

Abstract
To better understand the poor conservation of the helicase binding domain of primases (DnaGs) among the eubacteria, we determined the crystal structure of the Helicobacter pylori DnaG C-terminal domain (HpDnaG-CTD) at 1.78 Å. The structure has a globular subdomain connected to a helical hairpin. Structural comparison has revealed that globular subdomains, despite the variation in number of helices, have broadly similar arrangements across the species, whereas helical hairpins show different orientations. Further, to study the helicase-primase interaction in H. pylori, a complex was modeled using the HpDnaG-CTD and HpDnaB-NTD (helicase) crystal structures using the Bacillus stearothermophilus BstDnaB-BstDnaG-CTD (helicase-primase) complex structure as a template. By using this model, a nonconserved critical residue Phe534 on helicase binding interface of DnaG-CTD was identified. Mutation guided by molecular dynamics, biophysical, and biochemical studies validated our model. We further concluded that species-specific helicase-primase interactions are influenced by electrostatic surface potentials apart from the critical hydrophobic surface residues.
INTRODUCTION
Replication of chromosomal DNA is generally a universal process that requires a high degree of accuracy and precision to maintain fidelity in the transmission of genetic material from one generation to the next (1–4). This unique process involves multiprotein complexes that help to check the inevitable errors associated with DNA replication (5–7). Interference with any of these protein-DNA and protein-protein interactions may lead to numerous problems, including unviable offspring. Eubacterial DnaG primase is a single-stranded DNA (ssDNA)-dependent RNA polymerase responsible for the synthesis of oligonucleotide primers needed for DNA replication (8). Primase is recruited once or twice on the leading strand, in contrast to the lagging strand, where it is recruited several times (9, 10). DnaG primase also plays an important role in tuning the synthesis (11, 12). The eubacterial DnaG primase has three domains. The N-terminal domain (NTD) is involved in template DNA recognition and contains a zinc binding domain, and the central catalytic domain synthesizes oligonucleotide primers. The only known function of the C-terminal domain (CTD), also known as the helicase binding domain (HBD), is to interact with helicase at the replication fork. Of these domains, the CTD is least conserved (13). The HBD/CTD is sufficient to bind and stimulate the activities of DnaB helicase (3, 14). The stability of the interaction between DnaG primase and DnaB helicase varies among eubacteria. In Escherichia coli the interaction has been reported to be weak (9, 15, 16), whereas in Bacillus stearothermophilus the interaction is so strong that it can be purified on a gel filtration column (17). We have recently reported a moderate level of interaction between these proteins in Helicobacter pylori (14). The full-length structure of DnaG primase has yet to be determined, although the structures of individual domains have been reported. Primase C-terminal domain crystal and solution structures are known from E. coli (10) and B. stearothermophilus (13). The crystal structures of the zinc binding domain alone and together with RNA polymerase domain structure were determined in B. stearothermophilus (18) and Aquifex aeolicus (19), respectively. RNA polymerase domain and its complex with ssDNA crystal structures for E. coli were recently published (20, 21). Recently, a medium resolution helicase-primase complex structure was reported, and this study provided insight into the interaction pattern of the DnaG CTD with DnaB helicase (22). Since the helicase-binding domains in primases are poorly conserved, high-resolution structures from different organisms will be helpful for understanding the mechanism of interaction between DnaG and DnaB. H. pylori infection is present in half of the world's human population (23). It causes diverse diseases of the stomach, from chronic gastritis to mucosa-associated lymphoid tissue lymphomas (24, 25). Interestingly, recent work has shown that this organism may also have a role in autoimmune thrombocytopenia (26), Guillain-Barre syndrome (27), and in Alzheimer's disease (28) and in strokes (29). These diseases and especially ailments, such as persistent diarrhea, peptic ulcer, and gastric cancer, may be mitigated if a way can be found to eradicate H. pylori. Since the present therapeutic approach targeting this organism is not very effective, and the conditions in developing countries are suitable for this organism to flourish, a better therapeutic approach is urgently needed. Since the initiation of replication is a crucial step in reproduction of an organism, the structural and functional studies of this target process is important for future drug development.
The proteins involved in DNA replication and repair in H. pylori have been reviewed recently (30). Several DNA replication proteins, such as the initiator proteins DnaA and Hob-A (31–34), the replicative helicase DnaB and its unique dodecameric architecture (35–38), and the single-stranded DNA-binding protein (SSB) (39), have been characterized in H. pylori. Recently, first bipartite origin of chromosome replication in Gram-negative bacteria was reported in H. pylori (40). HpDnaB is unique because it can bypass the EcDnaC function in vivo, suggesting that it has an independent helicase loader function (41). The HpDnaB helicase alone binds to DNA weakly, but when HpDnaG primase interacts with HpDnaB, it binds to DNA strongly. This switching of weak to strong binding occurs because of the NTD conformational change, which is essential for HpDnaB function (36). Furthermore, the crystal structure of the NTD of HpDnaB and its interaction with the primase DnaG have been reported (14). However, the mode of interaction between these proteins has not been studied further.
Here, we report the 1.8-Å resolution structure of the CTD of H. pylori DnaG. It is the best-resolution structure reported to date for DnaG-CTD. Comparison to other DnaG-CTD reveals that it is a flexible structure with a globe region (GR) and a helical hairpin region (HHR), where the HHR can adopt multiple orientations relative to the GR. HpDnaG-CTD structure is different from the other two structures of EcDnaG-CTD and BstDnaG-CTD in terms of the number of helices present, subtle critical residue conservation at tertiary level of protein (compared to the structures of E. coli and B. stearothermophilus), and electrostatic surface potential. We have shown the mode of helicase-primase interactions in H. pylori based on the proposed model. This HpDnaB-HpDnaG model is developed from crystal structures of HpDnaB-NTD reported earlier by our group and that of HpDnaG-CTD, which is being reported here. We have successfully validated our model by mutational studies. Our findings successfully highlighted the criticality of the nonconserved hydrophobic residue responsible for the stability of the helicase-primase complex formation and associated biochemical activities. Further analysis has shown the possible role of surface electrostatic potential in the interaction of helicase-primase in eubacteria. Overall, these results helped us substantially to understand the mechanism of helicase-primase interactions and its important role in DNA replication specifically in H. pylori. These findings may possibly be extrapolated to eubacteria.
MATERIALS AND METHODS
Cloning of full-length HpDnaG and HpDnaG-CTD (C-terminal domain) point mutants.
The (full-length) HpDnaG F534A mutant was generated by site-directed mutagenesis as reported previously (42), using forward and reverse primers (5′-CCTAAAAGCTCGCTCCCTGCTAGCGAAAAAATGATCTGT-3′ and 5′-ACAGATCATTTTTTCGCTAGCAGGGAGCGAGCTTTTAGG-3′, respectively) and pET30b-HpDnaG clone as a template. HpDnaG-CTD containing the F534A mutant was cloned into NheI/XhoI sites of the expression vector pET21c (Novagen, Madison, WI), similar to wild-type HpDnaG-CTD as described earlier (see Table ST1 in the supplemental material). The cloning of wild-type full-length HpDnaG and HpDnaG-CTD was performed as described previously by our group (14).
Purification of recombinant full-length HpDnaB, HpDnaG, and point mutant HpDnaG F534A.
HpDnaB was purified as described earlier (35). For HpDnaG, E. coli strain BL21(DE3) was transformed with the pET30b-HpDnaG construct. For expression, 1% of overnight grown culture of a single colony was added to 1 liter of Luria-Bertani (LB) broth (Hi-Media) containing kanamycin (50 μg/ml), 1 mM glucose, and 1 mM MgCl2. Cells were grown to an optical density at 600 nm (OD600) of 0.4 at 37°C. The cell culture was induced with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and shaken overnight at 22°C. The cells were harvested at 7,000 rpm for 7 min and stored at −80°C for later use. The first step of purification was carried out using Ni-NTA (Qiagen, Germany) affinity chromatography. The cell pellets stored at −80°C were thawed and mixed with lysis buffer (50 mM Tris-HCl [pH 8.0], 100 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10 mM imidazole, 5 mM spermidine, 5 mM MgCl2, 10 mM β-mercaptoethanol [βME], 100 μM phenylmethylsulfonyl fluoride [PMSF]). After the addition of lysozyme (0.1 mg/ml), the cell suspension was incubated at 4°C for 30 min. The cell suspension was sonicated (Branson Ultrasonic Systems) in an ice-water mixture at 25% of the amplitude with a pulse (three to four cycles) of 30 s each interspersed with a 1-min interval. Sonified cell lysate was treated with 0.1% Triton X-100, followed by incubation for 1 h on a rotating rocker at 4°C. The lysate was then centrifuged at 15,000 rpm for 45 min at 4°C. The filtered cell lysate was incubated with Ni-NTA Sepharose resin (GE Healthcare, Sweden) at 4°C for 45 min. The protein-bound beads were washed three times with wash buffer (50 mM Tris-Cl [pH 8.0], 100 mM sodium phosphate [pH 8.0], 300 mM NaCl, 30 mM imidazole, 10 mM βME, and 100 μM protease inhibitor PMSF). Protein was eluted using 1 ml of elution buffer (250 mM imidazole in wash buffer) at 4°C in the batch method. The concentrated protein was loaded on a gel filtration chromatography Superdex 200 10/300 GL column (GE Healthcare), which was previously equilibrated with buffer G (20 mM MES [pH 5.5], 100 mM NaCl, 100 mM PMSF, and 10 mM βME). Peak fractions were checked by SDS–10% PAGE and pooled together. For the point mutant, we followed the methodology described above.
