

Abstract
The development and relapse of many psychopathologies can be linked to both stress and prefrontal cortex dysfunction. Glucocorticoid stress hormones target medial prefrontal cortex (mPFC) and either chronic stress or chronic administration of glucocorticoids produces dendritic remodeling in prefrontal pyramidal neurons. Exposure to stress also causes an increase in the release of the excitatory amino acid glutamate, which binds to N-methyl-D-aspartate (NMDA) receptors, which are plentiful in mPFC. NMDA receptor activation is crucial for producing hippocampal dendritic remodeling due to stress and for dendritic reorganization in frontal cortex after cholinergic deafferentation. Thus, NMDA receptors could mediate stress-induced dendritic retraction in mPFC. To test this hypothesis, dendritic morphology of pyramidal cells in mPFC was assessed after blocking NMDA receptors with the competitive NMDA antagonist ±3-(2-carboxypiperazin-4yl)propyl-1-phosphonic acid (CPP) during restraint stress. Administration of CPP prevented stress-induced dendritic atrophy. Instead, CPP-injected stressed rats showed hypertrophy of apical dendrites compared with controls. These results suggest that NMDA activation is crucial for stress-induced dendritic atrophy in mPFC. Furthermore, NMDA receptor blockade uncovers a new pattern of stress-induced dendritic changes, suggesting that other neurohormonal changes in concert with NMDA receptor activation underlie the net dendritic retraction seen after chronic stress.
Keywords: dendritic plasticity, Golgi histology, morphometry, prelimbic cortex, restraint stress
Introduction
Stress can precipitate or exacerbate many psychological disorders, most notably depression, schizophrenia, and posttraumatic stress disorder (e.g., Brown and Harris 1989; Ventura et al. 1989), and can also disrupt cognitive and emotional behavior (Holmes and Wellman 2009). Prefrontal cortex has been implicated in many stress-related disorders (Baxter et al. 1989; Drevets et al. 1992; Carter et al. 2001; Takahashi et al. 2004) and is involved in many of the cognitive processes that are influenced by chronic stress, including fear conditioning and retrieval of extinction, attentional set shifting, spatial learning and recognition, and working memory (reviewed in Quirk and Mueller 2007; Holmes and Wellman 2009; McLaughlin et al. 2009).
Chronic stress produces profound changes in the morphology of neurons in both the infralimbic and the prelimbic regions of medial prefrontal cortex (mPFC) of male rats (Cook and Wellman 2004; Izquierdo et al. 2006; Radley et al. 2006). Interestingly, dendritic morphology of mPFC appears to be exquisitely sensitive to stress: Just 1 week of brief daily restraint reduces mPFC apical dendritic branch number and length (Brown et al. 2005).
Prefrontal cortex is a target for glucocorticoids involved in the stress response (Meaney and Aitken 1985). Furthermore, exposure to stressors results in a variety of neurochemical changes in prefrontal cortex. For instance, acute stress increases glutamate (Moghaddam 1993), dopamine (Abercrombie et al. 1989), and acetylcholine release (Mark et al. 1996) in mPFC and prolongs serotonergic regulation of γ-aminobutyric acid (GABA)ergic iPSCs in prefrontal pyramidal neurons via activation of protein kinase C (Tan et al. 2004). In addition, previous exposure to chronic stress decreases baseline levels of dopamine release in mPFC (Mizoguchi et al. 2000) but sensitizes mPFC dopamine release in response to a novel stressor (Gresch et al. 1994). Thus, stress-induced changes in mPFC may contribute to stress-induced alterations in behaviors mediated by it. Given the well-documented link between prefrontal dysfunction and schizophrenia (Black et al. 2004), depression (Brody et al. 2001), and posttraumatic stress disorder (Liberzon et al. 1999; Rauch et al. 2003; Bremner et al. 2005; Shin et al. 2005), understanding the mechanisms underlying stress-induced alterations in prefrontal cortex may elucidate neurobiological mechanisms of psychopathology.
Given that stress-induced elevations in the glucocorticoid corticosterone mediate the dendritic retraction seen in pyramidal neurons in hippocampal area CA3 (Magariños and McEwen 1995) and mPFC is a target for glucocorticoids (Meaney and Aitken 1985), corticosterone may mediate the stress-induced dendritic retraction seen in mPFC. Indeed, systemic administration of the glucocorticoid receptor blocker RU38486 prevents the apical dendritic retraction resulting from 10 days of daily restraint (Liu and Aghajanian 2008).
However, glucocortocoid receptors may not be the sole mediators of stress-induced dendritic retraction. For instance, glutamate release in mPFC is increased during acute stress (Moghaddam 1993). Furthermore, corticosterone modulates NMDA receptor–mediated Ca2+ influx in cultured hippocampal neurons (Takahashi et al. 2002) and NMDA-dependent long-term potentiation (LTP; Shors et al. 1989), and these effects are mediated by glucocorticoid receptors (Xu et al. 1998; Yang et al. 2004). In addition, NMDA receptors play a critical role in dendritic plasticity: NMDA receptors mediate corticosterone's effects on dendritic morphology in hippocampal CA3 neurons (Magariños and McEwen 1995). Indeed, we have found that NMDA receptor activation is critical for dendritic reorganization occurring in frontal cortex after cholinergic deafferentation (Garrett et al. 2006). Thus, glutamatergic transmission at NMDA receptors may play a role in stress-induced dendritic reorganization in mPFC. Consistent with this hypothesis, chronic administration of the stress hormone corticosterone downregulates expression of the NR2B subunit of the NMDA receptor in mPFC (Gourley et al. 2008).
Given that NMDA receptor activation is crucial for producing remodeling in the hippocampus due to stress and for reorganization in frontal cortex due to cholinergic deafferentation, NMDA receptors may also play a role in stress-induced dendritic remodeling in medial prefrontal cortex. We tested this hypothesis by assessing whether blocking NMDA receptors during chronic stress prevents stress-induced dendritic retraction in mPFC.
