

Abstract
Mycobacterium tuberculosis displays remarkable genetic stability despite continuous exposure to the hostile environment represented by the host's infected macrophages. Similarly to other organisms, M. tuberculosis possesses multiple systems to counteract the harmful potential of DNA alkylation. In particular, the suicidal enzyme O6-methylguanine-DNA methyltransferase (OGT) is responsible for the direct repair of O6-alkylguanine in double-stranded DNA and is therefore supposed to play a central role in protecting the mycobacterial genome from the risk of G·C-to-A·T transition mutations. Notably, a number of geographically widely distributed M. tuberculosis strains shows nonsynonymous single-nucleotide polymorphisms in their OGT-encoding gene, leading to amino acid substitutions at position 15 (T15S) or position 37 (R37L) of the N-terminal domain of the corresponding protein. However, the role of these mutations in M. tuberculosis pathogenesis is unknown. We describe here the in vitro characterization of M. tuberculosis OGT (MtOGT) and of two point-mutated versions of the protein mimicking the naturally occurring ones, revealing that both mutated proteins are impaired in their activity as a consequence of their lower affinity for alkylated DNA than the wild-type protein. The analysis of the crystal structures of MtOGT and MtOGT-R37L confirms the high level of structural conservation of members of this protein family and provides clues to an understanding of the molecular bases for the reduced affinity for the natural substrate displayed by mutated MtOGT. Our in vitro results could contribute to validate the inferred participation of mutated OGTs in M. tuberculosis phylogeny and biology.
INTRODUCTION
During its entire life, Mycobacterium tuberculosis is exposed to a variety of potential physical and chemical DNA-damaging stresses that could compromise the settling, containment, and reactivation of the infection (1). In order to maintain high genome stability, signaled by a remarkably low level of genetic diversity among isolates (2), it is therefore mandatory for M. tuberculosis to possess efficient systems to counteract the effects of such environmental and host-generated DNA-endangering assaults (3–6). In particular, during its long-term persistence inside infected macrophages, M. tuberculosis must deal with endogenous DNA-alkylating chemical species originated by the action of highly reactive oxidative and nitrosative radicals (7). The majority of living organisms attain alkylated-base repair by deploying different strategies, depending upon the chemical nature of the alkyl group, the entity of the damage, and the physiological condition of the cell: (i) the multistep excision of a short, lesion-containing strand followed by new DNA synthesis; (ii) the substitution of the modified base in toto; or (iii) the direct surgical removal of the alkyl-substituting group from the base by sacrificing one molecule of a DNA-protein alkyltransferase (8, 9), such as the O6-methylguanine-DNA methyltransferase (OGT) (EC 2.1.1.63). All OGT members studied until now invariably act through a suicidal mechanism (10), by performing the stoichiometric transfer of the O6-alkyl group from the modified guanine to a strictly conserved cysteine residue in the protein active site, which is hosted in the C-terminal domain of the protein. This covalent modification leaves OGT permanently inactivated and possibly more prone to degradation (11, 12) (Fig. 1).
Fig 1.

Schematic representation of transfer of O6-alkyl group from modified guanine to strictly conserved cysteine in the protein active site.
Paralleling observations of other bacteria (13), the mycobacterial OGT-encoding sequence (adaB; Rv1316c) is part of an adaptive response operon, which was recently functionally characterized (14). Gene inactivation experiments demonstrated that adaB (here referred to as OGT, in analogy to orthologs in other species) is not essential for infectivity and survival either in vitro or in the mouse model of M. tuberculosis infection (15, 16). However, several observations support the importance of OGT activity in protecting the mycobacterial GC-rich DNA from the promutagenic potential of guanine O6-methylation. First, the OGT gene expression profile changes during infection and in response to alkylating compounds (14, 17, 18), signaling a requirement for fine-tuned OGT modulation under stressful conditions; moreover, the heterologous expression of M. tuberculosis OGT (MtOGT) in the Escherichia coli KT233 ada-ogt-defective strain suppresses its MNNG (N-methyl-N′-nitro-N-nitrosoguanidine) sensitivity (14) and rescues the hypermutator phenotype.
Interestingly, two nonsynonymous single-nucleotide polymorphisms (nsSNPs) have been identified in the OGT genes of both M. tuberculosis Beijing strains and multidrug-resistant isolates (19–21), leading several authors to postulate that corresponding mutated OGTs may contribute to the success of these strains in terms of worldwide distribution; indeed, it has been suggested that a defective OGT might result in an increased mutation frequency and, thus, in a better capability of the bacterium to rapidly adapt to the host (14, 19).
Notably, both these nsSNPs result in amino acid substitutions at the poorly structurally conserved and functionally characterized N-terminal domain of OGT, thus limiting the possibility of predicting their effect on protein activity exclusively based upon preexisting information. Many years of intense investigations rendered a detailed picture of the catalytic mechanism used by these proteins as well as of the structural elements of the C-terminal domain playing essential roles in DNA binding and alkyl group removal (8, 10); in contrast, a more limited number of studies put the focus onto the N-terminal domain, which could play a role in the coordination of the catalytic cycle (22) and/or in mediating protein assembly at the site of damage upon DNA binding (23–25).
In the present work, we exploited a recently described fluorescence-based approach (26) to analyze the kinetics of the reaction performed by wild-type MtOGT and by two point-mutated variants of the protein mimicking the ones occurring in M. tuberculosis clinical isolates (i.e., MtOGT-R37L and MtOGT-T15S). The method relies on the use of a fluorescent derivative of O6-benzylguanine, an inhibitor of human O6-alkylguanine-DNA alkyltransferase (AGT), and allows the dissection of the OGT reaction in its DNA binding and alkylguanine transfer steps, thereby leading to the determination of the DNA binding affinity and the alkyltransferase reaction rate.
