

Abstract
Antifungal resistance of Candida species is a clinical problem in the management of diseases caused by these pathogens. In this study we identified from a collection of 423 clinical samples taken from Tunisian hospitals two clinical Candida species (Candida albicans JEY355 and Candida tropicalis JEY162) with decreased susceptibility to azoles and polyenes. For JEY355, the fluconazole (FLC) MIC was 8 μg/ml. Azole resistance in C. albicans JEY355 was mainly caused by overexpression of a multidrug efflux pump of the major facilitator superfamily, Mdr1. The regulator of Mdr1, MRR1, contained a yet-unknown gain-of-function mutation (V877F) causing MDR1 overexpression. The C. tropicalis JEY162 isolate demonstrated cross-resistance between FLC (MIC > 128 μg/ml), voriconazole (MIC > 16 μg/ml), and amphotericin B (MIC > 32 μg/ml). Sterol analysis using gas chromatography-mass spectrometry revealed that ergosterol was undetectable in JEY162 and that it accumulated 14α-methyl fecosterol, thus indicating a perturbation in the function of at least two main ergosterol biosynthesis proteins (Erg11 and Erg3). Sequence analyses of C. tropicalis ERG11 (CtERG11) and CtERG3 from JEY162 revealed a deletion of 132 nucleotides and a single amino acid substitution (S258F), respectively. These two alleles were demonstrated to be nonfunctional and thus are consistent with previous studies showing that ERG11 mutants can only survive in combination with other ERG3 mutations. CtERG3 and CtERG11 wild-type alleles were replaced by the defective genes in a wild-type C. tropicalis strain, resulting in a drug resistance phenotype identical to that of JEY162. This genetic evidence demonstrated that CtERG3 and CtERG11 mutations participated in drug resistance. During reconstitution of the drug resistance in C. tropicalis, a strain was obtained harboring only defective Cterg11 allele and containing as a major sterol the toxic metabolite 14α-methyl-ergosta-8,24(28)-dien-3α,6β-diol, suggesting that ERG3 was still functional. This strain therefore challenged the current belief that ERG11 mutations cannot be viable unless accompanied by compensatory mutations. In conclusion, this study, in addition to identifying a novel MRR1 mutation in C. albicans, constitutes the first report on a clinical C. tropicalis with defective activity of sterol 14α-demethylase and sterol Δ5,6-desaturase leading to azole-polyene cross-resistance.
INTRODUCTION
Over the past 2 decades, the prevalence of fungal infections has increased significantly due to the growing number of populations at high risk (1–4). Candida species are the most commonly isolated fungal pathogens causing morbidity and mortality in patients with impaired immunity (2, 3). In the United States, Candida spp. has been ranked as the fourth etiological agent causing bloodstream infections (4). Although Candida albicans remains the major species responsible for disseminated candidiasis, a large number of reports have documented infections caused by other Candida species (3, 4). Among non-C. albicans species, C. tropicalis represents the third or fourth most commonly isolated species of Candida worldwide (4–6). Nevertheless, C. tropicalis has been identified as the most prevalent species of the non-C. albicans group by different epidemiological investigations (7, 8). It ranked as second in Latin America (20%) and is more common than Candida glabrata in the Asian-Pacific region (5, 9).
Prolonged prophylaxis or treatment with antifungal agents has increased the incidence of clinical isolates resistant to one or more antifungals in previously susceptible strains (1, 10). C. albicans and C. tropicalis were for a long time regarded as species largely susceptible to fluconazole and amphotericin B, but reports over the few last years have shown development of resistance to fluconazole in some centers and clinical therapy failure (1, 10–12). Azole compounds represent the most widely used class of antifungal drugs to treat Candida infections (1, 12). The emergence of azole resistance in Candida species makes necessary the development of new effective antifungal strategies against drug-resistant strains (13). Several investigations have explored, at the molecular level, the mechanisms responsible for the acquisition of azole resistance in clinical isolates (1, 10, 12, 14–23). Azoles exert their action by inhibiting the enzyme lanosterol 14α-demethylase in yeasts and molds and thus interfere with the biosynthesis of ergosterol in the fungal cell membrane. Ergosterol depletion coupled with the accumulation of methylated sterol precursors has been shown to affect membrane integrity and the function of some membrane bound proteins. This results in inhibition of cell growth and finally cell death. Azoles differ in their affinities to their target, which may account for differences in their spectrum of activity among various fungi (13, 24). Likewise, variations in the structure of azoles are thought to be responsible for the cross-resistance patterns among Candida species. Resistance mechanisms have been elucidated principally in C. albicans and in C. glabrata (1, 10, 14, 18, 19, 25, 26). Different mechanisms are responsible for the development of azole resistance in major Candida species. For example, resistance can be mediated by increased efflux of azoles resulting from the overexpression of multiple drug resistance genes such as ATP-binding cassette (ABC) transporters and the major facilitator superfamily (MFS) membrane transporters (1, 10, 12, 19, 26). Another common resistance mechanism is the overexpression of ERG11, which encodes for the azole target enzyme lanosterol 14-α demethylase. In addition, ERG11 polymorphisms leading to amino acid substitutions can decrease the affinity of azoles to the target enzyme (12, 15, 23). Other studies showed that some enzymes involved in the ergosterol biosynthesis pathways, such as Erg3, have been associated with azole and polyene cross-resistance in C. albicans clinical isolates (16, 17, 27). Recently, hyperactive alleles with gain-of-function (GOF) mutations in zinc finger transcription factor genes have been discovered and lead to overexpression of the genes responsible for azole resistance in Candida species (14, 20–22, 28).
The vast majority of studies analyzing the molecular mechanisms against antifungal agents originate from clinical Candida isolated from American, European, and Asian continents. We examined here the molecular mechanisms of antifungal resistance of two clinical isolates belonging to two different Candida species. These isolates were identified from a collection of yeast clinical samples from Tunisian hospitals systematically screened for their susceptibility to agents such as azoles, amphotericin B, and candins (29). From these isolates, a specific C. albicans isolate revealed a new gain-of-function mutation in MRR1, leading to the overexpression of MDR1 and thus fluconazole resistance. One other isolate, identified as C. tropicalis, exhibited cross-resistance between azoles and amphotericin B, which is typical of defects in the ergosterol biosynthesis pathway.
MATERIALS AND METHODS
Strains and culture conditions.
The yeast strains used in the present study are listed in the Table 1. C. albicans (JEY355) and C. tropicalis (JEY162) clinical isolates were obtained from two different public health institutions in Tunisia (Hospital Habib Thameur and the National Bone Marrow Transplantation Center). JEY355 (Ridha Kelifa) and JEY162 (Emna Chaker) were recovered from oral swab and blood culture specimens, respectively. Strains were stored in 20% glycerol at −80°C and cultured on either YPD (1% Bacto peptone [Difco Laboratories, Basel, Switzerland], 0.5% yeast extract [Difco], 2% glucose [Fluka, Buchs, Switzerland], and 2% agar for plate [Difco]) or minimal defined medium consisting of YNB (0.67% yeast nitrogen base [Difco] plus 2% glucose [Fluka], solidified with 2% agar [Difco] as required). YPD agar plates containing 200 μg/ml of nourseothricin (clonNAT; Werner BioAgents) were used as a selective medium for growth of nourseothricin-resistant isolates. To obtain transformant strains as nourseothricin sensitive in which the SAT1 flipper was excised, the transformants were processed as described previously (16) and grown in YPD with 2% maltose to induce MAL2 promoter controlling CaFLP expression. YNB with appropriate amino acid and bases was used as selective medium after transformation of yeast strains (10). YNB agar plate containing 5-fluoroorotic acid (5-FOA) at 100 μg/ml with 50 μg of uridine/ml was prepared for regeneration of the ura3 genetic marker. Bacterial strain Escherichia coli DH5α was used as a host for the construction and propagation of all plasmids. DH5α cells were grown in Luria-Bertani (LB) broth (Difco) or on LB plates, which were supplemented with ampicillin (0.1 mg/ml) or chloramphenicol (0.025 mg/ml) when required.
Table 1.