Materials and Methods
Animals
Adult male Sprague–Dawley rats (192–233 g, approximately 52 days old at the initiation of the experiment; Harlan Sprague–Dawley) were either exposed to 1 week of daily restraint stress or left unstressed. Within each group, rats received either no injection or an injection of either CPP (10 mg/kg, intraperitoneal [i.p.]) or vehicle (saline; 1 mL/kg, i.p.). This resulted in 6 groups: Unstressed + Uninjected (n = 6), Unstressed + Vehicle (n = 8), Unstressed + CPP (n = 8), Stressed + Uninjected (n = 6), Stressed + Vehicle (n = 6), and Stressed + CPP (n = 7). All rats were weighed and injected daily. Immediately after weighing and injection, stressed rats were placed in plastic semicylindrical restrainers (6.35 cm diameter × 15.24 cm length, modified so the tail piece locks into place; Braintree Scientific) in their home cages for 3 h, a manipulation that produces significant increases in plasma corticosterone levels and dendritic retraction in mPFC (Cook and Wellman 2004; Garrett and Wellman 2009). Unstressed rats were returned to the vivarium. All rats were housed in a vivarium on a 12:12-h light/dark cycle (lights on at 6:30 AM) with an ambient temperature of 23–25 °C, with free access to food and water. All experimental procedures occurred between 9:00 AM and 6:00 PM, were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the Bloomington Institutional Animal Care and Use Committee.
Histology and Dendritic Analysis
On the last day of restraint, tissue was processed using Glaser and Van der Loos' modified Golgi stain (Glaser and Van der Loos 1981), which allows for visualization of whole neurons, including processes. Animals were overdosed with urethane and then perfused with 0.9% saline. Brains were removed and immersed in Golgi–Cox solution (a 1:1 solution of 5% potassium dichromate and 5% mercuric chloride diluted 4:10 with 5% potassium chromate). When staining was complete (12 days; determined in pilot animals by developing test sections at regular intervals and assessing the presence of dendrites trailing off into a series of dots; see Coleman and Flood 1987), brains were dehydrated and then infiltrated with a graded series of celloidins before being embedded in 8% celloidin (8% [v/v] parlodion in 1:1 absolute ethanol:ether). Coronal sections were cut at 160 μm on a sliding microtome (American Optical 860). Free-floating sections were then alkalinized in 19% ammonia, developed in D19 (Kodak), fixed in Ilford rapid fixer (diluted 1:4 with distilled water), dehydrated through a graded series of ethanols, cleared in xylene, mounted, and coverslipped (see Glaser and Van der Loos 1981)
Pyramidal neurons in layer II–III of the Cg1–3 area of mPFC were drawn (Fig. 1B). The Cg1–3 area of mPFC (anterior cingulate and prelimbic cortex) is readily identified by its position on the medial wall of rostral cortex, and its location dorsal to infralimbic cortex, which is markedly thinner than the Cg1–3 area and has fewer, less well-defined layers (Zilles and Wree 1995). Within Cg1–3, layer II–III is readily identifiable in Golgi-stained material based on its characteristic cytoarchitecture. Its position is immediately ventral to the relatively cell-poor layer I (which also contains the distal dendritic tufts of layer II–III pyramids) and immediately dorsal to layer IV; in mPFC, this boundary is pronounced because of the greater cell-packing density and smaller somata of pyramidal cells in layer II–III relative to layer IV (Cajal 1995; Zilles and Wree 1995).
Figure 1.
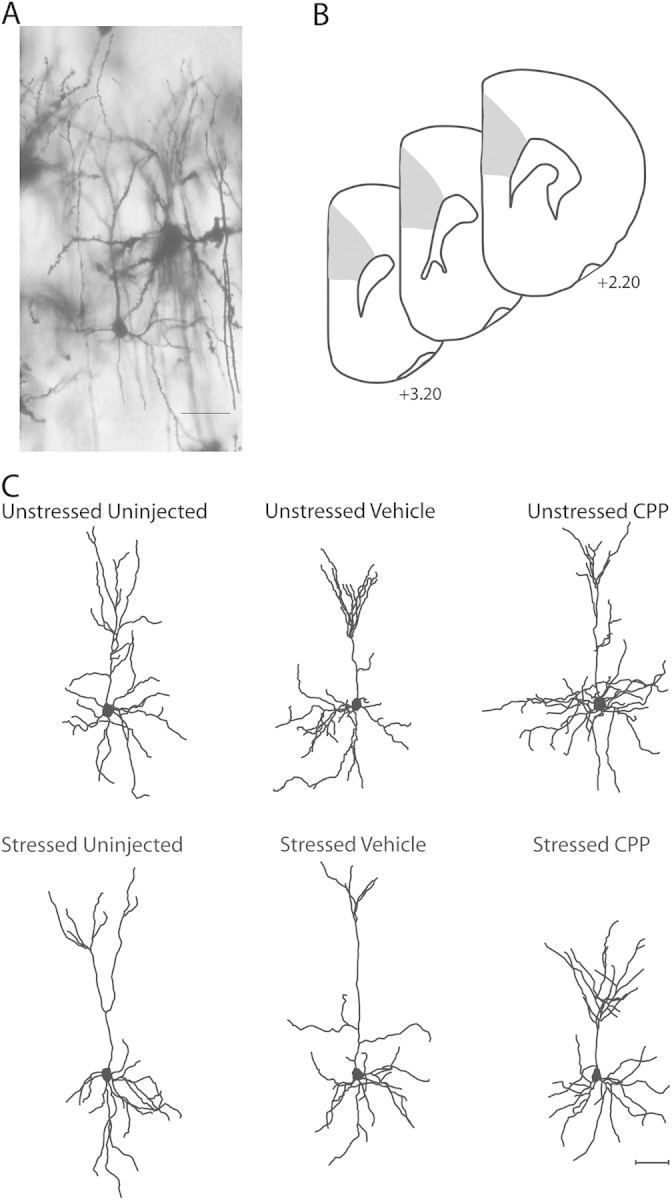
(A) Digital light micrograph of Golgi-stained neuron in layers II–III of mPFC in an unstressed vehicle-injected rat. Scale bar = 50 μm. (B) Schematic diagram of coronal section though rat prefrontal cortex. The portions of area Cg1–3 from which neurons were sampled is shown (shaded areas). Coordinates indicate positions relative to bregma (Paxinos and Watson 1998). (C) Computer-assisted reconstructions of Golgi-stained neurons in layers II–III of mPFC in unstressed (top) and stressed (bottom), uninjected (left), vehicle-injected (middle), and CPP-injected (right) rats. Neurons are representative of apical dendritic lengths near their respective group means. Scale bar = 50 μm.