The results of our biochemical studies reveal that although neither mutation affects the intrinsic alkyl-guanine-transferase reaction rate, both variants display reduced DNA binding affinity although to different extents, with MtOGT-R37L showing a 10-fold-lower affinity for alkylated double-stranded DNA (dsDNA) than the wild-type protein. Moreover, the crystal structures of MtOGT and its R37L mutant reported here give a contribution to the overall description of this structurally conserved class of proteins and provide a framework to investigate the possible molecular bases of the observed DNA binding defect.
MATERIALS AND METHODS
Chemicals.
All reagents were obtained from Sigma-Aldrich unless otherwise specified.
Construction of expression vectors.
The open reading frame coding for M. tuberculosis O6-methylguanine methyltransferase (Rv1316c) was isolated by PCR using the clone MTCY130 (Institute Pasteur, Paris, France) as the template, the Hot-Star PCR system (Qiagen), and primers MtOGTfwd and MtOGTrev (see Table S1 in the supplemental material). The NcoI-BamHI double-digested PCR product was then ligated into the similarly digested pET16b vector (Novagen) by standard techniques (27), resulting in the pET-MtOGT construct. Plasmids encoding the T15S and R37L point mutants of MtOGT (pET-MtOGT-T15S and pET-MtOGT-R37L) were obtained by using the pET-MtOGT construct as the DNA template, the two primers pairs T15Sfwd/T15Srev and R37Lfwd/R37Lrev (see Table S1 in the supplemental material), and the QuikChange II site-directed mutagenesis kit (Stratagene). In each construct, the region encoding the wild-type protein or its point-mutated variant was verified by sequencing (Eurofins MWG Operon).
Expression and purification of wild-type MtOGT and point mutants.
The following procedure was invariably used to express and purify the wild-type protein and the relative point-mutated versions used in biochemical analyses. E. coli strain BL21(DE3) bacteria (Novagen), freshly transformed with one of the three expression constructs, were spread onto LB agar plates with 50 μg/ml ampicillin and grown at 37°C overnight. The next day, colonies were scraped and inoculated in 1 liter of selective ZYP-5052 medium (28) to reach a starting optical density at 600 nm (OD600) of 0.1. This culture was further grown at 37°C for 3 h and then brought to 17°C for 16 h with vigorous shaking to autoinduce the expression of the recombinant protein. A bacterial pellet was obtained by centrifugation (11,000 × g for 15 min at 4°C), washed once in phosphate-buffered saline (PBS), dissolved in 50 ml of buffer A (20 mM Tris-HCl, pH 7.8), and disrupted by ultrasonication. Upon the addition of a protease inhibitor cocktail, the insoluble material of the lysate was removed by centrifugation (14,000 × g for 45 min at 4°C), and the recombinant protein was purified from the supernatant by fast protein liquid chromatography (FPLC) (Akta Basic Instrument; GE Healthcare), using, in sequence, HiTrapQ, MonoQ, HiTrap heparin, and Superdex 200 prepacked chromatography media (GE Healthcare), as further detailed in the supplemental material (see Fig. S1 in the supplemental material). During the entire procedure, the recombinant protein was monitored by standard SDS-PAGE analysis (29), and the protein concentration was determined by the Bradford assay (30), using bovine serum albumin as the standard.
Activity assays.
To measure the alkyltransferase activity of wild-type MtOGT and relative mutants, a fluorescence assay using SNAP-Vista Green reagent (New England BioLabs) (here referred to as VG for brevity) was used, as previously described (26) (see Fig. S2A in the supplemental material). Protein bands from SDS-PAGE were visualized by direct gel imaging (VersaDoc 4000; Bio-Rad), and their fluorescence intensities (FIs), corresponding to the reacted protein, were corrected for the actual amount of protein loaded into each lane by measuring the intensity of bands after Coomassie brilliant blue R250 staining. Activity assays at different pH values were performed with 1× Fluo reaction buffer containing 50.0 mM different buffering species, allowing the determination of the pH optimum of the reaction used in all subsequent experiments (data not shown).
Enzyme kinetics analysis.
Assuming a 1:1 substrate-enzyme stoichiometry and irreversible binding, incubation of a fixed amount of protein (5.0 μM) with VG (in the range of 0.1 to 20.0 μM) for 1 h at 25°C gave corrected FI data, which were fitted by a linear equation whose slope gave a direct reference value of FI/μM protein, which was then used to estimate the amount of covalently modified protein (expressed in picomoles) in time course experiments. The latter assays were performed at 25°C using different protein/VG molar ratios and taking 10.0-μl aliquots at time intervals, as detailed in Fig. S2B in the supplemental material. Plots of the picomoles of reacted protein versus time were fitted by exponential equations to determine the apparent rates for covalent modification (kobs). These values were plotted versus the VG doses ([VG]) by using the following hyperbolic equation:
| (1) |
where k and KVG are the rate of covalent linkage and the dissociation constant for the free enzyme and free VG reagent during the first collision step (before covalent modification), respectively (see Fig. S2C in the supplemental material). In order to calculate the apparent KVG values (KVGapp) of proteins for VG as a function of the double-stranded methylated DNA (dsDNAmet) concentration, time course experiments such as those described above were performed in the presence of increasing fixed doses of dsDNAmet (see Table S1 and Fig. S3 in the supplemental material). Data were fitted according to the following linear equation:
| (2) |
where KDNA is the dissociation constant of the protein for the dsDNAmet substrate. The results of these enzyme kinetics analyses are summarized in Table 1.
Table 1.