Strains used in this study
| Strain | Parent strain | Genotype | Source or reference |
|---|---|---|---|
| C. albicans | |||
| SC5314 | MRR1/MRR1 | 30 | |
| DSY4216 | SC5314 | MRR1/mrr1Δ::FRT-SAT1-FRT | This study |
| DSY4219 | DSY4216 | MRR1/mrr1Δ::FRT | This study |
| DSY4221 | DSY4219 | mrr1Δ::FRT-SAT1-FRT/mrr1Δ::FRT | This study |
| DSY4278 | DSY4221 | mrr1Δ::FRT/mrr1Δ::FRT | This study |
| DSY291 | Clinical isolate | MRR1/MRR1V877 | 20 |
| DSY294 (C43) | Clinical isolate | 1 | |
| DSY296 (C56) | Clinical isolate | 1 | |
| DSY2285 | Clinical isolate | 31 | |
| DSY2286 | Clinical isolate | 31 | |
| JEY355 | Clinical isolate | MRR1V877F/MRR1V877F | This study |
| JEY429 | DSY4278 | mrr1Δ::FRT/mrr1Δ::FRT::MRR1V877F | This study |
| JEY430 | DSY4278 | mrr1Δ::FRT/mrr1Δ::FRT::MRR1V877 | This study |
| JEY431 | JEY429 | mrr1Δ::FRT::MRR1V877F/mrr1Δ::FRT::MRR1V877F | This study |
| JEY432 | JEY430 | mrr1Δ::FRT::MRR1V877/mrr1Δ::FRT::MRR1V877 | This study |
| VSY2 | Clinical isolate | erg3-1/erg3-1 | 16 |
| JEY424 | VSY2 | erg3-1/erg3-1::Cterg3::FRT | This study |
| JEY425 | VSY2 | erg3-1/erg3-1::CtERG3::FRT | This study |
| C. tropicalis | |||
| DSY140 | ATCC 750 | ade2/ade2 gal1/gal1 ura3Δ::cat/ura3Δ::cat | 32 |
| JEY162 | Clinical isolate | Cterg11/Cterg11 Cterg3/Cterg3 | This study |
| JEY450 | DSY140 | CtERG3/CtERG3::Cterg3::hisG-URA3-hisG | This study |
| JEY437 | DSY450 | CtERG3/CtERG3::Cterg3::hisG | This study |
| JEY439 | JEY437 | CtERG3::Cterg3::hisG/CtERG3::Cterg3::SAT1 | This study |
| JEY440 | JEY439 | CtERG3::Cterg3::hisG/CtERG3::Cterg3::SAT1 CtERG11/CtERG11::Cterg11::hisG-URA3-hisG | This study |
| JEY441 | JEY440 | CtERG3::Cterg3::hisG/CtERG3::Cterg3::SAT1 CtERG11/CtERG11::Cterg11::hisG | This study |
| JEY442 | JEY440 | CtERG3::Cterg3::hisG/CtERG3::Cterg3::SAT1 CtERG11::Cterg11/CtERG11::Cterg11::hisG-URA3-hisG | This study |
| JEY445 | JEY442 | CtERG3::Cterg3::hisG/CtERG3::Cterg3::SAT CtERG11::Cterg11::hisG-URA3-hisG/CtERG11::Cterg11::hisG | This study |
| JEY311 | Clinical isolate | CtERG3/CtERG3 CtERG11/CtERG11 | This study |
| JEY433 | JEY311 | CtERG3/CtERG3 CtERG11/CtERG11::Cterg11::SAT1 | This study |
| JEY434 | JEY433 | CtERG3/CtERG3 CtERG11::Cterg11/CtERG11::Cterg11::SAT1 | This study |
DNA sequencing and analysis of MRR1, ERG11, CtERG3, and CtERG11.
Genomic DNAs from C. albicans JEY355 and C. tropicalis JEY162 were isolated as described previously (10) and were used as templates to amplify by PCR MRR1 and ERG11 of JEY355 and C. tropicalis ERG3 (CtERG3) and CtERG11 of JEY162 and JEY311 using the primers listed in Table 2. The PCR products were sequenced using an ABI Prism 3130 XL automated DNA sequencer (Perkin-Elmer/Applied Biosystems, Foster City, CA) with a a BigDye terminator cycle sequencing kit (version 1.1; Applied Biosystems) according to the manufacturer's protocol.
Table 2.
Primers used in this study
| Primer | Sequence (5′–3′) |
|---|---|
| MRR-Xho | GATTCTCGAGAATTGGCAGCTCTTATGTTA |
| MRR-SacI | ATTGCCGAGCTCTGTTTATCAAATTGATTTC |
| MRR-SpH | CTTTGGCATGCTTGATAATGTGCCTCTAGA |
| MRR-SacII | CTATAGCCGCGGTGGCAATTGACATTTTTA |
| MRR178_SacI | AACAGAGCTCTTTACCCCAATCCAAACAC |
| MRR-SacIId | GGAAAAAACCGCGGAACGATATACTACATACATC |
| MRR1-1 | GCTATTAATTCCTATTCC |
| ZCF36SEQ7R | CACTCAACGTAATAGTGACTTC |
| MRR1-8 | CAG AGC CCC AAG TTC GG |
| ZCF36SEQ7 | GAAGTCACTATTACGTTGAGTG |
| ZCF36SEQ1 | ATTACAATGTGTCCCACACAGG |
| MRR1-2 | CAACACTGCCTTTAGGGTTGA |
| ZCF36SEQ2 | CAGTTTACTTTATCCATTTATGCC |
| ZCF36SEQ6 | CATCCTTGTATTCCGTTTCACC |
| ZCF36SEQ3 | AGTTCCATTTATAGAAGAAGGC |
| ZCF36SEQ8 | CTTGATTGATAAGAGTTTGGATC |
| MRR1-3 | TCGTAATATGCCAGTGAAAATG |
| ZCF36SEQ4 | GTTGGAATTGCAGCTGTATCC |
| MRR-SacIId | GGAAAAAACCGCGGAACGATATACTACATACATC |
| ERG11_KpnI | GAGCATGGGTACCGGCGCGCGATTGTACGTGG |
| ERG11_XhoI | GTAACGCTCGAGTGAACAAGGTTGGGTAGTAA |
| ERG11-P3 | CTTACACTCTATGGACA |
| ERG11-ORF-F | ATTGTTGAAACTGTCATTG |
| ERG11-7 | CAGCAGAAACATCAGATA |
| ERG11-2 | CATGGGGTTGCCAATGTT |
| ERG11-6 | GAGCAAATGAACGGTCAA |
| ERG11-5 | CATATGCATTCTGAGAGT |
| ERG11-3B | CCCATTAAGAATCCCTGAA |
| ERG11-4 | CTGCTGGTTCAGTAGGTA |
| CtERG3-KpnI | TCTTGGTACCAGTATTACAGTGCACACAC |
| CtERG3-XhoI | AATCTCGAGGCTACTTTCATATCAAACGC |
| CtERG_1504 | TGTGGGACAGATTAGGTAG |
| CtERG_1263 | TTTGCATCCCATGCTTTC |
| CtERG_1013 | GGCAACTAGAGCCATTCC |
| CtERG_778 | CAGAAATCTACGGTTTAGC |
| CtERG_1502C | AGTGGTGAATTGACCATAG |
| CtERG_1207C | GGCCAATGTAACCATCTG |
| CtERG_1014C | CCAAACCGATTTCCAAAG |
| CtERG_712C | GCAAGGGAGAAATTTGAAG |
| CtERG11-ApaI | ACAATGGGCCCAAAACACGGTGAAGAAAT |
| CtERG11-XhoI | TTTTCTCGAGTGGTTGAAAAATTTTCTG |
| CtERG_401C | CATCAATGGCAGTATCAAC |
| CtERG_1113 | ACGCTGCTCAAAGAAAG |
| CtERG_1121C | GAGCAGCGTCACGTCTC |
| CtERG_1490 | CACATGCCATTGCATTC |
| CtERG_1499C | ATGGCATGTGCATTCTC |
| CtERG_1759 | ATTCGGTGGTGGTAGAC |
| CtERG_1920C | GCAGGTTCTAATGGTAAGG |
| CtERG_732C | TGGGTATAAGCTTCTTCAG |
| ERG11-ORF-F | ATTGTTGAAACTGTCATTG |
| ERG11-ORF-R | CCCCTAATAATATACTGATCTG |
| ACT1-ORF-F | GCATCACACTTTTTACAAT |
| ACT1-ORF-R | AAACATAATTTGAGTCATCTTT |
| ERG11-P2a | TTTGTCCCTTAGTGTTACACA |
| ACT1-P2a | TTGCTCCAGAAGAACATCCAGT |
| CtERG11_PyesF | ACTACTAGCAGCTGTAATACGACTCACTATAGGGAATATTAAGCTTATGGCTATTGTTGATACTGCCATT |
| CtERG11_PyesR | AGGGTTAGGGATAGGCTTACCTTCGAAGGGCCCTCTAGACTCGAGAACCATACAAGTATCTCTCTTTTCC |
| CtERG3-NcoI-NotI | CAAGGCGGCCGCCCATGGTTTGATCTTAGTAGTAG |
| CtERG3-NotI | ACAAGCGGCCGCGGTAATAAAATACAATCAAG |
| CtERG3-SacI | ATCTGAGCTCAAGTTAAAAATTTAACTC |
These primers were labeled with 6-FAM ( 6-carboxyfluorescein) at the 5′ end and TAMRA (tetramethylrhodamine) at the 3′ end and were used as TaqMan probes in qRT-PCR with primers ERG11-ORF-F/ERG11-ORF-R and ACT1-ORF1-F/ACT1-ORF-R.