Purification of recombinant HpDnaG-CTD and point mutant (F534A) HpDnaG-CTD.
The recombinant plasmid containing the CTD of HpDnaG insert was transformed into BL21(DE3) cells. Transformed BL21 cells were grown from a single colony in 2% LB medium containing 100 μM ampicillin at 37°C to an OD600 of 0.5. The culture was induced with 1 mM IPTG. Cells were allowed to shake further at 30°C for 6 h. The cells were harvested at 7,000 rpm for 7 min and stored at −80°C for later use. The cell pellets were resuspended in a buffer containing 30 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM imidazole, and 100 μM PMSF. The cells were homogenized and were subjected to three cycles of freeze-thaw. Freezing was done by dipping the 50-ml new centrifugation tube containing not more than 20 ml of suspended cells into the liquid nitrogen for 5 to 7 min (avoid old tube to prevent sudden breakage). Then frozen cells were allowed to thaw at 37°C in water bath till the frozen suspension attained fluidity in buffer (5 to 6 min maximum). The cells suspension was sonified (Branson Ultrasonic Systems) on ice-water mixture at 25% of the amplitude with a pulse (three to four cycles) of 30 s each interspersed with a 1-min interval. The sonified cell lysate was centrifuged at 18,000 rpm for 30 min. The sonified soup treated with 0.1% Triton-X was incubated for 45 min on rocker. The lysate was filtered and clarified lysate was passed through an Ni-NTA column (GE Healthcare) equilibrated with a buffer A (30 mM Tris-HCl [pH 7.5], 150 mM NaCl, 4 mM βME, and 20 mM imidazole). After a wash step with a gradient of imidazole from 20 to 130 mM, bound protein was eluted with buffer B (30 mM Tris-HCl, 150 mM NaCl, 4 mM βME, 4% glycerol, and 400 mM imidazole). The protein fractions were pooled, concentrated (using Amicon Ultracentrifugal filters [Millipore]), and purified further on a Hi-Load Superdex-75 16/60 column (GE Healthcare) equilibrated with buffer G (30 mM Tris-HCl [pH 7.5], 150 mM NaCl, 4 mM βME). The protein fractions were pooled and concentrated to ∼8 mg/ml.
The homogeneity of protein was checked with (12.5%) SDS-PAGE and dynamic light scattering (DLS). The DLS measurements were performed on SpectroSize300 from Nano Biochemistry Technology, Hamburg, Germany. The concentrated protein was used for crystallization, biochemical, and biophysical assays. The point mutant HpDnaG-CTD F534A was also purified as described above.
Overexpression and purification of the selenomethionine-labeled C-terminal domain of primase.
The gene sequence corresponding to the C-terminal domain (amino acid residues 413 to 559) of DnaG primase (HpDnaG-CTD) cloned in pET-21(c) vector as described before (14) was used for the overexpression in E. coli BL21(DE3) cells. Selenomethionine (Se-Met)-labeled HpDnaG-CTD was prepared to provide phases necessary for structure determination (see below). Se-Met-labeled protein was purified under reducing conditions to prevent the formation of a mixture of reduced and oxidized Se-Met-labeled protein. Protein labeling was carried out using media supplied by Molecular Dimensions (United Kingdom) for Athena Enzyme systems. The concentration of selenomethionine was maintained at about 25 mg/liter. Initially, the primary culture was grown in LB medium overnight. The next morning cells were harvested by centrifuging at 3,500 rpm for 8 min. The pellet obtained was resuspended in the complete selenomethionine media, and the process was repeated once again to remove any traces of LB medium. After inoculation of the secondary selenomethionine complete medium, and after about 4 h when the OD600 reached 0.5 at 37°C, cells were induced with 1 mM IPTG and shaken for another 8 h at 30°C. Cells were harvested at 7,000 rpm for 7 min and stored at −80°C for downstream processing. Further methodology of purification was similar to that for the native HpDnaG-CTD, as described above.
Crystallization, data collection, and structure determination.
Purified protein was crystallized using the hanging-drop vapor diffusion method in 24-well Linbro plates against a reservoir solution containing 18% PEG 5000MME, 4 mM βME, and 100 mM Tris-HCl (pH 6.8 to 8.4). Three microliters of (∼5 mg/ml) protein and 3 μl of reservoir solution were mixed and allowed to equilibrate at 16°C. The crystals were flash frozen in a cryoprotectant solution containing 30% PEG 400 mixed with mother liquor. The X-ray data for Se-Met-labeled crystals were collected at the BM14 synchrotron beamline (ESRF, Grenoble, France) at a selenium peak wavelength of 0.97860 Å. The data sets were indexed and scaled using HKL2000 (43). A partial structure was solved using the single wavelength anomalous scattering protocol of Auto-Rickshaw of the EMBL-Hamburg automated crystal structure determination platform (44). The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs of the CCP4 suite (45). Anomalous data were used to calculate FA values using the program SHELXC (46). Based on an initial analysis of the data, the maximum resolution for substructure determination, and initial phase calculation was set to 2.0 Å. All of the four heavy atoms expected (two selenomethionine for each of the two molecules of the asymmetric unit) were found using the program SHELXD (47). The correct hand for the substructure was determined using the programs ABS (48) and SHELXE (49). Initial phases were calculated after density modification using SHELXE. The initial phases were improved using density modification and phase extension to a 1.78-Å resolution using the program RESOLVE (50). Further, the model was built using the autobuild program ARP/wARP (51). The missing regions were built manually using the COOT graphics package, and refinement was carried out with REFMAC5 (52). The structure was further improved after iterative model building and refinement using COOT (53) and REFMAC5 (52), respectively. The final crystal structure was well refined with excellent electron density and validated by PROCHECK (54) of CCP4 suite. The figures were generated using PyMOL (55). The final refinement statistics are shown in Table 1. Coordinate and structure factor files for the crystals of HpDnaG-CTD have been deposited with the Protein Data Bank under accession code 4EHS.
Table 1.
Data collection and refinement statistics of HpDnaG-CTD
| Parameter | Resulta |
|---|---|
| Data collection statistics | |
| X-ray source | BM14, ESRF, France |
| Wavelength (Å) | 0.97860 |
| Space group | P212121 |
| Unit cell dimensions | |
| a, b, c (Å) | 48.9, 61.4, 82.4 |
| α, β, γ (˚) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 50.0–1.78 (1.81–1.78) |
| Rsym or Rmerge (%) | 6.7 (33.2) |
| I/σI | 57.1 (5.05) |
| Completeness (%) | 99.5 (94.20) |
| Redundancy (%) | 12.6 (9.2) |
| Mosaicity | 0.31 |
| Refinement statistics | |
| Resolution range (Å) | 50.0–1.78 |
| No. of unique observations | 24,493 |
| Redundancy (%) | 6.7 (4.9) |
| Avg I/σ(I) | 42.3 (3.8) |
| Rwork/Rfree | 19.5/24.9 |
| No. of atoms | |
| Protein | 1,994 |
| βME/water | 16/212 |
| Avg B factor (Å2) | |
| RMSD | |
| Bond length (Å) | 0.019 |
| Bond angle (°) | 2.184 |
| Ramachandran plot (%) | |
| Most favored regions | 92.3 |
| Additionally allowed regions | 7.2 |
| Generously allowed regions | 0.5 |
| Disallowed regions | 0 |
Values in parentheses are for the highest-resolution shell.
Electrostatic surface charge distribution calculations.
The electrostatic surface charge distribution was calculated using the ABPS (56) plugin in PyMOL. The negative electrostatic surface is shown in red, and the positive surface in shown in blue; all surfaces are drawn at ±3 e/kBT.
Helicase assay.