Kinetic constants of the reaction catalyzed by MtOGT and mutated proteins
| Protein | dsDNAmet concn (μM) | Mean k (s−1) ± SD | Mean KVG (μM) ± SD | Mean KDNA (μM) ± SD |
|---|---|---|---|---|
| MtOGT | 0 | 0.12 ± 0.02 | 1.82 ± 0.4 | 0.24 ± 0.11 |
| 0.63 | 0.14 ± 0.03 | 3.53 ± 0.5 | ||
| 1.00 | 0.10 ± 0.03 | 5.29 ± 0.9 | ||
| 1.25 | 0.14 ± 0.02 | 8.68 ± 0.8 | ||
| MtOGT-T15S | 0 | 0.10 ± 0.01 | 1.73 ± 0.5 | 0.48 ± 0.26 |
| 0.63 | 0.11 ± 0.01 | 2.29 ± 0.4 | ||
| 1.00 | 0.13 ± 0.02 | 3.46 ± 0.5 | ||
| 1.25 | 0.12 ± 0.03 | 5.40 ± 0.7 | ||
| MtOGT-R37L | 0 | 0.07 ± 0.02 | 3.17 ± 0.5 | 2.16 ± 0.23 |
| 0.63 | 0.09 ± 0.02 | 4.23 ± 0.7 | ||
| 1.00 | 0.07 ± 0.01 | 4.55 ± 0.6 | ||
| 1.25 | 0.08 ± 0.02 | 5.11 ± 0.5 |
Electrophoretic mobility shift assay (EMSA).
A 40-bp 6-carboxytetramethylrhodamine (TAMRA)-labeled double-stranded DNA probe (0.1 μM) was incubated at 25°C for 10 min with different amounts of proteins in the range of 0.0 to 120.0 μM in a total volume of 10.0 μl in 1× binding buffer (20.0 mM Tris-HCl [pH 7.5], 50.0 mM KCl, 0.1 mM dithiothreitol [DTT], and 10% glycerol). Samples were loaded onto an 8% polyacrylamide-bisacrylamide native gel in 1× Tris-borate-EDTA (TBE). Signals were visualized by direct gel imaging using a green light-emitting diode (LED)/605-nm-band-pass filter as excitation/emission parameters, respectively.
Crystallization.
Initial conditions for MtOGT crystallization were identified by means of a robot-assisted (Oryx4; Douglas Instruments), sitting-drop-based spare-matrix strategy using screen kits from Hampton Research. The best crystals were obtained by mixing 2 μl of a protein solution at 5 mg/ml with an equal volume of a reservoir solution containing 0.1 M HEPES (pH 7.5), 6% (wt/vol) polyethylene glycol 8000 (PEG 8000), and 6% (wt/vol) ethylene glycol and equilibrating the drop against 800 μl of the reservoir solution at 4°C in sitting-drop format. Crystals used in X-ray diffraction experiments grew to maximum dimensions of 0.2 mm in about 2 weeks. Similarly, crystallization of the MtOGT-R37L mutant enzyme was obtained by mixing 2 μl of the protein solution at 4 mg/ml with an equal volume of crystallization buffer (0.1 M HEPES [pH 7.5], 12% [wt/vol] PEG 8000, 4% ethylene glycol) and equilibrating the drop against 800 μl of the reservoir at 4°C in sitting-drop format, yielding 0.2-mm crystals in about 2 weeks.
Structure determination.
For X-ray data collection, crystals of both wild-type MtOGT and MtOGT-R37L were taken directly from the crystallization droplet, rapidly equilibrated in a solution containing the crystallization buffer and 15% glycerol as a cryoprotectant, and flash-frozen at 100 K under liquid nitrogen. A complete data set of the wild-type MtOGT crystal was collected at 100 K to a 1.8-Å resolution using synchrotron radiation (λ = 0.933 Å) at the ID14-EH1 beam line (European Synchrotron Radiation Facility [ESRF], Grenoble, France), equipped with an ADSC detector. Analysis of the collected diffraction data set allowed us to assign the crystal to the orthorhombic space group P21212, with the cell dimensions a = 59.13 Å, b = 81.75 Å, and c = 38.16 Å, containing 1 molecule per asymmetric unit, with a corresponding solvent content of 52%. For all data collections, diffraction intensities were evaluated, integrated, and scaled by using the CCP4 suite of programs (31). The structure of MtOGT was solved by molecular replacement using the program Phaser (32) and the structure of E. coli O6-methylguanine-DNA methyltransferase AdaC protein as the search model (Protein Data Bank [PDB] accession number 1SFE) (33). The resulting electron density map was of high quality and allowed automatic tracing by the program ARP/wARP (34), which was also used for adding solvent molecules. The program Coot (35) was used for manual rebuilding, and the program PHENIX (36) was used for crystallographic refinement.
A complete data set of the MtOGT-R37L crystal was collected at 100 K to a 2.8-Å resolution using synchrotron radiation (λ = 0.99 Å) at the ESRF ID23 beam line equipped with an ADSC detector. Since the analysis of the diffraction data set showed the MtOGT-R37L crystal to be isomorphic with those of the wild-type protein, we used the refined set of atomic coordinates of the MtOGT protein (in which water/ligand molecules and Arg37 side-chain atoms were omitted) for MtOGT-R37L structure determination; model building and crystallographic refinement were performed following the same procedure described above for the wild-type protein. Data collection, phasing, and refinement statistics are given in Table 2. Structural superpositions were performed with the Superpose program of the CCP4 suite (31). Figures 3 through 6 were generated with PyMol (37).
Table 2.
Data collection, phasing, and refinement statistics
| Parameter | Valuea |
|
|---|---|---|
| MtOGT | MtOGT-R37L | |
| Data collection | ||
| Space group | P21212 | P21212 |
| Wavelength (Å) | 0.933 | 0.99 |
| Resolution (Å) | 1.8 | 2.8 |
| Total no. of reflections | 70,150 | 16,209 |
| No. of unique reflections | 17,690 | 4,572 |
| Mean I (SD) | 15.8 (4.4) | 10.2 (3.6) |
| Completeness (%) | 99.5 (100) | 96.2 (100) |
| Multiplicity | 4.0 (4.0) | 3.5 (3.6) |
| Rmerge (%) | 4.8 (27.2) | 8.2 (31.6) |
| Rmeas (%) | 5.5 (31.3) | 9.4 (36.4) |
| Rpim (%) | 2.7 (15.3) | 4.5 (17.5) |
| Refinement | ||
| Rfactor/Rfree (%) | 21.8/26.4 | 18.6/26 |
| No. of protein atoms | 1,251 | 1,258 |
| No. of ligand atoms | 19 | |
| No. of water molecules | 141 | 48 |
| RMSD bond (Å) | 0.007 | 0.009 |
| RMSD angles (°) | 1.016 | 1.151 |
| Avg B (Å2) | ||
| Protein | 34.3 | 63.4 |
| Solvent | 40.5 | 50.2 |
| Residues in Ramachandran plot areas (%) | ||
| Preferred | 96.3 | 98 |
| Allowed | 3.7 | 2 |
Values in parentheses refer to the highest-resolution shell.