Plasmid constructions.
To construct a plasmid containing MRR1, the downstream region (the 3′ untranslated region [3′-UTR]) of the MRR1 open reading frame (ORF) from C. albicans SC5314 genomic DNA was amplified with the primers MRR-XhoI and MRR-KpnI (Table 2) containing XhoI and KpnI restriction sites and inserted into pBluescript II SK(+) (Stratagene) to yield pJE6. MRR1 flanked by 500 bp was amplified from genomic DNAs of JEY355 and DSY291 using the primers MRR178-SacI and MRR-SacIId (Table 2) containing SacI and SacII restriction sites. The resulting PCR product was digested with SacI and SacII and was cloned into the vector pJE6 to obtain pJE7 (MRR1 from JEY355: MRR1V877F) and pJE8 (MRR1 from DSY291: MRR1V877). To introduce the SAT1 flipper cassette, pSFS2A was digested by SacII and XhoI, and the resulting fragment was inserted between corresponding sites into pJE7 and pJE8 to obtain pJE9 and pJE10, respectively.
For MRR1 deletion, pSFSU1, which was previously used to inactivate URA3 in C. albicans (33), was digested sequentially by SphI/SacII and XhoI/SacI to introduce the 5′- and 3′-UTRs of MRR1 amplified by the primer pairs MRR-SpH/MRR-SacII and MRR-Xho/MRR-SacI, respectively. The obtained plasmid (pDS1581) was digested with SphI/SacI to release the entire disruption cassette to transform C. albicans SC5314. Selection was performed onto nourseothricin-containing YPD medium, and the SAT1 dominant marker was regenerated as described above.
To construct plasmid containing CtERG3, a method with three fragment ligation and pBluescript II SK(+) as vector was used. Regions 500 bp upstream (5′-UTR) and downstream (3′-UTR) of the CtERG3 ORF from JEY311 were amplified by PCR by using the primer pairs CtERG3-KpnI/CtERG3-NcoI-NotI and CtERG3-NotI/CtERG3-SacI. After digestion of PCR products (KpnI/NotI and NotI/SacI) and of the vector (KpnI/SacI), products were ligated to yield pJE3. The plasmid pJE1 was derived from pVS10 (16) in which CtERG3 amplified from genomic DNA of JEY162 with the primers CtERG3-NcoI and CtERG3-XhoI replaced C. albicans ERG3-2. Cterg3 inserted in pJE1 was subcloned into pJE3 to yield pJE4 and contained the SAT1 flipper cassette as a genetic marker. To use uridine auxotrophy as complementing marker, the plasmid pJE16 was constructed as follows. pMB-7 (30), which contains URA3 flanked by direct repeats of the Salmonella enterica serovar Typhimurium hisG gene, was digested with PstI and BglII, and the PCR product of pJE3 obtained with the primers pJE3-BamHI and pJE3-PstI were ligated at BamHI and PstI sites to obtain pJE15. The Cterg3 ORF was amplified from JEY162 genomic DNA with the primers CtERG3-KpnI and CtERG3-XhoI as described above and digested by KpnI and XhoI, whereas pJE15 was digested by KpnI and SalI. The purified fragment of CtERG3 (Cterg3 allele) was cloned into the KpnI/SalI sites of plasmid pJE15 to generate pJE16.
To construct a plasmid containing CtERG11, pSFS2A was used to introduce a 3′-UTR (500 bp) of CtERG11 amplified from genomic DNA of JEY311 with the primers CtERG11-SacI and CtERG11-SacII. The product was digested by SacI and SacII and ligated to yield pJE11. The Cterg11 allele from JEY162 was obtained by PCR with the primers CtERG11-ApaI and CtERG11-XhoI, digested with ApaI and XhoI, and cloned into the ApaI/XhoI sites of plasmid pJE11 to generate pJE12. For the use of uridine auxotrophy, plasmid pMB-7 was used as a vector. The plasmids pJE12 and pMB-7 were digested with BglII/XhoI and BglII/SalI, respectively. The useful fragments were ligated to yield pJE14. In order to exclude mutations present in all alleles generated by PCR, recombinant plasmids were sequenced.
Yeast strain transformation.
C. albicans and C. tropicalis strains were transformed by a lithium acetate procedure as reported previously (19) with corresponding linear DNA fragments. To screen C. albicans transformants, ∼100 cells were spread and grown for 48 h at 35°C on YPD plates containing 15 μg of nourseothricin/ml. C. tropicalis transformants were selected in YNB medium as described above.
Complementation of CtERG3 in C. albicans.
Plasmids pJE1 and pJE2 derived from pVS10 harbored the CtERG3 ORFs amplified by primers CtERG3-NcoI/CtERG3-XhoI (see above) from JEY162 and JEY311, respectively. These alleles were flanked by 5′- and 3′-UTRs for homologous recombination in C. albicans. The clinical VSY2 isolate (16) containing the defective erg3-1 allele was used as wild-type strain and transformed by 5 μl of ApaI/SacI-digested pJE1 and pJE2 in order to perform homologous recombination at the ERG3 locus. The sterols from transformant strains (JEY424 and JEY425) and wild-type strains (SC5314, VSY2, JEY162, and JEY311) were extracted by the alcoholic potassium hydroxide method as described previously (34). The ergosterol content was measured by scanning spectrophotometry between 240 and 310 nm with a NanoDrop ND-1000 UV-Vis spectrophotometer. The presence of ergosterol and the late sterol intermediate in the extracted sample resulted in a characteristic four-peak curve. The absence of detectable ergosterol in extracts was indicated by the absence of typical ergosterol absorption at ∼280 nm.
C. tropicalis strain constructions.
C. tropicalis transformant strains were obtained using the two selectable markers (SAT1-flipper cassette and URA3 blaster) as follows. DSY140 was transformed with pJE16 digested by KpnI/SacI to generate JEY450. Ura− cells from JEY450 were obtained by plating onto 5-FOA YNB to result in JEY437. JEY437 was next transformed with pJE4 digested by KpnI/SacI to obtain JEY439 into which both endogenous CtERG3 alleles are replaced by mutant alleles. Since Cterg3 specifically harbors the restriction site BsmI, the integration of Cterg3 alleles could be verified by BsmI digestion of PCR products obtained by the primers CtERG_778 and CtERG_1502C. The presence of the loss-of-function mutation in mutant alleles in JEY439 was confirmed by sequencing of the same PCR products. JEY439 was used to generate transformant strains harboring Cterg11 alleles. This was performed by transformation of JEY439 with ApaI/SacI-digested pJE14 to yield JEY440 (Ura+) and JEY441 (Ura−) after marker regeneration. The homozygous mutant JEY442 (Cterg11/Cterg11) was obtained by exposure of the heterozygote strain JEY440 (CtERG11/Cterg11) in YPD medium containing amphotericin B (AMB; 10 μg/ml). JEY445 was generated by transformation of JEY441 with ApaI/SacI-digested pJE14 to obtain mutant allele homozygosity at the ERG11 locus (Cterg11/Cterg11). The clinical isolate JEY311 was transformed with ApaI/SacI-digested pJE12 to yield JEY433 (CtERG11/Cterg11). This strain was next exposed to AMB (10 μg/ml) to obtain mitotic recombination in order to yield mutant alleles homozygosity at the CtERG11 locus (Cterg11/Cterg11). Since the Cterg11 ORF was truncated by 132 bp compared to the wild type, the presence of mutant Cterg11 alleles in transformants was verified by comparing size differences via gel electrophoresis of PCR products obtained with the primers CtERG_1113 and CtERG_1920C.