The double-stranded DNA substrate for the helicase assay was prepared as described previously (35). The helicase assay was carried out in a 20-μl reaction mixture containing 20 mM Tris-HCl (pH 8.0), 8.0 mM dithiothreitol (DTT), 2.5 mM MgCl2, 2.0 mM ATP, 80 μg of bovine serum albumin (BSA)/ml, 10 mM KCl, 4% sucrose, 10 fmol of helicase substrate, and various amounts of wild-type HpDnaB, wild-type HpDnaG, and mutant HpDnaG F534A proteins. The reaction mixture was incubated at 37°C for 30 min. The reaction was stopped by the addition of 5 μl of 5× stop buffer (1.25% SDS, 75 mM EDTA, 25% glycerol) and loaded onto a 10% native PAGE gel to resolve the products. The gel was dried and exposed to X-ray film (Kodak) and developed subsequently.
ATPase assay.
The substrate-dependent ATPase assay was carried out using a coupled pyruvate kinase/lactate dehydrogenase-linked assay with some minor changes as described previously (37, 57). In brief, the substrate-dependent ATPase activities of HpDnaBwt were determined alone or in combination with HpDnaG-CTD or HpDnaG-CTD F534A mutant. The reactions (70 μl) were carried out at 25°C in the reaction buffer containing 20 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 100 mM KCl, 8 mM DTT, 4% sucrose, and 80 μg of BSA/ml. The reaction mixture was further supplemented with 250 mM NADH, 2 mM phosphoenolpyruvate, 2.5 U of pyruvate kinase, 1.6 U of lactate dehydrogenase, and various concentrations of the ATP. The above reaction mixture was incubated for 3 min at room temperature prior to the addition of the respective protein (1.3 μM each). After the addition of the protein, the reaction kinetics were monitored by measuring the decrease in the absorbance at 340 nm continuously for first 3 min by using the DU-800 spectrophotometer (Beckman Coulter). The conversion of ATP to ADP is stoichiometrically coupled to the oxidation of NADH and was calculated using the equation: rate of ATP hydrolysis (mM/time) = Δ OD340/Δ time × 6.22 × cuvette path length. Where cuvette path length is 1 cm, 6.22 is the extinction coefficient for NADH at 340 nM. The rate of ATP hydrolysis against different substrate concentrations (ATP) for the enzymes described above was plotted using GraphPad Prism curve-fitting software.
Surface plasmon resonance (SPR).
To determine the difference in binding affinities of the HpDnaG-CTD and its Phe534Ala mutant, for HpDnaB, Autolab SPR was used at the Advanced Instrumentation Research Facility, Jawaharlal Nehru University, New Delhi, India. Here, the surface (self-assembled monolayer of 11-mercaptoundecanoic acid [MUA] on a gold surface; Autolab) was first activated with N-hydroxysuccinimide (NHS; 0.05 M)/N-ethyl-N-(diethylaminopropyl) and carbodiimide (EDC; 0.2 M). Both HpDnaG-CTD and HpDnaG-CTD F534A mutant were immobilized (separately on two different chips) to the activated sensor surface at a concentration of 50 μg/ml in filtered (0.22-μm pore size) and degassed 10 mM sodium acetate buffer (pH 5.0). The chip has two channels; channel one was used for immobilization of ligand and channel two was used as blank (the signals of the analyte with a ligand-free surface). After ligand immobilization, the surface was blocked with 100 mM ethanolamine at pH 8.5, followed by regeneration using 50 mM NaOH. The running buffer constituents were the same as those recommended for HBS BIAcore running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% P-20 surfactant [pH 7.4]). The association kinetics for HpDnaG-CTD was monitored for 300 s, followed by dissociation for the next 300 s, whereas for the HpDnaG-CTD F534A mutant, association was monitored for 180 s and dissociation was monitored for the next 300 s. HpDnaB samples of various concentrations were prepared in running buffer and injected at the rate of 20 μl/min across the sensor surface. The different protein concentrations of HpDnaB used against HpDnaG-CTD were 500, 250, 125, 75, and 40 nM, whereas for the HpDnaG-CTD F534A mutant, the concentrations of HpDnaB used were 1,000, 750, 500, 250, and 125 nM. Signal changes on the activated/blocked control panel were subtracted from the DnaG-DnaB binding interactions using in-line reference signal, and the subtracted sensorgrams were analyzed. The surface was regenerated with buffer consisting of a two pulses (manually delivered) of 75 mM NaOH. All of the data were recorded at 25°C. The data analysis was performed using Autolab SPR Kinetic Evaluation software.
Construction of the helicase-primase complex model in H. pylori.
The structure of the N-terminal domain of HpDnaB (HpDnaB-NTD), involved in primase binding, has been reported earlier by our group (22). The current work reveals the structure of the C-terminal domain of HpDnaG, which interacts with the HpDnaB-NTD. These two structures were used to model the HpDnaB-HpDnaG (helicase-primase) interaction site in H. pylori using the structure of the BstDnaB-BstDnaG-CTD (helicase-primase) complex as a template (19). The HpDnaB-NTD was superimposed on the BstDnaB-NTD domain and HpDnaG-CTD GR was superimposed on the BstDnaG-CTD GR of the complex. In order to superimpose the CTD helical hairpin structures, the helical hairpin of HpDnaG-CTD was rotated by hand ∼55°C, keeping the linker between helical hairpin and the lobe as the center of rotation, to obtain a least-root-mean-square deviation (RMSD) between BstDnaG-CTD and HpDnaG-CTD structures. This modification maximizes interactions between primase and helicase. The structure of the modeled complex was relaxed to eliminate bad contacts using molecular energy minimization in the AMBER molecular dynamics package (58).
Molecular dynamics simulation for binding affinity estimation.
Molecular dynamics simulations were performed on the models of the HpDnaB-DnaG complex and of the HpDnaB-DnaG-CTD F534A mutant complex using the AMBER 9.0 package (58). To create the in silico mutant, phenylalanine-534 was changed to alanine using Chimera (59). The input files for both systems were prepared using the tleap program from the AMBER suite and the AMBER99SB force field (60). Prior to the energy minimization and dynamics runs, the models were neutralized by the addition of Na+ counter ions and solvated with waters in a TIP3P water box up to 12 Å from the surface of the protein. First, the solvated complex was subjected to short minimizations using 500 steps of steepest descent, followed by 1,000 steps of conjugate gradient. The system was equilibrated by first heating it for 50 ps, while keeping all of the atoms of the protein complex fixed with a 50-kcal/mol harmonic potential. This calculation was followed by 50 ps of density equilibration with weak restraints on the HpDnaB-DnaG solvated complex and 500 ps of constant pressure equilibration at 300K. A final 2-ns production run was then performed. Long-range coulombic interactions were handled using the particle mesh Ewald summation. Both of the simulations shared common parameters, including the SHAKE algorithm (61), to constrain the bonds to hydrogen atoms with default parameters and an 8-Å cutoff. The trajectories of the complex structure were written out every 10 ps. The binding free energies were calculated by using the MM/GBSA (62) method as implemented in AMBER 11 (58). The trajectories were analyzed using the Ptraj module of the AMBER suite.
RESULTS
HpDnaG-CTD exists as a dimer in solution.
The gel filtration chromatography profile of the HpDnaG C-terminal domain (HpDnaG-CTD) on a calibrated Superdex G75 16/60 column showed a major peak at 68.3 ml (Fig. 1A). The low-molecular-mass gel filtration calibration kit containing five proteins in the range of 14.4 to 66 kDa and Blue Dextran 2000 were subjected to gel filtration chromatography under the same experimental conditions, and a standard curve was plotted using the log molecular mass of the proteins against the elution profile (Fig. 1B). The molecular mass of the HpDnaG-CTD was found to be ∼34 kDa. These results indicate that HpDnaG-CTD exists in a dimeric state in solution, since the molecular mass of the monomeric form is ∼17 kDa. The dynamic light scattering (DLS) measurements also validates the dimeric state of the HpDnaG-CTD (see Fig. S1 in the supplemental material). The proteins obtained from the peak fractions were subjected to SDS-PAGE analysis, followed by Coomassie blue staining (Fig. 1C), which showed the protein to be >95% pure.
Fig 1.

(A) HpDnaG-CTD is a dimer in solution. Size-exclusion chromatography of the C-terminal domain of primase (HpDnaG-CTD) passed through a HiLoad 16/60 Superdex 75 column, and 1-ml fractions were collected. The elution volume (68.3 ml) and the elution pattern of the protein are displayed. (B) The molecular mass of the eluted DnaG-CTD, deduced from standard plot, is about ∼34 kDa, and corresponds to the dimeric state of the protein. (C) SDS-PAGE showing fractions purified by gel filtration. The proteins are separated on SDS–12.5% PAGE and stained with Coomassie brilliant blue. Lane M shows the molecular markers; lanes P1 and P2 are gel filtration fractions.