Fig 3.

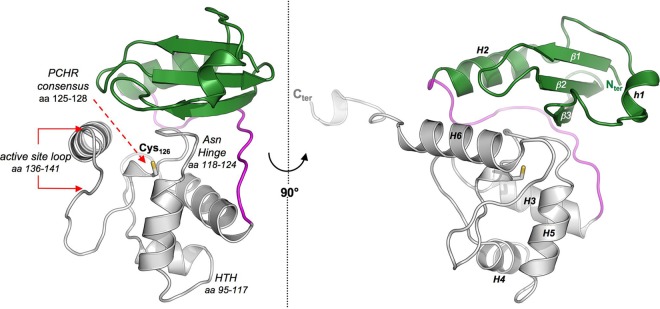
Crystal structure of MtOGT. Shown are cartoon representations of the MtOGT structure, as observed from two different points of view. In both images, the N-terminal domain, the C-terminal domain, and the connecting loop are colored in green, gray, and magenta, respectively. The catalytic cysteine residue (Cys126) is invariably drawn as sticks. Functional element labeling and secondary structure element numbering appear in the image at the left and at the right, respectively. aa, amino acids.
Fig 6.

Schematic representation of the possible functional consequences of the replacement of Arg37 with Leu in MtOGT. (i) Direct interference in the dealkylation reaction; (ii) reduced capability of establishing protein-protein interactions and tight monomer packing during cooperative DNA binding; (iii) direct participation of Arg37 in DNA binding. The N-terminal domain, the domain-connecting loop, and the C-terminal domain of the wild-type protein are colored green, magenta, and white, respectively. The MtOGT-R37L structure is uniformly rendered in red; the modeled dsDNA appears in yellow.
Protein structure accession numbers.
The atomic coordinates and structure factors of MtOGT and MtOGT-R37L have been deposited in the Protein Data Bank (http://www.rcsb.org/) under accession numbers 4BHB and 4BHC, respectively.
RESULTS AND DISCUSSION
Expression and purification of wild-type and point-mutated MtOGT variants.
In order to perform a molecular characterization of the M. tuberculosis OGT protein, the corresponding open reading frame was subcloned into the pET16b vector within the NcoI/BamHI sites of the polylinker to eliminate the standard polyhistidine coding region. Once transformed into E. coli strain BL21(DE3), the resulting pET-MtOGT expression construct drives the synthesis of an untagged version of the protein (MtOGT) (predicted molecular mass of 17,844 Da; 165 amino acid residues; Ile in position 2 replaced by Val as a consequence of the adopted subcloning strategy) that could be reproducibly purified at a high yield by FPLC-based standard techniques. pET-MtOGT was also used as the template for PCR-based site-directed mutagenesis to generate the expression vectors for the production in E. coli of two point-mutated variants of the enzyme (MtOGT-T15S and MtOGT-R37L), which mimic those naturally occurring in M. tuberculosis strains bearing nsSNPs at codon 15 (ACC mutated to AGC) or codon 37 (CGC mutated to CTA) of their OGT coding sequence (19–21). We obtained pure and highly homogeneous preparations of both MtOGT-R37L and MtOGT-T15S, and we observed that, similar to other OGTs, our recombinant proteins behave as monomers in solution (see Fig. S1 in the supplemental material). These observations indicate that the R37L and T15S amino acid substitutions do not affect the overall stability of the corresponding mutated proteins and do not alter their quaternary structure in the absence of ligands.
Biochemical characterization of MtOGT.
From a biochemical point of view, OGT proteins are at the same time the catalyst and one of the stoichiometric reagents of the alkyl group transfer from the modified base to the reactive cysteine (Cys126 in MtOGT) in the active site, resulting in permanent inactivation of the protein (10). We exploited this feature to characterize the kinetics of the reaction performed by MtOGT and its variants by adopting a recently developed procedure (26) that makes use of derivatives of O6-benzylguanine (a well-known inhibitor of AGTs), which acts by covalent transfer of the benzylic group to the active-site cysteine (see Fig. S2A in the supplemental material). The method allows the determination of the MtOGT catalytic activity from the direct measurement of the covalent protein-inhibitor fluorescent complex. Specifically, the fluorescein-labeled O6-benzylguanine derivative SNAP-Vista Green (VG) was used to measure the activities of MtOGT, MtOGT-T15S, and MtOGT-R37L by quantifying the intensity of the signal emitted by the VG-modified protein analyzed by SDS-PAGE. In all three cases, the fluorescence intensity of the band showed a linear dependence on the VG reagent concentration, reaching a plateau at a 1:1 protein/VG ratio, thus confirming the predicted stoichiometry and the suicide nature of the reaction catalyzed by MtOGT, regardless of the presence of either the T15S or R37L mutation (data not shown).
Notably, this analysis also revealed that the MtOGT active site is in principle capable of housing bulky base adducts, as shown previously for the Sulfolobus solfataricus protein (26); in addition, it confirmed the robustness of the fluorescent VG-based assay and its applicability for the careful characterization of OGT proteins in general as a reliable alternative to classical and laborious assays for DNA-alkyltransferase activity.
Time course experiments, performed with different protein/VG ratios, allowed us to determine the kinetic constants for the covalent modification of MtOGT, MtOGT-T15S, and MtOGT-R37L. The values obtained in the presence of the synthetic substrate VG were similar for all the three proteins (Table 1), suggesting that neither mutation affects the alkyltransferase reaction rate.