Pyramidal neurons were defined by a distinct, single apical dendritic tree extending from the apex of the soma toward the pial surface of the cortex, 2 or more basilar dendritic trees extending from the base of the soma and dendritic spines (Fig. 1A). Neurons selected for reconstruction did not have truncated branches and were unobscured by neighboring neurons and glia, with dendrites that were easily discriminable by focusing through the depth of the tissue. In 3 sections evenly spaced through the rostral–caudal extent of Cg1–3, all pyramidal neurons meeting these criteria were identified. Six neurons per animal (3 from each hemisphere) were then randomly selected from all of these identified neurons and reconstructed. All neurons were drawn at a final magnification of 600× and morphology of apical and basilar arbors was quantified in 3 dimensions using a computer-based neuron tracing system (Neurolucida, MBF Biosciences) with the experimenter blind to condition.
To assess overall changes in dendritic morphology, the number and length of apical and basilar dendrites were analyzed using 2-way analysis of variance (ANOVA) (drug treatment × stress) followed by appropriate planned comparisons. In addition, the number and length of terminal branches were compared across groups using 2-way ANOVA followed by appropriate planned comparisons (drug treatment × stress). For all analyses, planned comparisons consisted of F tests done within the context of the overall ANOVA (Hays 1994), comparing either drug treatments in unstressed rats or unstressed versus stressed rats within each drug treatment group.
Results
Weight Data
It is well established that chronic stressors such as restraint or immobilization attenuate normal weight gain (e.g., Martí et al. 1994; Cook and Wellman 2004; Garrett and Wellman 2009). Consistent with this, our results show that at the end of our manipulation, average weight of the stressed rats was 13% less than that of the unstressed rats, across all drug treatment groups. Two-way ANOVA showed a significant effect of stress (F1,36 = 162.11, P ≤ 0.05), which varied across the week (stress × day interaction, F3,108 = 109.34, P ≤ 0.05). There was no significant effect of drug or interaction of stress and drug (all Fs2,36 ≤ 2.04, ns), and this did not vary across the week (all Fs6,108 ≤ 2.39, ns). Planned comparisons revealed that within each drug treatment group, weight was significantly reduced on days 3, 5, and 7 in the stressed rats (Fig. 2; for uninjected rats, all Fs1,10 ≥ 42.52, P ≤ 0.05; for vehicle and CPP-injected rats, all Fs1,13 ≥ 13.88, P ≤ 0.05).
Figure 2.

Mean weight change (weight on day X–weight on day 1) on days 1, 3, 5, and 7 of restraint stress. Unstressed rats, across all drug treatment groups, gained weight throughout the week. However, stressed rats across all drug treatment groups had significantly attenuated weight gain on days 3, 5, and 7. Vertical bars represent standard error of the mean (SEM) values. Across all appropriate stressed and unstressed pairs the difference in weight gain is significant.
Dendritic Analyses
Apical Dendrites
In all treatment groups, complete impregnation of numerous cortical pyramidal neurons was apparent and both Cg1–3 and layers II–III were readily identifiable (Fig. 1A). To rule out potential artifactual differences in morphology due to differential sampling from layers II and III, the distance from the soma to the cortical surface was measured for each neuron. Average distance to the cortical surface was then compared across groups using a 2-way ANOVA. Average distance to the cortical surface did not vary with drug treatment (F2,36 = 0.34, ns) or stress (F1,36 = 2.70, ns; drug × stress interaction, F2,36 = 0.40, ns). Thus, the neurons were sampled from equivalent laminar depths.
To assess overall changes in apical dendrites, total branch number and length were compared across groups. Two-way ANOVA revealed significant effects of drug treatment (F2,36 = 8.77, P ≤ 0.05) and stress (F1,36 = 4.13, P ≤ 0.05) on apical branch number. Furthermore, the effect of stress varied with drug treatment (drug × stress interaction, F2,36 = 12.82, P ≤ 0.05). Planned comparisons revealed that within the unstressed groups there were no significant differences across uninjected, vehicle-injected, and CPP-injected groups (uninjected vs. vehicle or CPP, Fs1,12 ≤ 0.88, ns; vehicle vs. CPP, F1,14 = 0.01, ns). Thus, NMDA receptor blockade alone did not alter apical branch number. The effect of stress, however, varied with drug treatment. In uninjected and vehicle-injected rats, stress decreased apical branch number by 26% (F1,10 = 28.60, P ≤ 0.05) and 27% (F1,13 = 7.74, P ≤ 0.05), respectively, relative to unstressed rats. However, in CPP-injected rats, stress increased apical branch number by 22% and 21% relative to unstressed CPP-injected (F1,13 = 5.64, P ≤ 0.05) and uninjected rats (F1,11 = 5.52, P ≤ 0.05), respectively (see Figs. 1C and 3A).
Figure 3.