HpDnaG-CTD structure determination.
Molecular replacement with BstDnaG-CTD and EcDnaG-CTD failed to give any solution, likely due to the low homology and structural similarity between these structures. Se-Met-labeled protein crystals were used to get anomalous data and experimental phases. The best crystals of the Se-Met-labeled protein were obtained at pH 7.4 in about 28 days at 16°C. The crystals diffracted X-rays to 1.78 Å at BM14 of ESRF. The crystals belonged to the P212121 space group, with two molecules per asymmetric unit, and the solvent content was found to be relatively low at 32%. After the initial phases were improved upon density modification and phase extension up to the resolution 1.78 Å using the program RESOLVE (50), almost 80% of the model was built into the electron density with the help of the autobuild program ARP/wARP (51), yielding an Rfactor of 23% and an Rfree of 28%. The remaining structure was built manually. Initially, water molecules were picked up by ARP/WARP and later checked manually. Refinement was carried out with REFMAC5 (52), and the structure was improved with iterative model building using the COOT graphics package (53). The final model has an Rfactor of 19.5% and an Rfree of 24.9%, with a very good electron density (Fig. 2A). In an asymmetric unit two molecules of HpDnaG-CTD are arranged in a “head-to-toe” manner relative to one another, and a few van der Waals interactions between the monomers stabilize the dimer (Fig. 2B and C). Electron density corresponding to the N-terminal residues (413–437) of this domain was not visible in either monomer, indicating that this region is disordered. Two Cys residues (483 and 540), in both monomers, were found to form disulfide bonds with βME, which was added to prevent the oxidation of Se-Met-labeled protein during crystallization. In an asymmetric unit there are total of 245 residues, 4 βME, and 201 water molecules. The Ramachandran statistics are fine with 92.3% in the favored region, 7.2% in the additionally allowed region, 0.5% in the generously allowed region, and no residues in a disallowed region.
Fig 2.
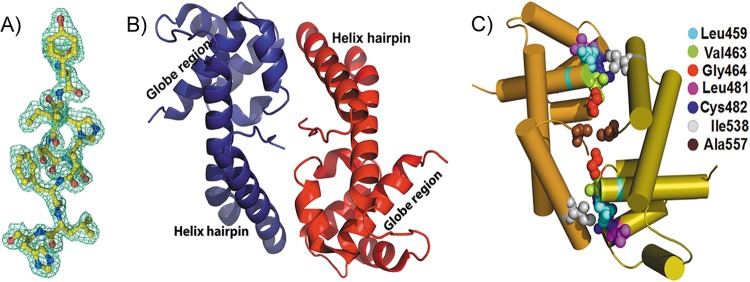
Crystal structure of HpDnaG-CTD. (A) Portion of the molecule shown with a 2Fo-Fc electron density map at 1σ calculated at a 1.8-Å resolution. (B) Ribbon diagram showing the crystal structure of the HpDnaG primase-CTD. The protein consists of alpha helices connected by loops. The overall structure has one globe/core region connected to the helical hairpin. There are two molecules in an asymmetric unit forming a dimer in a “head-to-toe” fashion. (C) Diagram showing the crucial interacting hydrophobic residues between the two monomers of HpDnaG-CTD in the crystal structure, making the dimeric interface.
Comparison of HpDnaG-CTD with other structures.
The HpDnaG-CTD (1.78-Å) structure has significantly higher resolution compared to the other two crystal structures determined for E. coli (2.8 Å) and B. stearothermophilus (2.9 Å). As indicated above, the crystal structure that we determined contains two molecules of HpDnaG-CTD in an asymmetric unit. The HpDnaG-CTD structure consists primarily of alpha helices and contains no beta sheets. The structure is formed by six helices, where the first four helices form the core/globe region (GR) and the last two helices form the helical hairpin region (HHR). The hairpin size may be species dependent which in turn makes structural overlay of this region less meaningful. The structural superposition of HpDnaG-CTD with the two other available DnaG-CTD structures of E. coli and B. stearothermophilus was done with the help of LSQMAN (63) to look for the differences. The GR of HpDnaG-CTD was superposed with the GR of BstDnaG-CTD (crystal structure, PDB ID 2R6A [22], and nuclear magnetic resonance [NMR] structure, PDB ID 1Z8S [13]) and the GR of EcDnaG-CTD (PDB ID 1T3W [10]) (Fig. 3). The RMSDs with respect to BstDnaG-CTD crystal, BstDnaG-CTD NMR, and EcDnaG-CTD crystal were found to be 1.771 Å (57 Cα atoms), 1.741 Å (57 Cα atoms), and 1.425 Å (37 Cα atoms of EcDnaG-CTD), respectively. The total structural alignment of HpDnaG-CTD using RAPIDO (64) yielded RMSD of 8.77Å (for 106 Cα atoms), 8.81Å (for 98 Cα atoms), and 15.69Å (for 66 Cα atoms), respectively (see Table ST2 in the supplemental material). The extent of superposition suggests that the GR of HpDnaG-CTD is structurally more similar to BstDnaG-CTD than to that of EcDnaG-CTD. Overall, the arrangement of helices in HpDnaG-CTD is similar to that in BstDnaG-CTD, with a core or GR connected to a HHR by a flexible linker. The number of alpha helices present in each structure also differs in these CTDs: the HpDnaG-CTD has 6 helices, and the EcDnaG-CTD has 7 helices, whereas BstDnaG-CTD has 8 helices (see Fig. S2 and S3 in the supplemental material). Comparison of the EcDnaG-CTD and HpDnaG-CTD structures shows the large deviation not only in the core or the GR but also in the HHR. In fact, it is the HHR that gives rise to the large structural variability. The HHR can adopt multiple orientations relative to the GR. The flexibility is also evident from the relatively high B-factor in the linker and HHR with respect to the GR (see Table ST3 in the supplemental material). The helical hairpin is very short in EcDnaG-CTD compared to other structures, but the length of the connecting helix between the globe and the first helix of hairpin in EcDnaG-CTD is very long (Fig. 3). Interestingly, multiple sequence alignment (MSA) of amino acid sequences and structural alignment did not yield any significant homology (see Fig. S2 and S4 in the supplemental material): although there are few homologous residues in all three sequences or structure belonging to the core of GR region or stabilizing the helical hairpin structure, but there is no conservation on the helicase binding surface.
Fig 3.

Superposition of the HpDnaG-CTD crystal structure with the crystal structure of EcDnaG-CTD and BstDnaG-CTD (solution and crystal) structures. Because of its flexibility, the helical hairpin region (HHR) may have different orientations when in a free or bounded state. The figure shows how the globe region (GR) of H. pylori, when superposed, gives insight into the differences in the shapes and orientations of HHR. The EcDnaG-CTD HHR is small and not flexible, but BstDnaG-CTD HHR is flexible. (A) Superposition of HpDnaG-CTD crystal structure (green) with EcDnaG-CTD (yellow). (B) Superposition of HpDnaG-CTD crystal structure (green) with BstDnaG-CTD crystal structure (cyan) shows the difference of about 60° in hairpin orientations. The angle is measured between Phe534, Arg522 of HpDnaG-CTD, and Arg574 of BstDnaG-CTD. (C) Superposition of HpDnaG-CTD crystal structure (green) with BstDnaG-CTD solution or NMR model (blue) highlights the flexibility of hairpin as seen by huge angular difference of about 130° in opposite side of helicase binding axis. (D) Green helical hairpin, the native HpDnaG-CTD crystal structure was rotated by about 55°. It is shown in cyan to model the bound state in the model (as shown in Fig. 4 in red). Owing to the flexibility of HHR, the large rotation of helical hairpin of HpDnaG-CTD was performed to model the complex with BstDnaB-BstDnaG-CTD (helicase-primase) complex as a template.
Importance of Phe534 for the stability of the H. pylori helicase-primase complex.