Previous work with the S. solfataricus OGT protein showed that the assay can also be applied to determine the activity of OGT proteins toward their natural substrate (i.e., alkylated DNA) in competition experiments (26). We thus repeated the time course experiments described above in the presence of increasing amounts of a double-stranded DNA fragment bearing an internal O6-methylated guanine (dsDNAmet), which is expected to compete with the VG reagent in the alkyltransferase reaction. According to equation 2, a linear plot of the KVG values as a function of the dsDNAmet concentration (see Fig. S3 in the supplemental material) allowed the calculation of the dissociation constant (KDNA) of MtOGT and of the two point mutants for this substrate. Our data indicate that both mutations affect the protein affinity for methylated DNA albeit to different extents: whereas the MtOGT-R37L point mutant exhibited a 10-fold-lower affinity for dsDNAmet (KDNA = 2.16 ± 0.23 μM) than the wild-type protein (KDNA = 0.24 ± 0.11 μM), this difference appeared less pronounced for MtOGT-T15S (Table 1). Thus, although neither mutation results in impaired intrinsic alkyltransferase activity, both MtOGT variants, and MtOGT-R37L in particular, exhibit reduced affinity for an alkylated DNA molecule.
To investigate the reasons for the reduced activity of the mutated MtOGTs toward their physiological substrate, we studied direct protein-DNA associations using a fluorescent dsDNA probe (see Table S1 in the supplemental material) in an EMSA-based analysis. Figure 2 shows that wild-type MtOGT binds dsDNA in a cooperative manner with a plateau at a DNA/protein molar ratio of 1:150 and a dissociation constant value (K) of approximately 7 μM, a value comparable to that determined for human AGT by using the same method (38). In contrast, MtOGT-T15S and MtOGT-R37L showed a significant reduction in dsDNA binding, with a plateau at DNA/protein molar ratios of 1:300 and 1:600 (K values of approximately 14 μM and 26 μM), respectively.
Fig 2.

DNA binding activity of wild-type MtOGT and point-mutated proteins. (A) Band shift analysis of MtOGT (wild type [WT]), MtOGT-T15S (T15S), and MtOGT-R37L (R37L) proteins. Lane 1, 1 pmol of fluorescent double-stranded DNA probe (DNA); lanes 2 to 9, increasing amounts of protein (P) incubated in the presence of the probe at the indicated DNA/protein molar ratios. The white and black arrowheads point to the free and protein-bound probes, respectively. (B) Plot of the DNA-bound protein fractions at the DNA/protein molar ratios indicated in panel A. K, dissociation constant (μM).
Taken together, our data indicate that although neither mutation affects specifically the kinetics of the alkyl group transfer from the synthetic VG substrate to the protein, both mutations significantly decrease the protein's capability of associating with DNA molecules and, consequently, the protein's ability to efficiently perform alkylated-DNA repair. To assess whether this catalytic defect translates in vivo into a “hypermutator” phenotype, possibly beneficial to the fitness of the bacillus under circumstances of selective pressure, will require further microbiological studies.
Structural analysis of MtOGT.
The N-terminal domain of the DNA-protein alkyltransferase family members features a low degree of both sequence and length conservation between different species, thus precluding an in silico mapping of the residues affected by the nsSNPs under study based exclusively upon information currently available in the Protein Data Bank (see Fig. S4 in the supplemental material). Therefore, to shed light on the molecular basis of the functional defect characterizing the MtOGT variants, we determined the crystal structures of the wild-type protein and of the MtOGT-R37L mutant at 1.8-Å and 2.8-Å resolutions, respectively, revealing in both cases one monomer in the asymmetric unit.
An excellent electron density was visible for the entire MtOGT protein chain (residues 2 to 165) and allowed the modeling of two molecules of glycerol used as the cryoprotectant. Since the crystals of MtOGT-R37L were isomorphic with those of MtOGT, we used the model of the wild-type protein to solve the structure of the mutant. The stereochemistry of the refined MtOGT and MtOGT-R37L structures was assessed with the program PROCHECK (39), revealing 96% and 98% of residues in the preferred region of the Ramachandran plot, respectively (Table 2).
MtOGT overall structure.
Similar to structures of other archaeal (40, 41) and eubacterial (33) OGTs as well as of human AGT in its ligand-free (42) and alkylated (43) states or in complex with modified dsDNA (25, 44), both MtOGT and MtOGT-R37L fold into a roughly globular molecular architecture built up by two domains connected by a long loop and ending in a 10-residue-long tail. In both structures, the tail adopts a fully extended conformation, pointing straight outwards from the bulk body of the protein (Fig. 3) and contacting the C-terminal domain of the adjacent symmetry mate within the crystal lattice (see Fig. S5 in the supplemental material).
The N-terminal domain consists of an antiparallel three-stranded β-sheet and connecting loops (residues 2 to 27) sandwiched between a mainly randomly coiled region (residues 28 to 46) containing a single helical turn at its middle (residues 35 to 39) (h1 in Fig. 3) on one side and a structurally conserved α-helix on the opposite one (residues 47 to 59) (H2 in Fig. 3).
The C-terminal domain (residues 75 to 155) adopts the prototypical all-α-fold and houses the highly conserved functional elements that previous biochemical and mutational studies (45, 46) demonstrated to be required to perform efficient catalysis. Referring to the MtOGT primary sequence, these motifs are (i) the helix-turn-helix (HTH) motif, which is responsible for DNA binding at its minor groove and bears Arg109, whose structural equivalent in human AGT (i.e., Arg128) has been demonstrated to act as a temporary substitute for the modified base upon its flipping out from the regular base stacking (25, 44); (ii) the “Asn hinge” building up one wall of the deep ligand binding cavity that accepts the modified base for repairing; (iii) the strictly conserved PCHR consensus motif surrounding the catalytic cysteine (Cys126); (iv) the active-site loop that participates in the correct positioning of the alkylated base inside the ligand binding pocket; and (v) the structurally conserved H6 helix that, by shielding the ligand binding cavity on the opposite side of the Asn hinge, contributes essential residues for completing both the catalytic triad (His127 and Glu153) and the modified-base-bonding network (Lys146) (Fig. 3, left) (25, 33, 40–44).