(A) Mean apical branch length and number for unstressed and stressed uninjected, vehicle-injected, and CPP-injected rats. Overall apical branch length and number did not vary with drug treatment across unstressed groups. However, stress decreased apical branch and number and in uninjected and vehicle-injected rats and increased branch length and number in CPP-treated rats. Vertical bars represent SEM values. Asterisks (*) indicate P ≤ 0.05 relative to unstressed rats within drug treatment group; hash (#) indicates P ≤ 0.07 relative to unstressed rats within drug treatment group; and dagger (†) indicates P ≤ 0.05 relative to unstressed uninjected rats. (B) Mean terminal branch length and number for apical dendrites in unstressed and stressed uninjected, vehicle-injected, and CPP-injected rats. Terminal branch length and number did not vary across unstressed groups. However, terminal branch length and number decreased in stressed uninjected rats while increasing in stressed CPP-treated rats. Vertical bars represent SEM values. Asterisks (*) indicate P ≤ 0.05 relative to unstressed rats within drug treatment group; hash (#) indicates P ≤ 0.09 relative to unstressed rats within drug treatment group; and dagger (†) indicates P ≤ 0.05 relative to unstressed uninjected rats.
Although there was no significant overall effect of drug treatment (F2,36 = 1.94, ns) or stress (F1,36 = 0.70, ns) on apical length, there was a significant interaction of drug and stress (F2,36 = 5.92, P ≤ 0.05). As with apical branch number, planned comparisons revealed that within the unstressed groups there were no significant differences across uninjected, vehicle-injected, and CPP-injected rats (uninjected vs. vehicle or CPP, Fs1,12 ≤ 2.78, ns; vehicle vs. CPP, F1,14 = 0.03, ns). The effect of stress, however, varied across drug treatments: in uninjected rats, stress decreased apical branch length 20% relative to unstressed rats (F1,10 = 9.37, P ≤ 0.05). A comparable reduction (22%) in stressed vehicle-injected rats approached significance relative to unstressed vehicle-injected rats (F1,12 = 3.70, P ≤ 0.07) and was significant relative to unstressed uninjected rats (F1,11 = 14.60, P ≤ 0.05). In CPP-injected rats, on the other hand, there was a 22% increase in apical branch length from unstressed CPP-injected rats (F1,13 = 5.10, P ≤ 0.05), with lengths comparable with those seen in unstressed uninjected controls (F1,11 = 1.88, ns). Thus, stress produced apical dendritic proliferation rather than debranching in CPP-injected rats (Figs. 1C and 3A).
Because terminal branches may show more plasticity than other dendritic compartments, terminal branch number and length were compared as well. For apical dendrites, 2-way ANOVA revealed a significant effect of drug treatment on terminal branch number (F2,36 = 42.72, P ≤ 0.05), but no significant overall effect of stress combined across all drug groups (F1,36 = 2.77, ns). However, the effect of stress varied across drug treatment groups (F2,36 = 10.33, P ≤ 0.05). Planned comparisons revealed that within the unstressed groups there were no significant differences across uninjected, vehicle-injected, and CPP-injected groups (uninjected vs. vehicle or CPP, Fs1,12 ≤ 0.01, ns; vehicle vs. CPP, F1,14 = 0.01, ns). In both uninjected and vehicle-injected rats, stress decreased terminal branch number by 24% (F1,10 = 14.76, P ≤ 0.05) and 21% (F1,13 = 6.85, P ≤ 0.05), respectively, relative to unstressed uninjected and vehicle-injected rats. In the CPP-injected group, there was a 22% increase in apical terminal branch number relative to either unstressed CPP-injected (F1,13 = 5.96, P ≤ 0.05) or unstressed uninjected controls (F1,11 = 5.17, P ≤ 0.05; see Figs. 1C and 3B). Similarly, 2-way ANOVA revealed a significant effect of drug treatment on terminal branch length (F2,36 = 3.45, P ≤ 0.05), but no significant overall effect of stress combined across all drug groups (F1,36 = 2.76, ns). However, there was a significant interaction of drug and stress (F2,36 = 7.59, P ≤ 0.05). Within the unstressed groups there were no significant differences across uninjected, vehicle-injected, and CPP-injected groups (uninjected vs. vehicle or CPP, Fs1,12 ≤ 1.88, ns; vehicle vs. CPP, F1,14 = 0.01, ns). In uninjected rats, stress decreased terminal branch length 28% relative to unstressed rats (F1,10 = 14.96, P ≤ 0.05). Although in the vehicle-injected group, a 20% decrease in terminal branch length failed to reach significance relative to unstressed vehicle-injected rats (F1,13 = 3.28, P ≤ 0.09), the decrease in terminal branch length was significant relative to unstressed uninjected controls (F1,11 = 19.43, P ≤ 0.05). In CPP-injected rats, there was a 23% increase in terminal branch length relative to unstressed CPP-injected rats that approached significance (unstressed vs. stressed CPP, F1,13 = 4.29, P ≤ 0.06; unstressed uninjected vs. stressed CPP, F1,11 = 0.09, ns). Thus, stress decreased the length of apical terminal branches in uninjected controls, and NMDA receptor blockade prevented this effect (Figs. 1C and 3B).
Basilar Dendrites
Whereas there was no significant main effect of either drug treatment (F2,36 = 0.24, ns) or stress (F1,36 = 1.43, ns) on basilar branch number, there was a significant interaction of drug and stress (Fig. 4A; F2,36 = 4.67, P ≤ 0.05). Planned comparisons revealed that within the unstressed groups there were no significant differences between uninjected and vehicle-injected groups (F1,12 = 2.16, ns), or between uninjected and CPP-injected groups (F1,12 = 1.34, ns). However, unstressed CPP-injected rats showed a small (13%) but significant decrease in branch number relative to unstressed vehicle-injected rats (F1,14 = 8.74, P ≤ 0.05). Thus, NMDA receptor blockade decreased basilar branch number. Furthermore, while stress did not significantly alter basilar branch number in either uninjected or vehicle-injected rats (uninjected, F1,10 = 0.29, ns; vehicle injected, F1,13 = 1.77, ns), in CPP rats, stress increased basilar branch number by 19% relative to unstressed CPP-injected rats (F1,13 = 12.63, P ≤ 0.05). Basilar branch number did not significantly differ in stressed CPP-injected rats relative to unstressed uninjected controls (F1,11 = 3.73, ns; see Fig. 4A).