Complementation of CtERG11 in Saccharomyces cerevisiae.
The S. cerevisiae strains used in the present study were DSY3886 derived from Y40122 [MATa ura3-52 leu2Δ1 his3Δ200 GAL2 CMVp (tetR′-SSN6)::LEU2 trp1::Tta] and DSY3961 with its endogenous ERG11 under the control of doxycycline (ERG11::kanMX-tetO7) and in which PDR5 was inactivated (35).
CtERG11 expression in S. cerevisiae.
CtERG11 ORFs flanked by pYES2/CT regions for homologous recombination in S. cerevisiae were amplified by PCR using the primers CtERG11_PyesF and CtERG11_PyesR. pYES2/CT contains a polyhistidine (His6) tag for protein tagging at the C-terminal end. Strain DSY3961 was transformed with 5 μl of XhoI/HindIII-digested pYES2/CT and 5 μl of C. tropicalis CtERG11 alleles previously amplified by PCR in order to perform homologous recombination in S. cerevisiae. Transformants were selected onto YNB-uracil and were screened in the same medium but containing galactose (2%) as the carbon source and doxycycline (2 μg/ml). CtERG11 genes were amplified and sequenced for verification.
Drug susceptibility testing.
The susceptibilities of yeast strains to fluconazole (FLC), voriconazole (VRC), and caspofungin (CAS) were determined according to the method recommended by Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST) for fermentative yeasts (36), with slight modifications. For some C. tropicalis strains, the medium RPMI 1640 was replaced by YPD, and the inoculum density was (0.5 to 2.5) × 103 CFU/ml. The MICs for AMB were determined by Etest (bioMérieux, Marcy l'Étoile, France) according to document M44-A of the Clinical and Laboratory Standards Institute. The interpretive breakpoints for susceptibility assays were as follows. For FLC, values of ≤2, 4, and ≥8 μg/ml correspond to susceptible (S), susceptible dose dependent (SDD), and resistant (R), respectively; for VRC, these values are ≤0.125 μg/ml (S), 0.25 to 0.5 μg/ml (SDD), and ≥1 μg/ml (R); and for AMB, these values are ≤1 μg/ml (S) and >1 μg/ml (R). C. albicans ATCC 90028 and C. tropicalis ATCC 750 were included as control strains.
Serial dilution assays.
The susceptibilities of C. albicans strains to different compounds were determined qualitatively by comparison of isolates to their respective isogenic parental strains by spotting serial dilutions of yeast cultures onto YPD medium agar plates containing different drug concentrations and incubated for 24 to 48 h at 35°C. The serial dilution assays was performed as described previously (37). The drug concentrations were optimized to allow growth differences between strains. The antifungal compounds were used from stock solutions and were prepared as follows. FLC was dissolved in distilled water at a concentration of 2.56 mg/ml, cyclosporine was dissolved in distilled water at 10 mg/ml, and brefeldin A was dissolved in distilled water at 10 mg/ml in dimethyl sulfoxide.
Sterol analysis.
C. tropicalis strains were grown overnight in 20 ml of YPD at 37°C and 200 rpm. The cells were harvested and washed with H2O, and nonsaponifiable lipids were extracted as reported previously (38). Samples were dried in a vacuum centrifuge, derivatized by the addition of 200 μl of anhydrous pyridine (Sigma) and 100 μl of 90% BSTFA [N,O-bis(trimethylsilyl)trifluoroacetamide]–10% TMS (trimethylsilyl; Sigma), and incubated at 80°C for 2 h. TMS-derivatized sterols were analyzed and identified using gas chromatography-mass spectrometry (GC-MS; Agilent 5975C Inert XL GC/MSD) with reference to retention times and fragmentation spectra for known standards. GC-MS data files were analyzed using Agilent software (MSD Enhanced ChemStation; Agilent Technologies, Ltd., Stockport, United Kingdom) to determine sterol profiles for all isolates and for integrated peak areas.
Western blot analysis.
To detect Mdr1 and Cdr1/Cdr2 in C. albicans, cell extracts were prepared by an alkaline extraction procedure as published (14). Mdr1 and Cdr1/Cdr2 immunodetections were performed by using two types of antibodies: primary rabbit anti-Mdr1 and anti-Cdr1/2 antibodies and secondary anti-rabbit-antibodies conjugated with horseradish peroxidase as described previously (14, 39). Immunodetection was realized with an Amersham ECL Prime detection reagent kit (Solution A: Luminol solution and Solution B: Peroxidase solution). The signals were revealed by chemiluminescence with an ImageQuant LAS 4000 Mini-System (GE Healthcare Life Sciences, Glattbrugg, Switzerland).
mRNA extraction and real-time reverse transcription-PCR (RT-PCR).
Total RNA was extracted from YPD broth cultures as described previously (14). Biological triplicates were prepared from JEY355 and SC5314 strains and from cells exposed to 10 μg of FLC/ml for 90 min at 30°C in the same medium. First-strand cDNAs were synthesized separately from 1 μg of total RNA in a 20-μl reaction mixture volume using the High-Fidelity reverse transcriptase (40). Quantitative PCRs were performed in duplicate as technical replicates using the StepOnePlus real-time PCR system (Applied Biosystems, Zug, Switzerland). The expression level of ACT1 was determined independently with the normalizing gene using iTaq Spermix with Rox from Bio-Rad (Cressier, Switzerland). The specific primers used as probes are listed in Table 2. The change in the fold expression was determined by calculating expression ratio according to the 2−ΔΔCT method.
Nucleotide sequence accession numbers.
The MRR1, ERG11, CtERG3, and CtERG11 gene sequences of C. albicans and C. tropicalis determined in the present study have been deposited in GenBank under accession numbers KC676659 to KC676665.
RESULTS
Susceptibility profiles of C. albicans strains.
Antifungal susceptibility testing showed that the clinical C. albicans isolate (JEY355) was resistant to FLC but not to VRC, CAS, and AMB (MICs of 8, <0.0078, 0.125, and 0.064 μg/ml, respectively) (Table 3). Since the major mechanism responsible for azole resistance in clinical C. albicans isolates is overexpression of membrane efflux pumps, the expression levels of multidrug transporters were measured in this isolate by immunodetection. The two main families of efflux proteins including the ATP-binding cassette (ABC) pumps and the major facilitator superfamily (MFS) transporters were investigated. As shown in Fig. 1, JEY355 showed constitutive overexpression of Mdr1 but not of Cdr1 and Cdr2. Cdr1 was detectable in all strains and was expressed at similar levels in DSY294, JEY355, and SC5314. As expected, Cdr1 and Cdr2 were highly expressed in the resistant clinical isolate DSY296, which was taken here as a positive control for Cdr1/Cdr2 overexpression. The expression of Mdr1 was not detectable in the wild-type strain SC5314 and isolate DSY2285, which is consistent with their azole susceptibility. JEY355 showed overexpression of Mdr1 without exposure to benomyl as MDR1-upregulating substance. Based on these data, the overexpression of MDR1 seems to be an important contributor to FLC resistance in JEY355.
Table 3.
MICs of C. albicans and C. tropicalis strains obtained by the EUCAST microdilution method
| Isolatea | MIC (μg/ml)b |
|||||||
|---|---|---|---|---|---|---|---|---|
| In RPMI 1640c |
In YPDd |
|||||||
| FLC | VRC | CAS | AMB | FLC | VRC | CAS | AMB | |
| SC5314 | 0.0625 | <0.0078 | 0.125 | 0.064 | ND | ND | ND | ND |
| DSY4278 | 0.0625 | <0.0078 | 0.125 | 0.064 | ND | ND | ND | ND |
| JEY355* | 8 | <0.0078 | 0.125 | 0.064 | ND | ND | ND | ND |
| JEY431 | 4 | <0.0078 | 0.125 | 0.064 | ND | ND | ND | ND |
| JEY432 | 0.0625 | <0.0078 | 0.125 | 0.064 | ND | ND | ND | ND |
| JEY162* | >128 | >16 | 0.5 | >32 | >128 | >16 | <0.0078 | >32 |
| JEY311* | 0.5 | 0.0312 | 0.125 | 0.19 | 1 | 0.0312 | <0.0078 | 0.19 |
| JEY433 | 128 | 8 | 0.25 | 0.25 | >128 | >16 | <0.0078 | 0.25 |
| JEY439 | >16 | >128 | 0.125 | 0.5 | <0.0312 | 0.0625 | <0.0078 | 0.5 |
| C. albicans ATCC 90028 | 0.0625 | <0.0078 | 0.0312 | 0.064 | 1 | <0.0078 | <0.0078 | 0.064 |
| C. tropicalis ATCC 750 | 0.5 | 0.0312 | 0.5 | 0.19 | 1 | 0.0312 | <0.0078 | 0.19 |
| JEY434 | ND | ND | ND | ND | >128 | >16 | <0.0078 | >32 |
| DSY140 | ND | ND | ND | ND | 1 | 0.0312 | <0.0078 | 0.064 |
| JEY442 | ND | ND | ND | ND | >128 | >16 | <0.0078 | >32 |
| JEY445 | ND | ND | ND | ND | >128 | >16 | <0.0078 | >32 |
*, Clinical isolate.