The observation that the orientation of HHR with respect to the GR varies considerably in different species suggests HHR to be flexible. This can be seen clearly in Fig. 3A, B, and C when HpDnaG-CTD's GR is superposed over GRs from EcDnaG-CTD (crystal structure), BstDnaG-CTD (crystal structure), and BstDnaG-CTD nuclear magnetic resonance (NMR) structures, respectively. The helical hairpin in HpDnaG-CTD shows the difference of 60° away from the bound form of crystal structure of BstDnaG-CTD (Fig. 3B), whereas the difference in orientation of the BstDnaG-CTD NMR model helical hairpin orientation is visible by about 130° away from the primase-binding surface of helicase (Fig. 3C); such flexibility in turn may allow the HHR to adopt a different orientation when bound to helicase than when not bound, which in turn may be important for the capacity of primase to interact with helicase during DNA replication. The HHR plays a very important role in the binding of primase with helicase at the fork, during DNA replication initiation. Previous NMR structural studies have proposed involvement of hydrophobic residues of the HHR in the interaction of DnaG-CTD with DnaB-NTD (13). Owing to the flexibility of HHR, the large rotation of this region in HpDnaG-CTD was done to model the complex (Fig. 3D) using BstDnaB/DnaG-CTD (helicase-primase) complex structure as a template. The green helical hairpin, which is native crystal structure, was rotated by about 55° and is shown in cyan to model the bound state. From our model (Fig. 4A), we identified a nonconserved large hydrophobic residue, Phe534, which is the only prominent surface-accessible hydrophobic residue of HpDnaG-CTD. Phe534 was found to stabilize the complex at a very crucial point of the helical hairpin loop, such that the whole hairpin is getting “hooked” by this residue. Small but insignificant sequence and structural homology (see Fig. S2 and S4 in the supplemental material) of HpDnaG-CTD with E. coli and B. stearothermophilus DnaG-CTDs confirms that Phe534 is not a conserved residue and also does not overlap with the stretch previously studied in E. coli and B. stearothermophilus (13, 65). The destabilization of the helicase-primase (HpDnaB-NTD/HpDnaG-CTD) complex model was predicted by molecular dynamics both in pre-equilibrium trajectories (Fig. 4B) and by the calculation of binding free energies of the native helicase-primase (NTD-HpDnaB/HpDnaG-CTD) and native helicase-mutant primase (HpDnaB-NTD/HpDnaG-CTD F534A) complexes. Using the MM/GBSA method as implemented in AMBER (see Materials and Methods), the calculated (in silico) free energy of binding between helicase (HpDnaB-NTD) and native primase (HpDnaG-CTD) is −9.2 kcal/mol and that between helicase (HpDnaB-NTD) and mutant primase (HpDnaG-CTD F534A) is −2.9 kcal/mol (see Table ST4 in the supplemental material). Interestingly, the structural alignment with EcDnaG-CTD and BstDnaG-CTD has shown that Phe534 is not a conserved residue (see Fig. S4 in the supplemental material). The binding affinities for both native and mutant HpDnaG-CTD in complex with HpDnaB were also measured in vitro, using surface plasmon resonance (SPR). Both native and mutant HpDnaG-CTDs were immobilized in equal amounts on two different chips under identical conditions as described in Materials and Methods. The wild-type HpDnaB was passed as an analyte with HBS buffer over both of the immobilized proteins. At first, when HpDnaB was passed at the same concentration over the two chips, there appeared to be a significant difference in response units (RU) with respect to both native and mutant HpDnaG-CTDs (Fig. 5A). After the initial results, different concentrations of HpDnaB were passed over the chips. The outcome of the SPR shows that the mutation of phenylalanine 534 to alanine causes an ∼16-fold decrease in the binding affinity; the Kd of the native protein (HpDnaG-CTD) is ∼32 nM, whereas that of the mutant protein (HpDnaG-CTD F534A) is ∼520 nM (Fig. 5B and C). Although the difference in binding affinity to the native (HpDnaG-CTD) and mutated (HpDnaG-CTD F534A) proteins is relatively large considering that only a single residue was mutated, there is still appreciable affinity between the helicase and the mutant primase. Perhaps other residues besides Phe534 also play a role in the interaction (see the Discussion). Nevertheless, it is usually the case that no more than 5% of the interface residues in a protein-protein interaction are important for binding (66). According to both in silico calculations and our in vitro experiment (SPR), Phe534 of primase (HpDnaG) helps to stabilize the helicase-primase (HpDnaB/HpDnaG) complex in H. pylori.
Fig 4.

In silico investigations of the H. pylori helicase-primase complex. (A) Model of the H. pylori helicase-primase complex. Experimentally determined structures of HpDnaB-NTD and HpDnaG-CTD, as well as that of the BstDnaB-BstDnaG-CTD complex, were used to generate the model of the complex. The model was further relaxed to eliminate bad contacts using molecular energy minimization with the help of the AMBER molecular dynamics package. The inset shows the crucial residues of the 9helical hairpin in contact with the primase. (B) RMSD analysis of the trajectories of native and mutant primase models. Time-dependent Cα-RMSD for all residues of the native (blue) and mutant (red) complex, indicating both mutant and native protein structures converged after 2-ns simulations.
Fig 5.
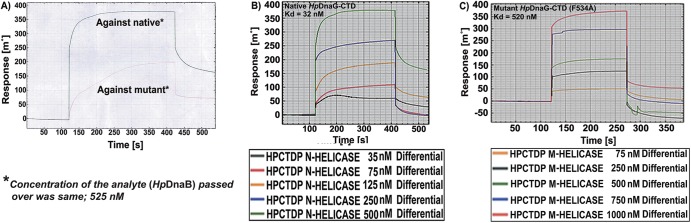
SPR sensogram. (A) Sensogram showing the difference in level of interaction of analyte (HpDnaB helicase) with HpDnaG-CTD native and mutant. The amounts of analyte passed in both cases were the same: 525 nM. (B and C) Sensorgram showing the different binding affinities of native HpDnaG-CTD and mutant HpDnaG-CTD for HpDnaB helicase. The legend shows the concentration of analyte (DnaB helicase used). The mutation of a single residue, Phe534 to Ala, on the helicase-binding surface resulted in a 16-fold decrease in the measured binding affinity.
Importance of Phe534 for the function of the H. pylori helicase-primase complex.
In addition to determining the effect of the Phe534Ala mutation on the binding affinity between the helicase (HpDnaB) and primase (HpDnaG), we also set out to determine whether the mutation also affects the functions of the proteins, in particular the helicase, ATPase, and DNA binding activities of HpDnaB. In addition to interacting with each other, helicases (DnaB) and primases (DnaG) also stimulate each other's activity. Primases stimulate NTPase, helicase, and (to some extent) DNA binding activities of helicase, while helicases regulate primase initiation activity and also the length of primers synthesized (14, 16, 17, 36, 67). Although, as described above, the stability of the complex varies across different species, the underlying biochemistry of the complexes appears to be same (22, 68). It has been shown earlier that the N terminus of HpDnaB interacts with the C terminus of HpDnaG by SPR and helicase assay (14). Finding the key residues for interaction between the C terminus of HpDnaG and the N terminus of HpDnaB is critical to understanding the formation of the complex. According to the above model, Phe534 at the C terminus of HpDnaG may be crucial for the protein-protein interaction (Fig. 6A). If such is the case, mutation of Phe534 would be expected to compromise the stimulation of helicase activity of HpDnaB by HpDnaG (primase). To test this hypothesis, helicase activities were measured. First, the mutant protein HpDnaG F534A was purified by gel filtration chromatography, along with the wild-type HpDnaG protein (Fig. 6B). After protein purification, a helicase assay was performed according to the protocol described in Materials and Methods. The results indicate that under otherwise similar conditions, stimulation of helicase activity is decreased by >50% in the presence of mutant HpDnaG F534A protein compared to the wild-type protein (Fig. 6C and D). This significant reduction in the stimulation of helicase (HpDnaB) activity by the mutated primase (HpDnaG) provides further strengthens the evidence for the involvement of Phe534 in the interaction between HpDnaB and HpDnaG.
Fig 6.

Purification of HpDnaG F534A mutant and its effect on HpDnaB wild-type helicase activity. (A) Schematic domain organization and site of point mutation in HpDnaG. (B) A gel filtration chromatogram of HpDnaG F534A mutant, as obtained by passing through Hi-Load Superdex-120 ml 16/60 column. Inset panel a shows the elution pattern from the gel filtration of HpDnaG F534A mutant, and inset panel b shows the results for HpDnaGwt (lane 2) and HpDnaG F534A mutant (lane 3) on an SDS-PAGE gel, along with that of the protein marker (lane 1). (C) Modulation of HpDnaB helicase activity in the presence of HpDnaGwt and HpDnaG F534A mutant. Different concentrations of HpDnaGwt and HpDnaG F534A mutant proteins were added in the HpDnaB helicase reaction as shown. The positions of the annealed probe and unwound oligonucleotides are shown in figure. The results indicate that the stimulation of HpDnaB helicase activity is reduced by ∼50% in the presence of the mutant protein compared to the HpDnaGwt protein. (D) The results of a quantitative analysis of the data from the gel in panel C are shown in the graph.