The active-site loop of MtOGTs is observed in a solvent-exposed conformation.
Our crystallographic analysis confirms the high degree of structural conservation displayed by the catalytic C-terminal domain among members of the protein-DNA alkyltransferase protein family. However, different from what was observed in all the crystal structures of OGTs, in both the MtOGT and MtOGT-R37L models the active-site loop is oriented toward the exterior of the protein body (Fig. 4A), adopting a conformation that closely resembles the one displayed by the equivalent region in the structures of alkyltransferase-like proteins (ATLs) (47) in complex with alkylated DNA (48, 49) (Fig. 4B).
Fig 4.
The active-site loop of MtOGT is observed in a solvent-exposed conformation. (A) Close-up views of equivalent regions of MtOGT (MTB) (both the wild-type protein and the R37L variant are shown at the left), E. coli AdaC (ECO) (PDB accession number 1SFE), and human AGT in the absence of ligand (HUM) (PDB accession number 1EH6), upon optimal structural superposition of their C-terminal domains. In each panel, residues of the region highlighted in the sequence alignment (top) and the catalytic cysteine are shown as sticks. The red arrow points to the OH group of the structurally equivalent Tyr residue. GOL, glycerol. (B) Cartoon representation of optimally superimposed MtOGT (green), the Schizosaccharomyces pombe alkyltransferase-like protein (SpATL) in the absence of ligands (yellow) (PDB accession number 3GVA; average RMSD = 1.8 Å), and SpATL in complex with O6-methylguanine dsDNA (magenta) (PDB accession number 3GX4; average RMSD = 1.5 Å) (the DNA strands appear as black ribbons). (C) Possible mode of binding of alkylated dsDNA to MtOGT (left), resulting upon optimal superposition of MtOGT on the structure of a Cys145Ser mutant of human AGT (*AGT) in complex with O6-methylguanine (O6-metG) dsDNA (right) (PDB accession number 1T38; average RMSD = 2.4 Å). Cys126 of MtOGT is observed at 3.8 Å from the methyl group of the O6-methylguanine. In panels B and C, residues discussed in the text appear as sticks, and the water molecule in panel C (W22) is rendered as a sphere.
In principle, such a solvent-exposed conformation of the MtOGT active-site loop still allows the HTH motif to peculiarly bind the substrate DNA at its minor groove, as disclosed by structural studies of AGT and ATLs (25, 44, 48, 49), and is fully compatible with the housing of the modified base inside the active site (Fig. 4C, left). In fact, by overlaying our structures upon that of an inactive variant of human AGT (catalytic Cys145 mutated to Ser) in complex with O6-guanine-methylated dsDNA (Fig. 4C, right) (25), we noted that the ligand molecule perfectly fits into the MtOGT active site, which appears fully accessible to the flipped-out base without requiring any side-chain repositioning. In this unprecedented conformation, the hydroxyl group of the highly conserved Tyr139 residue is correctly oriented to establish a hydrogen bond with the DNA sugar-phosphate backbone at the 5′ side of the modified base (Fig. 4C, left), apparently compensating for the absence at position 116 of the recognition helix H5 of a residue functionally equivalent to Arg135 of human AGT (Gly116 in MtOGT) (Fig. 4C). Although in this conformation, Tyr139 is no longer available for stacking against the alkylated base on the opposite side with respect to the recognition helix H5, the presence in the active site of Arg27 could contribute to anchoring the base by contacting its O6 position through a water-mediated hydrogen bond (Fig. 4C).
At present, we cannot univocally discern whether the conformation adopted by the active-site loop in our MtOGT structures represents a unique, distinctive trait of the mycobacterial proteins or is a consequence of crystal packing (see Fig. S5 in the supplemental material); nonetheless, the reported data provide further independent support for the proposed conformational plasticity of this region and its central role in the control of alkylated-DNA binding (25, 40–44, 48, 49).
MtOGT structures provide clues to explain the DNA binding impairment displayed by MtOGT-R37L.
Optimal superposition of the available OGT three-dimensional (3D) models from different species confirmed the limited structural conservation of their N-terminal domain (see Fig. S4 in the supplemental material). In this respect, the MtOGT structure allowed us to directly observe the intradomain contacts involving the mutated residues under study. In particular, Thr15 and Arg37 seem to play a central role in coordinating the network of contacts established between a small number of residues of strands β1 and β2 and the region bearing the h1 helix of the facing random coil. In fact, in the structure of MtOGT (Fig. 5A), the Thr15 OH group is observed within hydrogen-bonding distance of the Arg37 NH2 atom, which is engaged in an ion pair with Glu30; in turn, the latter residue coordinates a water molecule that is also in hydrogen-bonding contact with the Thr15 OH group. The observed hydrogen bond arrangement could provide a likely explanation for why the MtOGT-R37L protein displays a more reduced affinity toward alkylated DNA than does MtOGT-T15S. Indeed, in the structure of the MtOGT-R37L variant (Fig. 5B), the interactions involving Arg37 and the chemical identity of the local environment are lost, whereas the analysis of a model of the MtOGT-T15S mutant (Fig. 5C) suggests that these biochemical features remain unaltered, due to the presence in Ser15 of a hydroxyl group structurally equivalent to the one present in the substituted threonine.
Fig 5.

Possible structural role of Arg37. (A and B) Close-up views of the superposed N-terminal domain and part of the active site of wild-type MtOGT (A) and MtOGT-R37L (B), as observed in their respective crystal structures. (C) Equivalent regions of a structure-based model of the MtOGT-T15S variant. In all images, residues cited in the text appear as sticks, dotted lines indicate close interactions (distances appear in angstroms), and water molecules are rendered as spheres.