MICs for AMB were determined by Etest. ND, not determined.
EUCAST inoculum density, (0.5 to 2.5) × 105 CFU/ml.
CLSI inoculum density, (0.5 to 2.5) × 103 CFU/ml.
Fig 1.
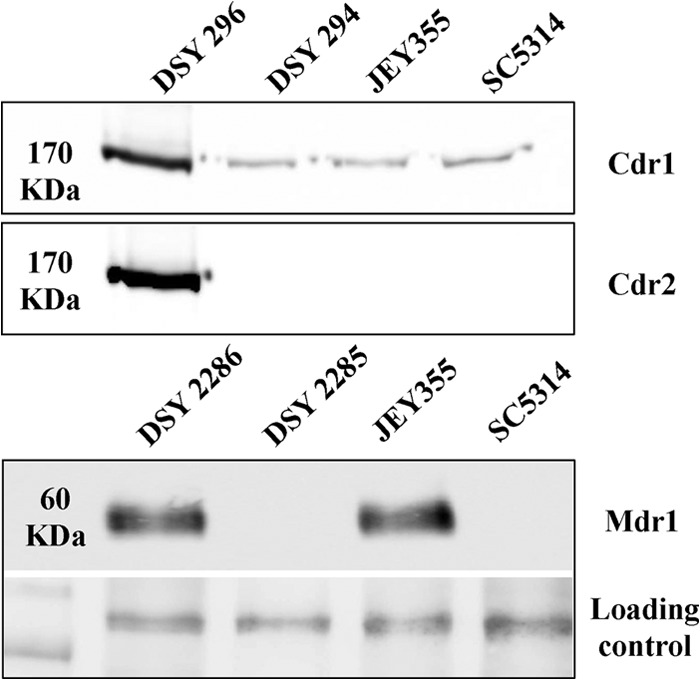
Expression of Cdr1, Cdr2, and Mdr1p in C. albicans isolates. Total protein extracts were prepared from DSY294 and DSY296, DSY2285 and DSY2286 (matched clinical isolates), and JEY355 (clinical isolate) and SC5314 (wild-type) as described in Materials and Methods, separated by SDS-PAGE, transferred to nitrocellulose membranes, and detected with specific antibodies. DSY294 and DSY296, control strains for Cdr1 and Cdr2 expression; DSY2285 and DSY2286, control strains for Mdr1 expression. Red Ponceau staining was used to control protein loading for Cdr1/Cdr2 detection (not shown). Immunodetection of unspecific signals was used as a loading control as indicated for Mdr1 detection. The positions of molecular mass standards in kilodaltons are shown on the left side.
MRR1 and ERG11 sequence analysis from the C. albicans isolate JEY355.
In order to determine the basis of MDR1 overexpression in JEY355, the zinc cluster transcription factor MRR1 controlling MDR1 expression was amplified and sequenced. The nucleotide sequence of MRR1 in JEY355 showed that the clinical isolate contained a single allele different from MRR1 of SC5314 at 29 positions, resulting in 5 amino acid changes and two tandem repeats of the NPQS sequence (Table 4). Interestingly, one of the MRR1 allele (MRR1V877) from the azole-susceptible isolate DSY291, as reported by Dunkel et al. (20), differed from MRR1 in JEY355 only by one nucleotide exchange (G2629T), which resulted in one amino acid substitution (V877F) in the encoded protein (MRR1V877F). The MRR1V877 allele from DSY291 was therefore used as a parental sequence to verify the role of the V877F change in the overexpression of MDR1.
Table 4.
Allelic polymorphisms observed in MRR1 and ERG11 for C. albicans strains
| Gene and isolate | Allele (GenBank accession no.) | Amino acid substitution(s)a |
|---|---|---|
| MRR1 | ||
| DSY291 | Allele 1 (EU497754) | – |
| Allele 2 (EU497755) | S16I, T73K, S171P, E1020Q, 2× NPQS | |
| JEY355 | Allele MRR1V877F (KC676659) | S16I, T73K, S171P, V877F, E1020Q, 2× NPQS |
| JEY429 | Allele MRR1V877F | S16I, T73K, S171P, V877F, E1020Q, 2× NPQS |
| JEY430 | One allele 2 from DSY291 | S16I, T73K, S171P, E1020Q, 2× NPQS |
| JEY431 | Two MRR1V877F alleles | S16I, T73K, S171P, V877F, E1020Q, 2× NPQS |
| JEY432 | Two alleles 2 from DSY291 | S16I, T73K, S171P, E1020Q, 2× NPQS |
| ERG11 | ||
| JEY355 | Allele 1 (KC676660) | E266D |
| Allele 2 (KC676661) | D116E, E266D |
–, Only amino acids that differ from those of Mrr1 of strain SC5314 are listed. The new gain-of-function mutation is indicated in boldface.
Sequence data for the coding region of ERG11 of JEY355 was compared to SC5314 (orf19.922). Two alleles were identified in JEY355. One of the ERG11 allele differed from the standard sequence by four nucleotides (T315C, C411T, C658T, and A798C), leading to one amino acid substitution (E266D). The other ERG11 allele showed 13 mutations, which resulted in two amino acid substitutions (D116E and E266D) in the encoded protein (Table 4). The amino acid substitutions in these alleles are also found in alleles from other azole-susceptible isolates (41) and thus the D116E and E266D substitutions are not likely to contribute to azole resistance in JEY355.
Constitutive MDR1 overexpression in JEY355 as a result of a gain-of-function mutation in MRR1.
The presence of a mutation in MRR1V877F suggests that this mutation was a gain-of-function (GOF) mutation responsible for MDR1 overexpression. To confirm this hypothesis, MRR1V877F and MRR1V877 from JEY355 and DSY291, respectively, were introduced into the mrr1Δ/Δ mutant DSY4278. The expression level of Mdr1 was tested in transformant strains by immunodetection (Fig. 2). The results demonstrate that the transformant strain JEY429 containing the MRR1V877F allele caused MDR1 overexpression compared to JEY430 harboring one MRR1V877 wild-type allele. In the wild-type strain SC5314, Mdr1 was not detected under standard growth conditions. The same result was observed in the homozygous mrr1Δ/Δ strain DSY4278. Mdr1 production could be induced as expected in C. albicans when cells were exposed to benomyl, except in DSY4278. JEY431 homozygous for MRR1V877F produced more Mdr1 compared to JEY429 heterozygous for MRR1V877F. Interestingly, Mdr1 reached similar detection levels when two MRR1V877F alleles were present, as is the case for JEY355 and JEY431. These levels reached those obtained by benomyl exposure in azole-susceptible strains. The expression of Mdr1 in JEY432 (homozygous for MRR1V877 from DSY291) was not detected, thus confirming that the MRR1V877 allele was not constitutively active. These results thus confirmed that the amino acid change V877F is a GOF mutation. The amino acid change V877F is likely the major cause of FLC resistance in JEY355.
Fig 2.