Figure 4.

(A) Mean basilar branch length and number for unstressed and stressed uninjected, vehicle-injected, and CPP-injected rats. Overall basilar branch length and number were decreased in unstressed CPP-treated rats, and this effect was reversed in stressed CPP-treated rats. Vertical bars represent SEM values. Daggers (†) indicate P ≤ 0.05 relative to unstressed uninjected rats; asterisks (*) indicate P ≤ 0.05 relative to unstressed rats within drug treatment group. (B) Mean terminal branch length and number for basilar dendrites in unstressed and stressed uninjected, vehicle-injected, and CPP-injected rats. Terminal basilar branch length and number decreased in unstressed CPP-treated rats; this effect was reversed in stressed CPP-treated rats. Vertical bars represent SEM values. Dagger (†) indicates P ≤ 0.05 relative to unstressed uninjected rats; double dagger (‡) indicates P ≤ 0.05 relative to unstressed vehicle-treated rats; and asterisks (*) indicate P ≤ 0.05 relative to unstressed rats within drug treatment group.
For basilar branch length, a significant effect of drug treatment (Fig. 4A; F2,36 = 3.70, P ≤ 0.05) but not stress (F1,36 = 2.66, ns) was present. Furthermore, the interactive effect of drug and stress was not significant (F2,36 = 2.39, ns). While unstressed uninjected and vehicle-injected rats did not differ significantly (F1,12 = 0.07, ns), the unstressed CPP-injected group showed decreases of 22% and 19% from unstressed uninjected and vehicle-injected groups, respectively (F1,12 = 12.85, P ≤ 0.05; F1,14 = 6.65, P ≤ 0.05, respectively). Moreover, while stress did not significantly alter basilar dendritic length in uninjected and vehicle-injected rats (uninjected, F1,10 = 0.45, ns; vehicle injected, F1,13 = 0.19, ns), in the CPP-injected group, stress increased basilar branch length 25% from unstressed CPP-injected controls (F1,13 = 18.44, P ≤ 0.05), with lengths comparable with that of unstressed uninjected controls (F1,11 = 0.15, ns). Thus, CPP treatment produced decreases in basilar branch number and length, and stress reversed this effect (Fig. 4A).
Although there was no significant main effect of drug treatment (Fig. 4B; F2,36 = 0.50, ns) or stress (F1,36 = 2.67, ns) on number of terminal branches on basilar dendrites, there was a significant interaction of drug and stress (F2,36 = 3.44, P ≤ 0.05). Unstressed CPP-injected rats showed a 12% decrease in terminal number from unstressed vehicle-injected rats (F1,14 = 8.96, P ≤ 0.05). Within unstressed groups, no other significant drug effects were present (uninjected vs. vehicle, F1,12 = 2.70, ns; uninjected vs. CPP, F1,12 = 0.86, ns). As with the overall analyses, the effect of stress varied with drug treatment. In the uninjected and vehicle groups, there was no significant difference in basilar terminal branch number in stressed relative to unstressed rats (F1,10 = 1.58, ns and F1,13 = 0.51, ns, respectively). In the CPP-injected group, stress increased basilar terminal branch number by 17% relative to unstressed CPP-injected rats (F1,13 = 13.27, P ≤ 0.05) and 11% relative to unstressed uninjected rats (F1,13 = 5.24, P ≤ 0.05; see Fig. 4B).
There was a significant effect of drug treatment (Fig. 4B; F2,36 = 3.16, P ≤ 0.05) but not stress (F1,36 = 2.69, ns) on terminal branch length. The interaction of drug and stress was not significant (F2,36 = 2.12, ns). Within the unstressed rats, whereas vehicle injections did not significantly alter terminal branch length relative to uninjected controls (F1,12 = 0.27, ns), CPP injection significantly decreased terminal branch length by 22% and 19% relative to uninjected and vehicle-injected rats, respectively (F1,12 = 12.76, P ≤ 0.05; F1,14 = 7.71, P ≤ 0.05). The effect of stress again varied across groups, with uninjected and vehicle-injected stressed rats showing no significant difference in terminal length from unstressed rats (F1,10 = 0.37, ns and F1,13 = 0.16, ns). In CPP-injected rats, there was a 26% increase in terminal branch length in stressed compared with unstressed rats (F1,13 = 14.76, P ≤ 0.05), resulting in terminal branch lengths in stressed CPP-injected rats comparable with uninjected controls. Thus, NMDA receptor blockade decreased basilar terminal branch number and length, while stress reversed this effect (see Fig. 4B).
Discussion
Many studies have documented the classic stress effect on morphology of mPFC in males as a reduction in the number and length of apical branches of pyramidal neurons (e.g., Cook and Wellman 2004; Radley et al. 2004; Brown et al. 2005; Garrett and Wellman 2009), and this effect was replicated in the present study. One week of daily restraint stress caused retraction of the apical dendritic arbor in uninjected and vehicle-injected controls, likely due at least in part to alterations in terminal branches, which showed similar patterns in response to stress. Interestingly, in vehicle-treated animals, while stress produced significant debranching, the stress-induced decrease in apical branch length was less robust than that seen in the uninjected rats. Inspection of the data (Fig. 3B) suggests that this was due to a small and nonsignificant decrease in apical branch length and increase in variability in unstressed vehicle-injected rats relative to uninjected controls. This is consistent with data indicating that even very mild stressors such as 10 min of daily restraint stress (Brown et al. 2005) or 3 weeks of daily vehicle injections (Wellman 2001) can produced apical dendritic retraction in mPFC and suggests that perhaps a subpopulation of individuals may be more susceptible to mild stress.