How might a nonconservative substitution of MtOGT Arg37, which maps far away from both the Cys126 reactive center and the currently identified DNA binding motifs, produce an impact on protein function, as revealed by our in vitro biochemical characterization? In principle, there are three possible—and not mutually exclusive—scenarios: the R37L mutation could (i) induce conformational changes of the N-terminal random-coiled region that can affect catalysis, (ii) alter the assembly of protein complexes at the damaged site during cooperative DNA binding, and (iii) impair an unexpected participation of the Arg37-bearing random coil to direct DNA binding (Fig. 6).
For the first scenario, it must be noted that by comparing the structures of wild-type MtOGT and the MtOGT-R37L variant (Fig. 5), we did not observe any dramatic conformational change affecting the protein backbone (average root mean square deviation [RMSD] = 0.3 Å). Moreover, even taking into account the more limited resolution of the MtOGT-R37L model, the only appreciable differences at the level of the N-terminal domain are represented by Glu30 and Arg27, whose side chains appear largely undefined in the structure of the mutated protein (Fig. 5B). Similarly, the low increase of the thermal factor values of the N-terminal random-coil residues is comparable for both the MtOGT and MtOGT-R37L models (not shown). Thus, our data do not indicate an increased conformational flexibility of this region in the mutated protein, possibly directly interfering with the catalysis.
Regarding the second scenario, seminal studies on human AGT revealed a participation of discrete regions of the N-terminal domain in the cooperative behavior displayed by the protein during DNA recognition (10, 23, 24), and our biochemical characterization shows a severe undermining of MtOGT DNA binding activity upon R37L mutation (Table 1 and Fig. 2); these observations prompted us to speculate that the lack of Arg37 might alter the possibility of establishing optimal protein-protein contacts required for a tight assembly of OGT monomers at the alkylated site. Consequently, although we did not observe any significant structural difference between the wild-type and mutated MtOGT, we cannot rule out that a repositioning of the N-terminal random coil could become evident only in the presence of DNA, therefore affecting the cooperativity of DNA binding.
The third hypothesis mentioned above, implying a direct involvement of the Arg37-bearing protein moiety in DNA binding, is strongly suggested by the results of our biochemical analysis but needs further site-directed mutagenesis analysis and structural investigations to be definitively assessed.
Overall, our results, besides revealing a high level of structural conservation among DNA-protein alkyltransferases, could provide a framework to orient future studies aimed at an understanding of the molecular mechanism leading to the observed functional defect shown by the analyzed MtOGT variants.
In conclusion, we showed that while the catalytic activity of the mutated MtOGT variants toward the synthetic substrate used is not diminished relative to the wild-type protein, the same mutations result in decreased affinity toward the physiological alkylated-DNA substrate. Consequently, the mutants under analysis are less efficient than the wild-type protein in carrying out the complete catalytic cycle required for alkylated-DNA direct repair in vitro. However, due to the critical contribution of the physiological context in determining whether or not a specific mutation in a protein impacts its function, definitive proof of a role of these MtOGT nsSNPs in altering alkylation damage sensitivity and mutation frequency of the corresponding M. tuberculosis strain will come only from future in vivo studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the European Community (project SysteMTb HEALTH-F4-2010-241587).
We acknowledge the European Synchrotron Radiation Facility (Grenoble, France) for provision of synchrotron radiation at beam lines ID14 and ID23.
Footnotes
Published ahead of print 5 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02298-12.
REFERENCES
- 1. Gorna AE, Bowater RP, Dziadek J. 2010. DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection. Clin. Sci. (Lond.) 119:187–202 [DOI] [PubMed] [Google Scholar]
- 2. Dos Vultos T, Mestre O, Rauzier J, Golec M, Rastogi N, Rasolofo V, Tonjum T, Sola C, Matic I, Gicquel B. 2008. Evolution and diversity of clonal bacteria: the paradigm of Mycobacterium tuberculosis. PLoS One 3:e1538. 10.1371/journal.pone.0001538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizrahi V, Andersen SJ. 1998. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 29:1331–1339 [DOI] [PubMed] [Google Scholar]
- 4. Warner DF, Mizrahi V. 2007. The survival kit of Mycobacterium tuberculosis. Nat. Med. 13:282–284 [DOI] [PubMed] [Google Scholar]
- 5. Dos Vultos T, Mestre O, Tonjum T, Gicquel B. 2009. DNA repair in Mycobacterium tuberculosis revisited. FEMS Microbiol. Rev. 33:471–487 [DOI] [PubMed] [Google Scholar]
- 6. Kurthkoti K, Varshney U. 2012. Distinct mechanisms of DNA repair in mycobacteria and their implications in attenuation of the pathogen growth. Mech. Ageing Dev. 133:138–146 [DOI] [PubMed] [Google Scholar]
- 7. Voskuil MI, Bartek IL, Visconti K, Schoolnik GK. 2011. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2:105. 10.3389/fmicb.2011.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dalhus B, Laerdahl JK, Backe PH, Bjoras M. 2009. DNA base repair—recognition and initiation of catalysis. FEMS Microbiol. Rev. 33:1044–1078 [DOI] [PubMed] [Google Scholar]
- 9. Shrivastav N, Li D, Essigmann JM. 2010. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis 31:59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pegg AE. 2011. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 24:618–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanugula S, Goodtzova K, Pegg AE. 1998. Probing of conformational changes in human O6-alkylguanine-DNA alkyl transferase protein in its alkylated and DNA-bound states by limited proteolysis. Biochem. J. 329:545–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu-Welliver M, Pegg AE. 2002. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 23:823–830 [DOI] [PubMed] [Google Scholar]