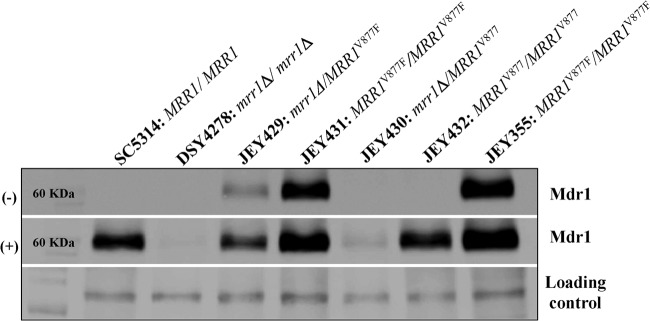
Expression of Mdr1 in C. albicans strains. Total proteins extracts were prepared from SC5314 (wild-type), DSY4278 (mrr1Δ/Δ), JEY429, JEY430, JEY431, and JEY432 (transformant strains), and JEY355 (clinical isolate) as described in Materials and Methods, separated by SDS-PAGE, transferred to nitrocellulose membranes, and detected with specific antibodies. MRR1, reference allele from SC5314; MRR1V877F, MRR1 allele from JEY355; MRR1V877, MRR1 allele from DSY291, a clinical susceptible isolate. Cells were incubated without (−) or with (+) benomyl (35 μg/ml) for 30 min as indicated. Immunodetection of unspecific signals was used as a loading control as indicated. The positions of the molecular mass standards in kilodaltons are shown on the left side.
Interestingly, JEY355 and JEY431 (homozygous for MRR1V877F) are resistant to FLC but not to VRC (Table 3). This result is, however, consistent with the role of Mdr1 as a specific mediator of FLC resistance as opposed to Cdr1/Cdr2 (1, 42). Serial dilution assays were used to better understand the role of the homozygous MRR1V877F GOF mutation in FLC resistance of JEY355. It is known that MRR1 GOF mutations render C. albicans resistant to many toxic compounds, including brefeldin A. As a complementary approach to FLC susceptibility, brefeldin A was used to reveal MDR1 overexpression, since it has been shown that this substance is specific for Mdr1 (19, 39). As shown in Fig. 3, C. albicans isolates harboring hyperactive MRR1V877F alleles were more resistant to brefeldin A than a wild-type strain. For FLC susceptibility testing, cyclosporine was added to the medium to circumvent the trailing phenomenon due to azole tolerance. No growth of strains with wild-type MRR1 alleles (SC5314, DSY4278, JEY430, and JEY432) was observed on a YPD plate containing FLC and cyclosporine. Interestingly, the same result was observed for JEY429 harboring one hyperactive allele (MRR1V877F). However, JEY431 (MRR1V877F/MRR1V877F) was able to grow on this medium, as did JEY355; the latter strain, however, grew better. These results confirmed that MRR1V877F conferred FLC resistance but also underline that two alleles should be present in order to achieve high resistance levels. The results also show that JEY355 may contain additional mediators of FLC resistance in complement to MRR1V877F alleles. This conclusion is based on the antifungal susceptibility testing showing that the clinical isolate JEY355 was more resistant to FLC (MIC = 8 μg/ml) than was the transformant JEY431 (MIC = 4 μg/ml).
Fig 3.

Serial dilution susceptibility assays of C. albicans strains onto YPD agar plates. Strains were spotted onto agar plates containing brefeldin A (A) and FLC with cyclosporine (B) and incubated at 35°C for 24 and 48 h.
Expression levels of ERG11 measured by quantitative reverse transcription-PCR (qRT-PCR).
The expression levels of ERG11 were measured in JEY355 in order to investigate the possible role of this gene as a complementary mediator of FLC resistance (Fig. 4). Surprisingly, ERG11 was found to be less expressed in JEY355 compared to the wild type (SC5314). FLC was, however, able to induce ERG11 expression in JEY355 to the levels observed in SC5314: ERG11 mRNA levels were 7- and 2-fold higher in JEY355 and SC5314, respectively, compared to untreated conditions. These data suggest that ERG11 expression may only play a marginal role in the FLC resistance of JEY355.
Fig 4.
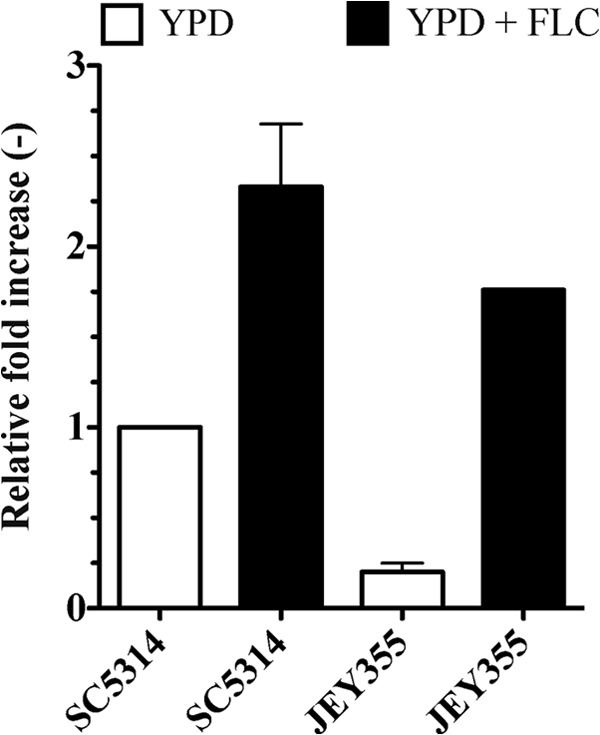
Expression levels of ERG11 in SC5314 and clinical isolate JEY355. Expression levels were measured by qRT-PCR in biological triplicates and technical duplicates. Error bars represent the standard deviations. Strains were exposed to FLC (10 μg/ml) for 90 min.
Susceptibility profiles of C. tropicalis strains.
Isolate JEY162 was the other azole-resistant isolate identified from the Tunisian hospital collection (29). In vitro antifungal susceptibility testing performed by the EUCAST method revealed that the clinical isolate JEY162 was cross-resistant to azoles and AMB (Etest assay) but was still susceptible to echinocandins (Table 3). The MIC values for FLC, VRC, AMB, and CAS were >128, >16, >32, and 0.5 μg/ml, respectively. In YPD broth medium, similar MIC values were observed for azoles (Table 3). These data indicated that CAS was still active in JEY162 and thus suggest that this agent could be still used in the case of azole-AMB cross-resistance of C. tropicalis.
CtERG11 and CtERG3 sequence analyses.
Given the cross-resistance profiles of JEY162, we undertook sterol profile analysis. As shown in Table 5, the major sterol present in this isolate was 14α-methyl fecosterol (71% of all sterols). The presence of this sterol in the absence of FLC treatment suggests a perturbation in both CtERG3 and CtERG11 activities. Effectively, only ergosta-7,22-dienol would be expected as a major sterol if only CtERG3 was affected. The sterol profile observed in JEY162 is also comparable to an erg3 mutant under azole treatment (27) and thus again suggests possible defect in sterol 14α-demethylation in this strain.
Table 5.
Sterol profiles determined by GC-MS of C. tropicalis isolates
| Sterol typea | % Total sterols (mean ± SD)b for isolate: |
||||||
|---|---|---|---|---|---|---|---|
| DSY140 | JEY162 | JEY439 | JEY442 | JEY445 | JEY311 | JEY434 | |
| m/z 480* | 9.1 ± 5.4 | ||||||
| m/z 482* | 5.4 ± 1.0 | ||||||
| Ergosta-dienol | 10.4 ± 2.1 | ||||||
| Zymosterol | 1.9 ± 0.7 | ||||||
| Ergosterol | 73.5 ± 6.1 | 84.1 ± 7.0 | |||||
| Ergosta-5,8-dienol | 4.4 ± 0.7 | ||||||
| Ergosta trienol | 2.5 ± 0.11 | ||||||
| Ergosta-7,22-dienol | 55.8 ± 2.8 | 1.1 ± 0.6 | |||||
| Fecosterol | 1.1 ± 0.6 | 7.2 ± 0.7 | |||||
| 14α-Methyl fecosterol | 64.1 ± 10.8 | 76.4 ± 2.7 | 65.3 ± 5.9 | 9.1 ± 2.1 | |||
| 4,14-Dimethyl zymosterol | 2.5 ± 0.7 | 1.9 ± 0.2 | 1.6 ± 0.1 | 2.7 ± 0.7 | |||
| Ergosta-8-enol | 5.2 ± 0.3 | ||||||
| Ergosta-5,7-dienol | 1.3 ± 1.2 | 7.5 ± 2.6 | |||||
| Episterol | 1.7 ± 0.6 | 14.3 ± 1.2 | 1.4 ± 1.2 | ||||
| Ergosta-7-enol | 6.4 ± 1.4 | ||||||
| m/z 482(2)* | 1.3 ± 0.3 | ||||||
| 14α-Methyl-ergosta-8,24(28)-dien-3α,6β-diol | 52.8 ± 5.8 | ||||||
| Obtusifoliol | 10.4 ± 3.5 | 18.6 ± 3.8 | 12.4 ± 1.4 | 15.9 ± 1.1 | 1.5 ± 1.4 | 13.5 ± 4.4 | |
| Eburicol | 3.7 ± 1.9 | 15.4 ± 7.0 | 9.6 ± 1.4 | 18.2 ± 6.1 | 6.0 ± 2.7 | ||
*, Sterol products with unknown structures.