NMDA receptor activation during stress likely contributes to the stress-induced apical dendritic retraction, as administration of the competitive NMDA receptor antagonist CPP during restraint stress prevented the stress-induced dendritic retraction. In fact, the combination of NMDA receptor blockade and stress produced apical dendritic proliferation.
While NMDA receptor blockade alone did not significantly alter apical dendritic morphology, NMDA receptor blockade caused basilar dendritic retraction. Furthermore, this retraction was reversed by chronic stress, consistent with the hypothesis that the increased glutamatergic transmission at NMDA receptors during stress (Bagley and Moghaddam 1997; Steciuk et al. 2000; Moghaddam and Jackson 2004) counteracted the effects of daily NMDA receptor blockade. Again, this drug- and stress-induced dendritic remodeling appeared to occur mainly in terminal branches.
Blockade of NMDA receptors during restraint stress prevented stress-induced apical dendritic retraction in mPFC. This is consistent with previous literature showing that NMDA receptors are crucial for stress-induced dendritic retraction in the hippocampus (Magariños and McEwen 1995), as well as studies showing a role for NMDA receptor activation in other types of cortical plasticity (Garrett et al. 2006). The effect of NMDA receptor blockade was likely not due to decreased corticosterone release, as the CPP-treated rats showed attenuation of weight gain that is characteristic of stressed animals (Martí et al. 1994; Cook and Wellman 2004; Brown et al. 2005).
Glucocorticoid receptor blockade has also been shown to prevent stress-induced dendritic retraction in mPFC (Liu and Aghajanian 2008). It is quite possible that stress-induced increases in corticosterone and its actions at glucocorticoid receptors are responsible for the role of NMDA receptors in stress-induced dendritic retraction. For instance, in vitro, pretreatment with corticosterone prolongs NMDA receptor–mediated Ca2+ influx. Application of the NMDA antagonist MK-801 terminates this effect (Takahashi et al. 2002), indicating that corticosterone prolonged Ca2+ influx via NMDA receptors. Furthermore, acute stress (inescapable shock) impairs NMDA-dependent LTP in hippocampus, and the extent of impairment is correlated with corticosterone concentrations (Shors et al. 1989). These effects of corticosterone on NMDA receptor function are dependent on glucocorticoid receptors. Stress-induced facilitation of LTD and impairment of LTP in hippocampal area CA1 in rats is prevented by glucocorticoid receptor blockade during stress (Xu et al. 1998; Yang et al. 2004). Thus, corticosterone's action at glucocorticoid receptors can modulate NMDA receptor function.
Interestingly, NMDA receptor blockade during stress did not merely block stress-induced dendritic retraction in mPFC. Instead, in the presence of NMDA receptor blockade, stress produced proliferation of both apical and basilar branches relative to control rats. This stands in marked contrast to the effects of NMDA receptor blockade on stress-induced dendritic remodeling in the hippocampus. NMDA receptor blockade during stress prevents hippocampal dendritic retraction but does not result in dendritic proliferation (Magariños and McEwen 1995). This different pattern of results in mPFC versus hippocampus suggests that, while some aspects of the mechanisms of stress effects on dendritic morphology in both structures may be shared, other mechanisms must be divergent.
The “uncovering” of a different pattern of dendritic remodeling in mPFC suggests that other neurochemical changes occurring during stress may produce unique stress-induced alterations that are only apparent in the absence of NMDA receptor activation. Many prefrontal neurotransmitter systems and molecular signaling pathways are altered by stress, for instance, the monoaminergic neurotransmitters serotonin (Robbins 2005; Maier et al. 2006; Holmes 2008; Lapiz-Bluhm et al. 2008), dopamine (Murphy, Arnsten, Jentsch, et al. 1996; Murphy, Arnsten, Goldman-Rakic, et al. 1996; Mizoguchi et al. 2000; Pani et al. 2000; Mizoguchi et al. 2004; Pascucci et al. 2007), and norepinephrine (Pascucci et al. 2007; Ramos and Arnsten 2007), and neurotrophic factors such as brain-derived neurotrophic factor (Duman and Monteggia 2006). Each could produce unique effects on dendritic morphology, with their combination producing net apical dendritic retraction. If one individual effect is blocked during stress, then this could result in a different pattern of stress effects, rather than no change relative to controls.
For instance, increases in the extracellular concentration of monoamines can produce dendritic proliferation: Administration of a chronic sensitizing cocaine regimen increases both apical and basilar dendritic branch length in mPFC (Robinson and Kolb 1999). Thus, the prefrontal elevations in dopamine during repeated stress may produce dendritic proliferation. During stress, without drug manipulation, this potential dopamine–dependent pathway might be overshadowed by other stress-induced alterations—e.g., repeated activation of NMDA receptors—that produce dendritic atrophy. Elimination of the regressive effects of NMDA receptor activation on dendritic morphology, for instance, by blocking NMDA receptor activation as in this study, might “uncover” the dendritic proliferation induced by chronic activation of the prefrontal dopaminergic system. Future studies using pharmacological manipulations of the dopaminergic system during chronic stress could test this hypothesis.
Finally, the systemic administration used in the present study does not allow us to identify the precise site of action of NMDA receptors in mediating prefrontal dendritic retraction. Chronic stress influences several corticolimbic structures that provide input to mPFC, most notably the hippocampus (e.g., Magariños and McEwen 1995) and amygdala (e.g., Vyas et al. 2002), and stress-induced dendritic remodeling in mPFC could result from altered input from these structures. Thus, whether NMDA receptors in mPFC mediate the stress-induced dendritic retraction or whether stress-induced activation of these receptors in other structures ultimately leads to prefrontal dendritic remodeling remains to be determined.