One of the hallmarks of DnaB helicases is the ATPase activity which is essential for its helicase activity. It will be interesting to see the effect of HpDnaG-CTD wild-type and mutant (F534A) proteins on the ATPase activity of HpDnaB. For this purpose, we performed NADH-LDH coupled ATPase assay in the presence of HpDnaB alone or in the combination with HpDnaG-CTD or HpDnaG-CTD F534A mutant protein. We found that both proteins (wild type and mutant) stimulated the ATPase activity of HpDnaB. However, the HpDnaG-CTD F534A mutant showed a slower kinetics ATP hydrolysis rate (∼30%) than the wild-type protein under our experimental conditions (Fig. 7B). The reduced helicase activity of HpDnaB in the presence of mutant protein may be attributed to the reduced ATPase activity of HpDnaB.
Fig 7.

Purification of HpDnaG-CTD F534A mutant and its effect on HpDnaB (wild type) ATPase activity. (A) Gel filtration chromatogram of HpDnaG-CTD F534A (mutant) obtained by passage through a Superdex-200 10/300 GL (GE Healthcare) column. Inset panel a shows the induction profile of HpDnaG-CTD F534A (indicated by an asterisk), along with marker (M), and inset panel b shows a Coomassie blue-stained gel corresponding to the peak fractions (15.85 ml) collected after gel filtration of HpDnaG-CTD F534A. (B) Stimulation of HpDnaB ATPase activity in the presence of HpDnaG-CTD or HpDnaG-CTD F534A mutant protein. ATPase assays were performed as described previously in Materials and Methods. ATP hydrolysis rates were calculated for the HpDnaB alone or in combination with HpDnaG-CTD or HpDnaG-CTD F534A mutant protein (each 1.3 μM). ATP hydrolysis rates were plotted against substrate concentration (ATP) as shown in figure. Alone HpDnaG-CTD (wild type or mutant protein) did not show any ATPase activity (data not shown) but both stimulated HpDnaB ATPase activity. However, the presence of HpDnaG-CTD F534A mutant protein showed overall decrease (∼30%) in ATP hydrolysis rate than HpDnaG-CTD. The Vmax and Km of each reaction are shown at the bottom.
Recently, we have shown that the C terminus of HpDnaG profoundly stimulates the DNA binding activity of HpDnaB, possibly by unmasking the DNA binding region of HpDnaB (36). This result prompted us to evaluate the effect of the HpDnaG-F534A mutant on the stimulation of DNA-binding activity of HpDnaB. For this purpose, we first introduced the Phe534Ala mutation in HpDnaG-CTD, followed by expression and purification of the protein using Ni-NTA and followed by gel filtration chromatography (Fig. 7A). Further, we performed an electrophoretic mobility shift assay using HpDnaB and various concentrations of native HpDnaG-CTD and the mutant HpDnaG-CTD F534A proteins. Although the presence of either the native or the mutant form of HpDnaG-CTD stimulated the DNA-binding activity of HpDnaB, the mutant protein showed only a marginal reduction in the stimulation of DNA-binding activity (∼10 to 15%) compared to the wild-type HpDnaG-CTD under our experimental conditions (data not shown). It is not clear at this point why the effect of mutation is marginal on the stimulation of HpDnaB DNA-binding activity, whereas the mutant shows a significant reduction in the helicase activity. The results presented above (SPR, helicase assay, ATPase assay, and the DNA binding assay) clearly show the importance of Phe534 in the stability of HpDnaB-HpDnaG (helicase-primase) complex and hence validates our model for this complex.
DISCUSSION
Previous studies conducted by others and our group have shown the central role of DnaG primase C-terminal domain (CTD) in the interaction with the N-terminal domain (NTD) of helicase (10, 14). Interestingly, primase CTD shows more diversity than its interacting partner, NTD helicase. The diversity occurs more at the primary sequence level than at the tertiary level. Nevertheless, the three-dimensional structures were not similar enough for us to determine the structure of HpDnaG-CTD by molecular replacement. Just as for the previous two structures of DnaG-CTD, this structure was also solved by experimental phasing. The multiple sequence alignment (MSA) did not offer any clue to the overall three-dimensional structural similarity. In fact, after solving the structure of HpDnaG-CTD, the three-dimensional structural alignment with EcDnaG-CTD and BstDnaG-CTD shows some conservation of the folds; this conservation occurs primarily in the globular region (GR), which seems to be less diverse than the helical hairpin region (HHR). In fact, it is the hairpin region that has the major role in the interaction with DnaB-NTD (helicase), and it shows a high degree of diversity at the primary, secondary, and tertiary levels. Interestingly, the Dali server (69) shows that HpDnaG-CTD has more structural similarity with DnaB-NTD rather than the BstDnaG-CTD and EcDnaG-CTD structures. The structure-based alignment rarely shows the alpha-helical boundaries to be actually aligned (see Fig. S2 in the supplemental material).
The EcDnaG-CTD and HpDnaG-CTD crystal structures determined thus far show that they exist as dimers but, interestingly, the dimerization surface and mode of dimerization is different. This dimerization of primase CTD may simply be a concentration-dependent dimerization and may not be biologically significant since full-length primase is generally seen as a monomer. However, the dimerization could be due to the flexibility of hairpin, since the crystal structures reported thus far has different dimeric interfaces and dimerization pattern. Moreover, the dimerization surface and helicase binding surface are on different sides of the protein (see Fig. S2 in the supplemental material). In the dimeric state, the helicase binding surface is not in a proper conformation to bind to helicase and may play some role in preventing nonspecific activity at the replication fork when not bound to helicase or may help to bind with some other proteins such as single-stranded DNA-binding protein (SSBs) (10).
The H. pylori helicase-primase complex (Fig. 4A) was modeled using the H. pylori crystal structures of HpDnaG-CTD (primase, reported here) and HpDnaB-NTD (helicase) solved by us previously (14) and using the cocrystal structure of BstDnaB-BstDnaG-CTD as a template (22). The analysis of the interacting residues from the HpDnaB-HpDnaG (helicase-primase) complex model revealed a hydrophobic interaction between phenylalanine 534 in the helical hairpin region of the HpDnaG-CTD (primase) and Leu82 and Phe86 of HpDnaB-NTD (helicase). In the HpDnaG (primase) monomer, Phe534 is solvent accessible; burial of this apolar residue upon association with HpDnaB (helicase) likely plays an important role in the stability of the complex. This point is supported by molecular-dynamics (MD) calculations, which revealed that the HpDnaB-HpDnaG (helicase-primase) complex would get destabilized upon mutation of Phe534 to alanine. The destabilization was indicated by MD both in pre-equilibrium trajectories (Fig. 4B) and by the calculation of binding free energies of the native HpDnaB-NTD/HpDnaG-CTD (helicase-primase) and HpDnaB-NTD/HpDnaG-CTD F534A (helicase-mutant primase) complexes (see Table ST2 in the supplemental material). The energy difference was enough to sense the instability of the HpDnaB-HpDnaG (helicase-primase) complex of the mutant with respect to the native protein. These binding affinity calculations were then tested experimentally by expressing a Phe534Ala mutant of HpDnaG-CTD and measuring its affinity for helicase by SPR. According to this analysis, the affinity of this mutant HpDnaG-CTD for HpDnaB (helicase) is 16 times less than the affinity of the native HpDnaG-CTD for HpDnaB (helicase). Taken together, these findings helped us to identify Phe534 as one of the key primase residues that may be involved in the interaction between primase and helicase in H. pylori. The importance of this phenylalanine may be specific to H. pylori since this residue is not conserved.