The sterol composition (i.e., the percentage of total sterols with the standard deviations of three replicate strains grown in YPD) is given. Minor sterols (constituting <1%) were not included in the table.
We therefore expected to observe some alterations in CtERG11 from JEY162. In parallel, we also undertook the cloning of CtERG3 from the same isolate, since it has been established in several reports that ERG11 defects cannot be present without corresponding ERG3 loss of function (43, 44). The CtERG11 and CtERG3 alleles from JEY162 and JEY311 (azole-susceptible clinical isolate from the Tunisian hospitals collection) were amplified and sequenced by using the primers listed in Table 2. A comparison to the wild-type sequences from C. tropicalis MYA-3404 (Broad Institute) strain revealed that JEY162 exhibited a deletion of 132 nucleotides in the CtERG11 ORF and two silent point mutations (C225T and A264G). This homozygous deletion between (+824 and +955 with respect to reference sequence) resulted in the absence of 44 amino acids and a D275V substitution that did not interrupt the ORF but may lead to a total loss of CtErg11 activity. In contrast, JEY311 (an azole-susceptible isolate) harbored three silent mutations (C225T, A264G, and C1554T), which suggests that CtERG11 is functional in this isolate. For CtERG3, our results indicate that only JEY162 harbored a single missense mutation (C774T) causing an amino acid substitution (S258F).
Functional complementation of CtERG11 alleles in S. cerevisiae.
The tetracycline regulatable system was used to determine the activity of CtErg11 of JEY162 by expressing the C. tropicalis CtERG11 alleles in an S. cerevisiae strain background in which the endogenous ERG11 gene is under the control of the Tet promoter (35). The addition of doxycycline in this strain shut off endogenous ERG11 expression, which results in the absence of growth. Any heterologous ERG11 gene could be expressed in this yeast genetic background to probe its functionality.
The spotting of positive colonies into selective medium with doxycycline, as shown in Fig. 5, demonstrates that, in the presence of CtERG11 from JEY311, DSY3961 could still restore growth, thus implying that this gene was functional. In the case of CtERG11 from JEY162, no growth was observed with doxycycline; thus, this allele (now referred as to Cterg11) could not be considered functional.
Fig 5.
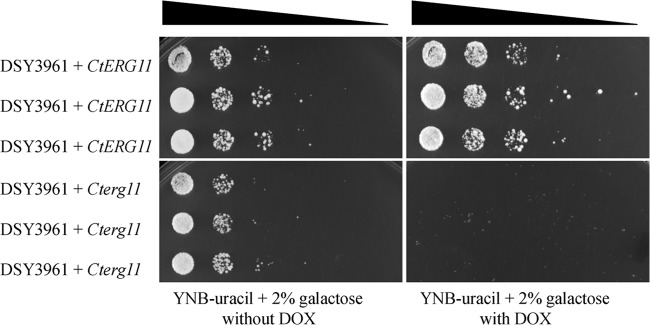
Serial dilution assays of cell suspensions from positive colonies into plates containing YNB-uracil with 2% of galactose in the absence or presence of 20 μg of doxycycline (DOX)/ml. DSY3961+CtERG11 and DSY3961+Cterg11 contained the CtERG11 alleles from JEY311 and JEY162, respectively.
CtERG3 from JEY162 is defective.
In order to verify whether or not CtERG3 from JEY311 was still functional, we used a previously described C. albicans erg3 mutant (VSY2) (16) and introduced in this strain CtERG3 alleles from an azole-susceptible C. tropicalis isolate (JEY311) and from JEY162 cloned in plasmids pJE1 and pJE2, respectively. The plasmids were designed to achieve gene replacement at the ERG3 locus in C. albicans. After transformation of these plasmids into VSY2, resulting in strain JEY424 (containing CtERG3 from JEY162) and JEY425 (containing CtERG3 from JEY311), the total sterols were extracted and analyzed for spectral properties. As shown in Fig. 6, both VSY2 and JEY424 exhibited identical sterol profiles, indicating the absence of ergosterol, whereas JEY425 exhibited a sterol profile typical for the presence of ergosterol. Thus, these data suggest that the single missense mutation (C774T) in CtERG3 present in JEY162 causing an S258F amino acid substitution results in a loss of function. We therefore referred to this allele as Cterg3. These data also confirmed that a loss of function in ERG11 is usually accompanied by defects in ERG3.
Fig 6.
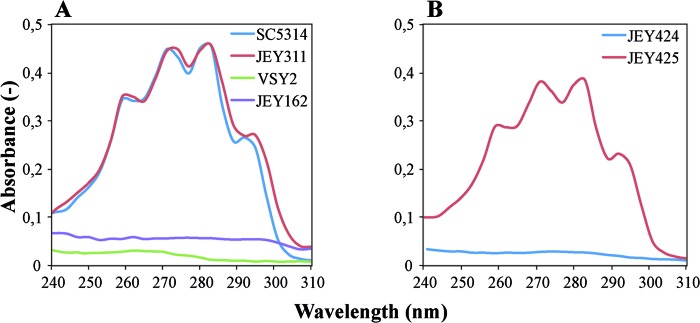
Sterol profiles of azole-susceptible and azole-resistant isolates. Sterols were extracted from cells grown in YPD, and the spectral profiles between 240 and 310 nm were determined by using a NanoDrop ND-1000 UV-Vis spectrophotometer. (A) VSY2 and SC5314, C. albicans isolates with nonfunctional and functional ERG3 alleles, respectively; JEY311, azole-susceptible C. tropicalis clinical isolate with wild-type CtERG3; JEY162, FLC- and AMB-resistant isolate with nonfunctional CtERG11 and CtERG3 alleles. (B) JEY424 and JEY425, isolates with nonfunctional and functional ERG3 alleles, respectively.
Reconstruction of ERG3 and ERG11 defects of JEY162 in a C. tropicalis wild-type isolate.
In order to verify that the ERG3 and ERG11 loss of functions were the major cause of azole-AMB cross-resistance in JEY162, we sequentially introduced the Cterg3 and Cterg11 alleles from JEY162 into the C. tropicalis strain DSY140 (ade2 ura3) that is derived from ATCC 750 (32). After verification of the introduction of defective alleles at both ERG3 and ERG11 loci, the obtained strains were analyzed for drug susceptibility and sterol profiles. We also attempted to obtain erg11 mutants by directly plating erg11/ERG11 heterozygotes onto AMB-containing medium, which was previously shown to facilitate loss of heterozygosity at the ERG11 locus to result in an erg11/erg11 homozygous state (34). As observed in Table 5, the strain JEY439 (Cterg3/Cterg3) contained mainly ergosta-7,22-dienol, a finding that is consistent with the sterol profile for an erg3 mutant. This strain showed intermediate MICs compared to its parent azole-susceptible isolate (DSY140) and JEY162 (Table 4). Strains JEY442 and JEY445 (Cterg3/Cterg3 and Cterg11/Cterg11) derived from JEY439 but by different selection approaches showed 14α-methyl fecosterol to be a major sterol and thus are very similar to the original sterol profile of the clinical strain JEY162. These sterol profiles are consistent with defect in both CtERG3 and CtERG11. Moreover, azoles and AMB MICs were identical between JEY162 and JEY442/JEY445, thus supporting the notion that only CtERG11 and CtERG3 defects are present in JEY162 to yield azole-AMB cross-resistance (Table 3).
After selection of AMB-resistant isolates from the Cterg11/CtERG11 heterozygote and verification of Cterg11 homozygosity, strain JEY434 was obtained. It is noteworthy that the frequency at which the AMB-resistant isolates appeared was between 10−5 and 10−6. This frequency is consistent with mitotic recombination events that were previously documented in similar cases of loss of heterozygosity in C. albicans (34). The sterol profile of this isolate surprisingly revealed 3,6-diol as a major sterol (Table 5). This specific sterol can usually be obtained in wild-type isolates exposed to azoles (45). In JEY434, this specific sterol was obtained even without azole exposure. The presence of 3,6-diol in JEY434 is consistent with Cterg11 deficiency; however, it contradicts other data indicating that erg11 mutants are only viable unless accompanied by ERG3 defects (44). In JEY434, we were not able to identify CtERG3 defects (data not shown). It should be noted that this is the first time that such a profile was detected in a fungal pathogen. At this stage, it cannot be excluded that AMB exposure of the erg11/ERG11 heterozygote as a driving force to obtain loss of heterozygosity at the ERG11 locus could be accompanied by secondary compensatory mutation(s).