Nonetheless, this study demonstrates that administration of the competitive NMDA receptor blocker CPP during daily restraint prevents chronic stress-induced dendritic retraction in mPFC, instead producing dendritic proliferation in response to stress. These data add to a growing body of evidence suggesting that in prefrontal cortex, the morphological consequences of stress are quite complex. For instance, as shown previously (e.g., Cook and Wellman 2004), stress produces net dendritic retraction in apical dendrites and no net change in basilar dendrites. In this study, we have further shown that NMDA receptor blockade in unstressed rats produces differential effects in basilar versus apical dendrites. Together, these data suggest that different mechanisms regulate the morphology of these 2 dendritic compartments. Furthermore, stress has different effects depending on whether or not the NMDA receptor is blocked, and even these differences are not uniform across the dendritic arbor. Indeed, others have shown differential patterns of dendritic remodeling across prefrontal subregions, with apical dendritic retraction in mPFC but proliferation in orbitofrontal cortex (Liston et al. 2006). Finally, while the present study focused on males, we have shown that stress-induced dendritic remodeling is sex dependent, with female rats showing stress-induced dendritic proliferation that is mediated at least in part by estradiol (Garrett and Wellman 2009). A subsequent study suggests further regional and circuit specificity to stress effects on prefrontal morphology in females (Shansky et al. 2010). Thus, while our data suggest that NMDA receptor activation during stress contributes to the dendritic retraction, it is likely that the mechanism underlying the effect of stress on medial prefrontal dendritic morphology is complex, with an intricate web of competing changes from activation of a variety of neurotransmitter systems, multiple circuits, and gonadal hormones producing dendritic remodeling.
Funding
Indiana METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc. and National Institutes of Mental Health (MHR03087794 to C.L.W.).
Acknowledgments
The authors thank Dr Dale R. Sengelaub and 2 anonymous reviewers for helpful comments on the manuscript. Conflict of Interest: None declared.
References
- Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Baxter LR, Schwartz JM, Phelps ME, Mazziotta JC, Guze BH, Selin CE, Gerner RH, Sumida RM. Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch Gen Psychiatry. 1989;46:243–250. doi: 10.1001/archpsyc.1989.01810030049007. [DOI] [PubMed] [Google Scholar]
- Black JE, Kodish IM, Grossman AW, Klintsova AY, Orlovskaya D, Vostrikov V, Uranova N, Greenough WT. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744. doi: 10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, Grillon C, Charney DS. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol Med. 2005;35:791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Saxena S, Mandelkern MA, Fairbanks LA, Ho ML, Baxter JLR. Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry. 2001;50:171–178. doi: 10.1016/s0006-3223(01)01117-9. [DOI] [PubMed] [Google Scholar]
- Brown GW, Harris TO. Depression. In: Brown GW, Harris TO, editors. Life events and illness. New York: Guilford; 1989. pp. 49–93. [Google Scholar]
- Brown SM, Henning S, Wellman CL. Short-term, mild stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex. 2005;15:1714–1722. doi: 10.1093/cercor/bhi048. [DOI] [PubMed] [Google Scholar]
- Cajal SRy. Histology of the nervous system. New York: Oxford University Press; 1995. [Google Scholar]
- Carter CS, MacDonald AW, 3rd, Ross LL, Stenger VA. Anterior cingulate cortex activity and impaired self-monitoring of performance in patients with schizophrenia: an event-related fMRI study. Am J Psychiatry. 2001;158:1423–1428. doi: 10.1176/appi.ajp.158.9.1423. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A functional anatomical study of unipolar depression. J Neurosci. 1992;12:3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Kim I, Wilson RE, Wellman CL. Effect of N-methyl-d-aspartate receptor blockade on plasticity of frontal cortex after cholinergic deafferentation in rat. Neuroscience. 2006;140:57–66. doi: 10.1016/j.neuroscience.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Wellman CL. Chronic stress effects on dendritic morphology in medial prefrontal cortex: sex differences and estrogen dependence. Neuroscience. 2009;162:195–207. doi: 10.1016/j.neuroscience.2009.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high-quality Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–125. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2008;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresch PJ, Sved AF, Zigmond MJ, Finlay JM. Stress-induced sensitization of dopamine and norepinephrine efflux in medial prefrontal cortex of the rat. J Neurochem. 1994;63:575–583. doi: 10.1046/j.1471-4159.1994.63020575.x. [DOI] [PubMed] [Google Scholar]
- Hays WL. Statistics. Fort Worth (TX): Harcourt Brace; 1994. [Google Scholar]
- Holmes A. Genetic variation in cortico-amygdala serotonin function and risk for stress-related disease. Neurosci Biobehav Rev. 2008;32:1293–1314. doi: 10.1016/j.neubiorev.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neurosci Biobehav Rev. 2009;33:773–783. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Wellman CL, Holmes A. Rapid dendritic retraction in medial prefrontal neurons and impaired fear extinction following exposure to uncontrollable stress. J Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapiz-Bluhm MD, Soto-Pina AE, Hensler JG, Morilak DA. Chronic intermittent cold stress and serotonin depletion induce deficits of reversal learning in an attentional set-shifting test in rats. Psychopharmacology (Berl) 2009;202:329–341. doi: 10.1007/s00213-008-1224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Amdur R, Jung TD, Chamberlain KR, Minoshima S, Koeppe RA, Fig LM. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999;45:817–826. doi: 10.1016/s0006-3223(98)00246-7. [DOI] [PubMed] [Google Scholar]
- Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R-J, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- Maier SF, Amat J, Baratta MV, Paul E, Watkins LR. Behavioral control, the medial prefrontal cortex, and resilience. Dialogues Clin Neurosci. 2006;8:397–406. doi: 10.31887/DCNS.2006.8.4/smaier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark GP, Rada PV, Shors TJ. Inescapable stress enhances extracellular acetylcholine in the rat hippocampus and prefrontal cortex but not the nucleus accumbens or amygdala. Neuroscience. 1996;74:767–774. doi: 10.1016/0306-4522(96)00211-4. [DOI] [PubMed] [Google Scholar]
- Martí O, Martí J, Armario A. Effects of chronic stress on food intake in rats: influence of stressor intensity and duration of daily exposure. Physiol Behav. 1994;55:747–753. doi: 10.1016/0031-9384(94)90055-8. [DOI] [PubMed] [Google Scholar]
- McLaughlin K, Baran S, Conrad C. Chronic stress- and sex-specific neuromorphological and functional changes in limbic structures. Mol Neurobiol. 2009;40:166–182. doi: 10.1007/s12035-009-8079-7. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH. [3H]Dexamethasone binding in rat frontal cortex. Brain Res. 1985;328:176–180. doi: 10.1016/0006-8993(85)91340-x. [DOI] [PubMed] [Google Scholar]
- Mizoguchi K, Ishige A, Takeda S, Aburada M, Tabira T. Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J Neurosci. 2004;24:5492–5499. doi: 10.1523/JNEUROSCI.0086-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui D-H, Tabira T. Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci. 2000;20:1568–1574. doi: 10.1523/JNEUROSCI.20-04-01568.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Jackson M. Effect of stress on prefrontal cortex function. Neurotox Res. 2004;6:73–78. doi: 10.1007/BF03033299. [DOI] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AF, Goldman-Rakic PS, Roth RH. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Natl Acad Sci U S A. 1996;93:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AF, Jentsch JD, Roth RH. Dopamine and spatial working memory in rats and monkeys: pharmacological reversal of stress-induced impairment. J Neurosci. 1996;16:7768–7775. doi: 10.1523/JNEUROSCI.16-23-07768.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani L, Porcella A, Gessa GL. The role of stress in the pathophysiology of the dopaminergic system. Mol Psychiatry. 2000;5:14–21. doi: 10.1038/sj.mp.4000589. [DOI] [PubMed] [Google Scholar]
- Pascucci T, Ventura R, Latagliata EC, Cabib S, Puglisi-Allegra S. The medial prefrontal cortex determines the accumbens dopamine response to stress through the opposing influences of norepinephrine and dopamine. Cereb Cortex. 2007;17:2796–2804. doi: 10.1093/cercor/bhm008. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2007;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WGM, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125:1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Ramos BP, Arnsten AF. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther. 2007;113:523–536. doi: 10.1016/j.pharmthera.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Segal E, Pitman RK, Carson MA, McMullin K, Whalen PJ, Makris N. Selectively reduced regional cortical volumes in post-traumatic stress disorder. Neuroreport. 2003;14:913–916. doi: 10.1097/01.wnr.0000071767.24455.10. [DOI] [PubMed] [Google Scholar]
- Robbins TW. Chemistry of the mind: neurochemical modulation of prefrontal cortical function. J Comp Neurol. 2005;493:140–146. doi: 10.1002/cne.20717. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb R. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Shansky RM, Hamo C, Hof PR, Lou W, McEwen BS, Morrison JH. Estrogen promotes stress sensitivity in a prefrontal cortex–amygdala pathway. Cereb Cortex. 2010;20:2560–2567. doi: 10.1093/cercor/bhq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, et al. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF. Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science. 1989;244:224–226. doi: 10.1126/science.2704997. [DOI] [PubMed] [Google Scholar]
- Steciuk M, Kram M, Kramer GL, Petty F. Immobilization-induced glutamate efflux in medial prefrontal cortex: blockade by (+)-Mk-801, a selective NMDA receptor antagonist. Stress. 2000;3:195–199. doi: 10.3109/10253890009001123. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Koeda M, Oda K, Matsuda T, Matsushima E, Matsuura M, Asai K, Okubo Y. An fMRI study of differential neural response to affective pictures in schizophrenia. Neuroimage. 2004;22:1247–1254. doi: 10.1016/j.neuroimage.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kimoto T, Tanabe N, Hattori T, Yasumatsu N, Kawato S. Corticosterone acutely prolonged N-methyl-D-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J Neurochem. 2002;83:1441–1451. doi: 10.1046/j.1471-4159.2002.01251.x. [DOI] [PubMed] [Google Scholar]
- Tan H, Zhong P, Yan Z. Corticotropin-releasing factor and acute stress prolongs serotonergic regulation of GABA transmission in prefrontal cortical pyramidal neurons. J Neurosci. 2004;24:5000–5008. doi: 10.1523/JNEUROSCI.0143-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura J, Neuchterlein KH, Lukoff D, Hardesty JD. A prospective study of stressful life events and schizophrenic relapse. J Abnorm Psychol. 1989;98:407–411. doi: 10.1037//0021-843x.98.4.407. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49:245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- Xu L, Holscher C, Anwyl R, Rowan MJ. Glucocorticoid receptor and protein/RNA synthesis-dependent mechanisms underlie the control of synaptic plasticity by stress. Proc Natl Acad Sci U S A. 1998;95:3204–3208. doi: 10.1073/pnas.95.6.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-H, Huang C-C, Hsu K-S. Behavioral stress modifies hippocampal synaptic plasticity through corticosterone-induced sustained extracellular signal-regulated kinase/mitogen-activated protein kinase activation. J Neurosci. 2004;24:11029–11034. doi: 10.1523/JNEUROSCI.3968-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilles K, Wree A. Cortex: areal and laminar structure. In: Paxinos G, editor. The rat nervous system. 2 ed. San Diego (CA): Academic Press; 1995. pp. 649–685. [Google Scholar]