Helicase and primase are known to coregulate each other's activities at the replication fork (14, 16, 17, 36, 67). A helicase assay, carried out to check for the effect of the Phe534Ala mutation of HpDnaG (primase) on the DNA unwinding ability of HpDnaB (helicase), gave a very strong indication of Phe534 being an important residue in the helicase-primase interactions in H. pylori. A large difference between native and mutant HpDnaG (primases) on their stimulation of HpDnaB's (helicase) DNA unwinding was seen. Interestingly, the effect of this mutation on ssDNA-binding activity of HpDnaB was not that significant. The dynamics of the interaction between the helicase and primase may be different at the replication fork rather than on the ssDNA itself. However, we noticed a moderate decrease (∼30%) in the stimulation of ATP hydrolysis rate of HpDnaB in the presence of HpDnaG-CTD F534A (mutant) compared to the native HpDnaG-CTD (Fig. 7). This may explain the difference in the stimulation of helicase activity and DNA-binding activity of HpDnaB in the presence of wild-type and mutant HpDnaG. Overall, these studies validated our model of the HpDnaB-HpDnaG (helicase-primase) complex. Moreover, our analysis based on helicase and primase surface electrostatic potential from E. coli, B. stearothermophilus, and H. pylori crystal structures (Fig. 8) has showed that the helicase and primase interaction (complex formation) in eubacteria may not be because of hydrophobic residues alone; charged residues too have a role to play. The pattern of charge distribution on the surface of helicase and primase from B. stearothermophilus is consistent with strong interactions between these molecules, whereas the presence of negatively charged surfaces (i.e., like charged surfaces) for both helicase and primase of E. coli helps explain their transient association. In the case of H. pylori, the helicase and primase surfaces are almost complementary with respect to their surface charge potentials, which contribute to the good binding affinity. This favorable charge interaction likely contributes to the capacity of the H. pylori complex to remain intact, albeit in a destabilized state, when primase residue 534 is mutated from Phe to Ala as described above. Based on the available structural data, including the present study, we can suggest a possible mechanism to explain regulation of helicase-primase activity at the replication fork. DnaG-CTD forms a dimer not only in H. pylori as reported here but also in E. coli, although weakly (10). The difference in the reported conformations of HHR, including the NMR structure of BstDnaG-CTD (13) and its complex with BstDnaB (helicase-primase crystal structure) (22), is indicative of a flexible HHR. In the case of HpDnaG-CTD and EcDnaG-CTD, the surfaces forming the (homo) dimer and the complex with DnaB (helicase) are different. Nevertheless, the interaction of primase subunits in trans when bound to helicase has been discussed in the literature (19). When the CTD of primase is not bound to the NTD of helicase, the helical hairpin may fold back to interfere with the RNA polymerase/catalytic domain or the Zn binding domain. This may, in turn, prevent nonspecific interaction with the ssDNA for primer synthesis. Based on our model, when DnaG-CTD comes into contact with helicase, the otherwise bent helical hairpin becomes straight for proper binding to helicase. This straightening of the helical hairpin of CTD away from the RNA polymerase domain or Zn binding domain may lead to the activation of primase by coming in contact with helicase.
Fig 8.

Electrostatic potential surface charge distribution on the primase and helicase interacting surfaces. (a to c′) Electrostatic potential surfaces of NTDs of helicase and CTDs of primase for EcDnaB (a) and EcDnaG (a′), BstDnaB (b) and BstDnaG (b′), and HpDnaB (c) and HpDnaG (c′), respectively. The NTD dimer of the BstDnaB-BstDnaG-CTD (helicase-primase) complex structure was used as a template to generate the model for the respective dimers of other helicases. Negative, neutral, and positive electrostatic potentials are displayed in the figure in red, white, and blue, respectively. The electrostatic surface charge distribution was calculated using ABPS plugin in PyMOL. The negative electrostatic surface is shown in red, and the positive surface is shown in blue; surfaces are drawn at ±3 e/kBT.
The recent work (38) on HpDnaB using electron microscopic and biophysical studies have shown interesting results about the dodecameric state, while our previous studies indicated HpDnaB to be a hexamer (35, 37) (see Fig. S5 in the supplemental material). In the dodecameric state, hexametric HpDnaB dimerizes through the N-terminal domains and in this conformation the primase binding surface may not be accessible (38). Although the helicase assay, ATPase assay and the SPR studies (see Results) clearly shows that HpDanG indeed interacts with HpDnaB in solution. Previously we have already shown that HpDnaB-NTD interacts with HpDnaG-CTD (14). In addition to this, we have successfully validated our proposed model of HpDnaB-HpDnaG complex experimentally, which clearly highlighted the interacting surfaces involved in the complex formation and the residues involved. It is plausible that the hexameric and dodecameric species of HpDnaB are in equilibrium and that the techniques used to measure this are not sensitive enough to look into the dynamics; moreover, these measurements may also have some inherent limitations (22, 38, 70). Perhaps the HpDnaG competes for the binding with HpDnaB. In vivo, at the replication fork after loading, dodecamer splits into two hexamers, and both hexamers travel in opposite directions on separate strands. Here, the hexamer must be binding to HpDnaG. Once the primase (HpDnaG) binds to the helicase (HpDnaB), its helicase activity gets activated and unwinding starts. The binding of DnaG-CTD on the DnaB-NTD leads to the unmasking of the strong DNA binding motif and hence stronger binding is there; however, soon after the displacement of the primase, the helicase goes back to the weak binding state and hence moves over to the next point on the fork (36). The below-optimal level of activities of unwinding of DNA duplex, movement of helicase, length of primer synthesized, and loop formation will in turn slow down the replication fork progression. The interaction pattern of primase and helicase seems to be unique (10, 14, 17) for an organism and therefore any change may affect not only the replication initiation but overall DNA replication.
Supplementary Material
ACKNOWLEDGMENTS
S.G. acknowledges Indo-German grant from DBT, DST-PURSE, and UGC RNW. S.K.D. acknowledges a Swarnajayanti Fellowship (Department of Science and Technology, Government of India), University Grant Commission (SAP), Indian Council for Medical Research (core grant), and DST-PURSE program for support. S.A.A.R. acknowledges CSIR, and V.V. acknowledges ICMR for a fellowship. We thank AIRF (Advance Instrumentation Research Facility) and JNU for providing the SPR facility.
Footnotes
Published ahead of print 12 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00091-13.
REFERENCES
- 1. Jacob F, Brenner S, Cuzin F. 1963. On the regulation of DNA replication in bacteria. Cold Spring Harbor Symp. Quant. Biol. 28:329–348 [Google Scholar]
- 2. Messer W. 2002. The bacterial replication initiator DnaA: DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol. Rev. 26:355–374 [DOI] [PubMed] [Google Scholar]
- 3. Davey MJ, Jeruzalmi D, Kuriyan J, O'Donnell M. 2002. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell. Biol. 3:826–835 [DOI] [PubMed] [Google Scholar]
- 4. Kunkel TA, Bebenek K. 2000. DNA replication fidelity. Annu. Rev. Biochem. 69:497–529 [DOI] [PubMed] [Google Scholar]
- 5. Marians KJ. 1992. Prokaryotic DNA replication. Annu. Rev. Biochem. 61:673–719 [DOI] [PubMed] [Google Scholar]
- 6. McHenry CS. 1988. The asymmetric dimeric polymerase hypothesis: a progress report. Biochim. Biophys. Acta 951:240–248 [DOI] [PubMed] [Google Scholar]
- 7. McHenry CS. 1991. DNA polymerase III holoenzyme: components, structure, and mechanism of a true replicative complex. J. Biol. Chem. 266:19127–19130 [PubMed] [Google Scholar]
- 8. Yoshida K, Inoue I. 2004. Peptide binding to Geminin and inhibitory for DNA replication. Biochem. Biophys. Res. Commun. 317:218–222 [DOI] [PubMed] [Google Scholar]
- 9. Tougu K, Peng H, Marians KJ. 1994. Identification of a domain of Escherichia coli primase required for functional interaction with the DnaB helicase at the replication fork. J. Biol. Chem. 269:4675–4682 [PubMed] [Google Scholar]
- 10. Oakley AJ, Loscha KV, Schaeffer PM, Liepinsh E, Pintacuda G, Wilce MC, Otting G, Dixon NE. 2005. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 280:11495–11504 [DOI] [PubMed] [Google Scholar]
- 11. Frick DN, Richardson CC. 2001. DNA primases. Annu. Rev. Biochem. 70:39–80 [DOI] [PubMed] [Google Scholar]
- 12. Kornberg A, Baker TA. 1992. DNA replication. W.H. Freeman, New York, NY [Google Scholar]
- 13. Syson K, Thirlway J, Hounslow AM, Soultanas P, Waltho JP. 2005. Solution structure of the helicase-interaction domain of the primase DnaG: a model for helicase activation. Structure 13:609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kashav T, Nitharwal R, Abdulrehman SA, Gabdoulkhakov A, Saenger W, Dhar SK, Gourinath S. 2009. Three-dimensional structure of N-terminal domain of DnaB helicase and helicase-primase interactions in Helicobacter pylori. PLoS One 4:e7515. 10.1371/journal.pone.0007515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu YB, Ratnakar PV, Mohanty BK, Bastia D. 1996. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc. Natl. Acad. Sci. U. S. A. 93:12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mitkova AV, Khopde SM, Biswas SB. 2003. Mechanism and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 278:52253–52261 [DOI] [PubMed] [Google Scholar]