- 13. Samson L, Cairns J. 1977. A new pathway for DNA repair in Escherichia coli. Nature 267:281–283 [DOI] [PubMed] [Google Scholar]
- 14. Yang M, Aamodt RM, Dalhus B, Balasingham S, Helle I, Andersen P, Tonjum T, Alseth I, Rognes T, Bjoras M. 2011. The ada operon of Mycobacterium tuberculosis encodes two DNA methyltransferases for inducible repair of DNA alkylation damage. DNA Repair (Amst.) 10:595–602 [DOI] [PubMed] [Google Scholar]
- 15. Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 16. Durbach SI, Springer B, Machowski EE, North RJ, Papavinasasundaram KG, Colston MJ, Bottger EC, Mizrahi V. 2003. DNA alkylation damage as a sensor of nitrosative stress in Mycobacterium tuberculosis. Infect. Immun. 71:997–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., III 2004. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279:40174–40184 [DOI] [PubMed] [Google Scholar]
- 18. Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ebrahimi-Rad M, Bifani P, Martin C, Kremer K, Samper S, Rauzier J, Kreiswirth B, Blazquez J, Jouan M, van Soolingen D, Gicquel B. 2003. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg. Infect. Dis. 9:838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olano J, Lopez B, Reyes A, Lemos MP, Correa N, Del Portillo P, Barrera L, Robledo J, Ritacco V, Zambrano MM. 2007. Mutations in DNA repair genes are associated with the Haarlem lineage of Mycobacterium tuberculosis independently of their antibiotic resistance. Tuberculosis (Edinb.) 87:502–508 [DOI] [PubMed] [Google Scholar]
- 21. Mestre O, Luo T, Dos Vultos T, Kremer K, Murray A, Namouchi A, Jackson C, Rauzier J, Bifani P, Warren R, Rasolofo V, Mei J, Gao Q, Gicquel B. 2011. Phylogeny of Mycobacterium tuberculosis Beijing strains constructed from polymorphisms in genes involved in DNA replication, recombination and repair. PLoS One 6:e16020. 10.1371/journal.pone.0016020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fang Q, Kanugula S, Pegg AE. 2005. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry 44:15396–15405 [DOI] [PubMed] [Google Scholar]
- 23. Adams CA, Melikishvili M, Rodgers DW, Rasimas JJ, Pegg AE, Fried MG. 2009. Topologies of complexes containing O6-alkylguanine-DNA alkyltransferase and DNA. J. Mol. Biol. 389:248–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams CA, Fried MG. 2011. Mutations that probe the cooperative assembly of O6-alkylguanine-DNA alkyltransferase complexes. Biochemistry 50:1590–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. 2004. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 11:714–720 [DOI] [PubMed] [Google Scholar]
- 26. Perugino G, Vettone A, Illiano G, Valenti A, Ferrara MC, Rossi M, Ciaramella M. 2012. Activity and regulation of archaeal DNA alkyltransferase: conserved protein involved in repair of DNA alkylation damage. J. Biol. Chem. 287:4222–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sambrook J, Fritsh EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Studier FW. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 29. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 30. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 31. Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 32. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moore MH, Gulbis JM, Dodson EJ, Demple B, Moody PC. 1994. Crystal structure of a suicidal DNA repair protein: the Ada O6-methylguanine-DNA methyltransferase from E. coli. EMBO J. 13:1495–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perrakis A, Morris R, Lamzin VS. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6:458–463 [DOI] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 36. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA: http://www.pymol.org/ [Google Scholar]
- 38. Spratt TE, Wu JD, Levy DE, Kanugula S, Pegg AE. 1999. Reaction and binding of oligodeoxynucleotides containing analogues of O6-methylguanine with wild-type and mutant human O6-alkylguanine-DNA alkyltransferase. Biochemistry 38:6801–6806 [DOI] [PubMed] [Google Scholar]
- 39. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291 [Google Scholar]
- 40. Hashimoto H, Inoue T, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Kai Y. 1999. Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase. J. Mol. Biol. 292:707–716 [DOI] [PubMed] [Google Scholar]
- 41. Roberts A, Pelton JG, Wemmer DE. 2006. Structural studies of MJ1529, an O6-methylguanine-DNA methyltransferase. Magn. Reson. Chem. 44(Spec No):S71–S82. 10.1002/mrc.1823 [DOI] [PubMed] [Google Scholar]
- 42. Wibley JE, Pegg AE, Moody PC. 2000. Crystal structure of the human O(6)-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 28:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daniels DS, Mol CD, Arvai AS, Kanugula S, Pegg AE, Tainer JA. 2000. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. EMBO J. 19:1719–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duguid EM, Rice PA, He C. 2005. The structure of the human AGT protein bound to DNA and its implications for damage detection. J. Mol. Biol. 350:657–666 [DOI] [PubMed] [Google Scholar]
- 45. Crone TM, Goodtzova K, Pegg AE. 1996. Amino acid residues affecting the activity and stability of human O6-alkylguanine-DNA alkyltransferase. Mutat. Res. 363:15–25 [DOI] [PubMed] [Google Scholar]
- 46. Xu-Welliver M, Pegg AE. 2000. Point mutations at multiple sites including highly conserved amino acids maintain activity, but render O6-alkylguanine-DNA alkyltransferase insensitive to O6-benzylguanine. Biochem. J. 347:519–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Margison GP, Butt A, Pearson SJ, Wharton S, Watson AJ, Marriott A, Caetano CM, Hollins JJ, Rukazenkova N, Begum G, Santibáñez-Koref MF. 2007. Alkyltransferase-like proteins. DNA Repair 6:1222–1228 [DOI] [PubMed] [Google Scholar]
- 48. Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. 2009. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature 459:808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilkinson OJ, Latypov V, Tubbs JL, Millington CL, Morita R, Blackburn H, Marriott A, McGown G, Thorncroft M, Watson AJ, Connolly BA, Grasby JA, Masui R, Hunter CA, Tainer JA, Margison GP, Williams DM. 2012. Alkyltransferase-like protein (Atl1) distinguishes alkylated guanines for DNA repair using cation-pi interactions. Proc. Natl. Acad. Sci. U. S. A. 109:18755–18760 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.