- 30.Li ZR, Shokes JE, Kounosu A, Imai T, Iwasaki T, Scott RA. Biochemistry. 2003;42:15003–15008. doi: 10.1021/bi035078h. [DOI] [PubMed] [Google Scholar]
- 31.Sucrow W, Brockmann R, Haupt HJ, Preut H. Liebigs Ann. Chem. 1984:1711–1718. [Google Scholar]
- 32.Berhe S, Slupe A, Luster C, Charlier HA, Jr, Warner DL, Zalkow LH, Burgess EM, Enwerem NM, Bakare O. Bioorg. Med. Chem. 2010;18:134–141. doi: 10.1016/j.bmc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Struebe F, Rath S, Mattay J. Eur. J. Org. Chem. 2011:4645–4653. [Google Scholar]
- 34.Honey MA, Blake AJ, Campbell IB, Judkins BD, Moody CJ. Tetrahedron. 2009;65:8995–9001. [Google Scholar]
- 35.Frigoli M, Mehl GH. Chem. Eur. J. 2004;10:5243–5250. doi: 10.1002/chem.200305682. [DOI] [PubMed] [Google Scholar]
- 36.Deng H, Fang Y. ACS Med. Chem. Lett. 2012;3:550–554. doi: 10.1021/ml300076u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson DR, Meyers MJ, Kurumbail RG, Caspers N, Poda GI, Long SA, Pierce BS, Mahoney MW, Mourey RJ. Bioorg. Med. Chem. Lett. 2009;19:4878–4881. doi: 10.1016/j.bmcl.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell H, Leblanc Y. J. Org. Chem. 1994;59:682–687. [Google Scholar]
- 39.Swann E, Barraja P, Oberlander AM, Gardipee WT, Hudnott AR, Beall HD, Moody CJ. J. Med. Chem. 2001;44:3311–3319. doi: 10.1021/jm010884c. [DOI] [PubMed] [Google Scholar]
- 40.Song Y, Buettner GR. Free Radical Biol. Med. 2010;49:919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernatek E. Acta Chem. Scand. 1953;7:677–681. [Google Scholar]
- 42.Lindemann H, Pickert W. Liebigs Ann. Chem. 1927;456:275–311. [Google Scholar]
- 43.Udd S, Jokela R, Franzen R, Tois J. Tetrahedron Lett. 2010;51:1030–1033. [Google Scholar]
- 44.Phillips RM, Naylor MA, Jaffar M, Doughty SW, Everett SA, Breen AG, Choudry GA, Stratford IJ. J. Med. Chem. 1999;42:4071–4080. doi: 10.1021/jm991063z. [DOI] [PubMed] [Google Scholar]