- 17. Bird LE, Pan H, Soultanas P, Wigley DB. 2000. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry 39:171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pan H, Wigley DB. 2000. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure 8:231–239 [DOI] [PubMed] [Google Scholar]
- 19. Corn JE, Pease PJ, Hura GL, Berger JM. 2005. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell 20:391–401 [DOI] [PubMed] [Google Scholar]
- 20. Keck JL, Roche DD, Lynch AS, Berger JM. 2000. Structure of the RNA polymerase domain of Escherichia coli primase. Science 287:2482–2486 [DOI] [PubMed] [Google Scholar]
- 21. Corn JE, Pelton JG, Berger JM. 2008. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat. Struct. Mol. Biol. 15:163–169 [DOI] [PubMed] [Google Scholar]
- 22. Bailey S, Eliason WK, Steitz TA. 2007. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318:459–463 [DOI] [PubMed] [Google Scholar]
- 23. Parsonnet J. 1995. The incidence of Helicobacter pylori infection. Aliment Pharmacol. Ther. 9(Suppl 2):45–51 [PubMed] [Google Scholar]
- 24. Cover TL, Blaser MJ. 1996. Helicobacter pylori infection, a paradigm for chronic mucosal inflammation: pathogenesis and implications for eradication and prevention. Adv. Intern. Med. 41:85–117 [PubMed] [Google Scholar]
- 25. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 26. Gasbarrini A, Franceschi F, Tartaglione R, Landolfi R, Pola P, Gasbarrini G. 1998. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet 352:878. [DOI] [PubMed] [Google Scholar]
- 27. Ghabaee M, Ghanbarian D, Brujeni GN, Bokaei S, Siavoshi F, Gharibzadeh S. 2010. Could Helicobacter pylori play an important role in axonal type of Guillain-Barre syndrome pathogenesis? Clin. Neurol. Neurosurg. 112:193–198 [DOI] [PubMed] [Google Scholar]
- 28. Kountouras J, Boziki M, Gavalas E, Zavos C, Grigoriadis N, Deretzi G, Tzilves D, Katsinelos P, Tsolaki M, Chatzopoulos D, Venizelos I. 2009. Eradication of Helicobacter pylori may be beneficial in the management of Alzheimer's disease. J. Neurol. 256:758–767 [DOI] [PubMed] [Google Scholar]
- 29. Whincup PH, Mendall MA, Perry IJ, Strachan DP, Walker M. 1996. Prospective relations between Helicobacter pylori infection, coronary heart disease, and stroke in middle aged men. Heart 75:568–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nitharwal RG, Verma V, Dasgupta S, Dhar SK. 2011. Helicobacter pylori chromosomal DNA replication: current status and future perspectives. FEBS Lett. 585:7–17 [DOI] [PubMed] [Google Scholar]
- 31. Zawilak A, Durrant MC, Jakimowicz P, Backert S, Zakrzewska-Czerwinska J. 2003. DNA binding specificity of the replication initiator protein, DnaA from Helicobacter pylori. J. Mol. Biol. 334:933–947 [DOI] [PubMed] [Google Scholar]
- 32. Zawilak-Pawlik A, Kois A, Stingl K, Boneca IG, Skrobuk P, Piotr J, Lurz R, Zakrzewska-Czerwinska J, Labigne A. 2007. HobA: a novel protein involved in initiation of chromosomal replication in Helicobacter pylori. Mol. Microbiol. 65:979–994 [DOI] [PubMed] [Google Scholar]
- 33. Natrajan G, Hall DR, Thompson AC, Gutsche I, Terradot L. 2007. Structural similarity between the DnaA-binding proteins HobA (HP1230) from Helicobacter pylori and DiaA from Escherichia coli. Mol. Microbiol. 65:995–1005 [DOI] [PubMed] [Google Scholar]
- 34. Natrajan G, Noirot-Gros MF, Zawilak-Pawlik A, Kapp U, Terradot L. 2009. The structure of a DnaA/HobA complex from Helicobacter pylori provides insight into regulation of DNA replication in bacteria. Proc. Natl. Acad. Sci. U. S. A. 106:21115–21120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soni RK, Mehra P, Choudhury NR, Mukhopadhyay G, Dhar SK. 2003. Functional characterization of Helicobacter pylori DnaB helicase. Nucleic Acids Res. 31:6828–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nitharwal RG, Verma V, Subbarao N, Dasgupta S, Choudhury NR, Dhar SK. 2012. DNA binding activity of Helicobacter pylori DnaB helicase: the role of the N-terminal domain in modulating DNA binding activities. FEBS J. 279:234–250 [DOI] [PubMed] [Google Scholar]
- 37. Nitharwal RG, Paul S, Dar A, Choudhury NR, Soni RK, Prusty D, Sinha S, Kashav T, Mukhopadhyay G, Chaudhuri TK, Gourinath S, Dhar SK. 2007. The domain structure of Helicobacter pylori DnaB helicase: the N-terminal domain can be dispensable for helicase activity, whereas the extreme C-terminal region is essential for its function. Nucleic Acids Res. 35:2861–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stelter M, Gutsche I, Kapp U, Bazin A, Bajic G, Goret G, Jamin M, Timmins J, Terradot L. 2012. Architecture of a dodecameric bacterial replicative helicase. Structure 20:554–564 [DOI] [PubMed] [Google Scholar]
- 39. Sharma A, Nitharwal RG, Singh B, Dar A, Dasgupta S, Dhar SK. 2009. Helicobacter pylori single-stranded DNA binding protein: functional characterization and modulation of H. pylori DnaB helicase activity. FEBS J. 276:519–531 [DOI] [PubMed] [Google Scholar]
- 40. Donczew R, Weigel C, Lurz R, Zakrzewska-Czerwinska J, Zawilak-Pawlik A. 2012. Helicobacter pylori oriC: the first bipartite origin of chromosome replication in Gram-negative bacteria. Nucleic Acids Res. 40:9647–9660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Soni RK, Mehra P, Mukhopadhyay G, Dhar SK. 2005. Helicobacter pylori DnaB helicase can bypass Escherichia coli DnaC function in vivo. Biochem. J. 389:541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 44. Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. 2005. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D Biol. Crystallogr. 61:449–457 [DOI] [PubMed] [Google Scholar]
- 45. Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D 50:760–763 [DOI] [PubMed] [Google Scholar]
- 46. Sheldrick GM, Hauptman HA, Weeks CM, Miller R, Usón I. 2001. International tables for macromolecular crystallography, vol F, p 333–345 Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 47. Schneider TR, Sheldrick GM. 2002. Substructure solution with SHELXD. Acta Crystallogr. D 58:1772–1779 [DOI] [PubMed] [Google Scholar]
- 48. Hao Q. 2004. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Crystallogr. 37:498–499 [Google Scholar]
- 49. Sheldrick GM. 2002. Macromolecular phasing with SHELXE. Z. Kristallogr. 217:644–650 [Google Scholar]
- 50. Terwilliger TC. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perrakis A, Morris R, Lamzin VS. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6:458–463 [DOI] [PubMed] [Google Scholar]
- 52. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 53. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 54. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291 [Google Scholar]
- 55. DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 56. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98:10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dar MA, Sharma A, Mondal N, Dhar SK. 2007. Molecular cloning of apicoplast-targeted Plasmodium falciparum DNA gyrase genes: unique intrinsic ATPase activity and ATP-independent dimerization of PfGyrB subunit. Eukaryot. Cell 6:398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Case TAD, Cheatham DA, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossvai I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. 2012. AMBER 12. University of California, San Francisco, CA [Google Scholar]
- 59. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 60. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 65:712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Darden T, Perera L, Li L, Pedersen L. 1999. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 7:R55–R60 [DOI] [PubMed] [Google Scholar]
- 62. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., III 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897 [DOI] [PubMed] [Google Scholar]
- 63. Kleywegt G, Jones TA. 1994. A super position. CCP4/ESF-EACBM Newsl. Protein Crystallogr. 31:9–14 [Google Scholar]
- 64. Mosca R, Schneider TR. 2008. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res. 36:W42–W46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chintakayala K, Larson MA, Griep MA, Hinrichs SH, Soultanas P. 2008. Conserved residues of the C-terminal p16 domain of primase are involved in modulating the activity of the bacterial primosome. Mol. Microbiol. 68:360–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ofran Y, Rost B. 2007. Protein-protein interaction hot spots carved into sequences. PLoS Comput. Biol. 3:e119. 10.1371/journal.pcbi.0030119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Johnson SK, Bhattacharyya S, Griep MA. 2000. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry 39:736–744 [DOI] [PubMed] [Google Scholar]
- 68. Thirlway J, Soultanas P. 2006. In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 188:1534–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Afonso JP, Chintakayala K, Suwannachart C, Sedelnikova S, Giles K, Hoyes JB, Soultanas P, Rafferty JB, Oldham NJ. 21 March 2013. Insights into the structure and assembly of the Bacillus subtilis clamp-loader complex and its interaction with the replicative helicase. Nucleic Acids Res. [Epub ahead of print.] 10.1093/nar/gkt173 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.