DISCUSSION
In this study, we identified molecular mechanisms contributing to the antifungal resistance of clinical Candida spp. isolated from Tunisian hospitals. During the last 2 decades, several studies showed that C. albicans isolates can develop resistance to azole drugs by various mechanisms, including the overexpression of efflux pump membrane proteins, which actively transport antifungal drugs out of the cell (1, 18, 19, 28), or mutations in the target enzyme, decreasing its affinity to azoles (15, 23). Zinc cluster proteins, a family of transcription factors that is unique to the fungal species (40), play a central role in the regulation of genes involved in drug resistance. The expression of CDR1 and CDR2 is controlled by the zinc cluster transcription factor designed Tac1 as Transcriptional Activator of CDR genes (14). The transcription factor Mrr1 was identified as a central regulator of MDR1 expression (18). MRR1 inactivation in azole-resistant isolates results in the loss of MDR1 expression and increased susceptibility to azole drugs (18). MDR1 can also be regulated by additional transcription factors, such as Cap1 and Mcm1 (46). ERG11 expression is controlled by the transcription factor Upc2 (47, 48). In particular, the identification of mutations in the TAC1, MRR1, and UPC2 genes revealed their role in the acquisition of antifungal resistance (49). In the present study, the upregulation of MDR1 in JEY355 suggested the presence of a GOF mutation in MRR1. The mutation at position V877 had not yet been reported. By restoring the MRR1V877F allele in the background of an mrr1Δ/Δ mutant strain, we could demonstrate that V877F substitution was a GOF mutation. The expression level of Mdr1 was increased compared to controls when two GOF alleles were present in C. albicans, and these levels approached those of the clinical isolate JEY355. However, drug susceptibility tests suggested that JEY355 may still contain not-yet-identified additional resistance mediators. We have not addressed additional Mdr1-dependent regulators such as Cap1/Mcm1 (46) in JEY355, since putative additional resistance mechanisms might not be Mdr1 dependent. In addition, given that ERG11 expression levels are intrinsically low in JEY355, it is not likely that its direct regulator, UPC2, contains GOF mutations that are usually associated with high expression levels (22).
Decreasing the affinity between Erg11 and FLC induced by mutations in ERG11 gene are frequent in FLC-resistant C. albicans. In contrast, upregulation of ERG11 causes resistance in a restricted number of strains (1, 15). The sequence analyses of ERG11 showed that JEY355 harbored two alleles with two amino acid substitutions, including D116E and E266D. These two substitutions were reported previously; however, they are not responsible for FLC resistance in C. albicans (50). The expression level of ERG11 mRNA determined by qRT-PCR in JEY355 and SC5314 showed that ERG11 was more expressed in SC5314 than in JEY355 under standard growth conditions. After the induction of ERG11 with FLC, ERG11 mRNA levels in JEY355 were more increased compared to SC5314. These findings reflect that azoles may favor a better response to FLC in JEY355 compared to SC5314 but could not demonstrate unambiguously that ERG11 participates in the azole resistance of JEY355.
Resistance to azole drugs in clinical C. tropicalis isolates has increased, and many reports show the emergence of the pathogenic yeast as major species responsible for causing infections in immunocompromised patients (7, 51, 52). However, only a few reports exist on the molecular understanding of antifungal resistance in C. tropicalis. Cases of acquired resistance to azoles in C. tropicalis have been associated with the overexpression of CtERG11 containing missense mutations (51, 52). Other studies documented that azole resistance correlated with the upregulation of multidrug transporters (53). The results of the present study show an unusual C. tropicalis clinical isolate (JEY162) originating from a yeast collection of hospital isolates from Tunisian hospitals with cross-resistance between azole and polyene drugs. Unfortunately, no correlation between drug exposure and resistance development could be established since no clinical records of antifungal treatments were available. The molecular investigations demonstrate that JEY162 harbored a deletion in CtERG11 and a missense mutation in CtERG3. A few similar examples of azoles and AMB cross-resistance exist in the literature. For example, Hull et al. (54) described in C. glabrata the occurrence of an ERG11 loss-of-function mutation resulting in azole/polyene cross-resistance. This isolate accumulated mostly lanosterol (ca. 75% of all sterols), 14α-methyl fecosterol, and 4,14α-dimethylzymosterol and low amounts of 14α-methyl 3β,6α-diol. This is in contrast to sterol profiles of JEY162 in which mostly 14α-methyl fecosterol was detected. In C. albicans, Martel et al. (27) described an isolate (CA108) with resistance to azoles and AMB with mutations in both ERG11 and ERG5. This mutant showed ergosta-5,7-dienol as a major sterol, which is a signature of ERG5 deficiency. In the study of Martel et al. (27), the ERG11 mutation resulting in an A114S substitution could not be unambiguously attributed to a loss of function. We previously published that erg11 mutants could also be obtained with AMB selection pressure starting from a C. albicans erg11/ERG11 heterozygote (34). The obtained mutant exhibited lanosterol/eburicol as major sterols and thus resembles the sterol profiles of the C. glabrata erg11 mutant identified by Hull et al. (54). Another C. albicans isolate (D10) was reported into which 14α-methyl 3β,6α-diol accumulated, together with lanosterol (44). In terms of the sterol profile, this isolate thus resembles to JEY434, which was demonstrated here to harbor Cterg11 defective alleles. Therefore, it is likely that D10 could also exhibit erg11 defects only.
In conclusion, the present study explored the molecular basis of antifungal resistance in two isolates originating from Tunisian hospitals. The data revealed a novel transcriptional activator mutation and also an unusual sterol mutant. The systematic molecular analysis of clinical isolates again highlights the diversity by which antifungal resistance can arise. Continuing efforts in these analyses are relevant for expanding the repertoire of all possible mutations associated with antifungal resistance in fungal pathogens.
ACKNOWLEDGMENTS
We thank Ridha Kelifa (Service des Laboratoires, Hôpital Habib Thameur, Tunis, Tunisia) and Emna Chaker (Laboratoire de Parasitologie-Mycologie, Hôpital la Rabta, Tunis, Tunisia) for providing Candida clinical isolates.
J.E. acknowledges the Secrétariat d'Etat à l'Éducation et à la Recherche SER de la Confédération Suisse and the Ministère de l'Enseignement Supérieur et Recherche Scientifique de la Tunisie for a training fellowship.
Footnotes
Published ahead of print 29 April 2013
REFERENCES
- 1. Sanglard D, Kuchler K, Ischer F, Pagani JL, Monod M, Bille J. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39:2378–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohkubo T, Sugawara Y, Takayama T, Kokudo N, Makuuchi M. 2012. The risk factors of fungal infection in living-donor liver transplantations. J. Hepatobiliary Pancreat. Sci. 19:382–388 [DOI] [PubMed] [Google Scholar]
- 3. Slavin MA, Sorrell TC, Marriott D, Thursky KA, Nguyen Q, Ellis DH, Morrissey CO, Chen SC. 2010. Candidaemia in adult cancer patients: risks for fluconazole-resistant isolates and death. J. Antimicrob. Chemother. 65:1042–1051 [DOI] [PubMed] [Google Scholar]
- 4. Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39:309–317 [DOI] [PubMed] [Google Scholar]
- 5. Pfaller MA, Boyken L, Hollis RJ, Messer SA, Tendolkar S, Diekema DJ. 2005. In vitro activities of anidulafungin against more than 2,500 clinical isolates of Candida spp., including 315 isolates resistant to fluconazole. J. Clin. Microbiol. 43:5425–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfaller MA, Diekema DJ, Rinaldi MG, Barnes R, Hu B, Veselov AV, Tiraboschi N, Nagy E, Gibbs DL. 2005. Results from the ARTEMIS DISK Global Antifungal Surveillance Study: a 6.5-year analysis of susceptibilities of Candida and other yeast species to fluconazole and voriconazole by standardized disk diffusion testing. J. Clin. Microbiol. 43:5848–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kothavade RJ, Kura MM, Valand AG, Panthaki MH. 2010. Candida tropicalis: its prevalence, pathogenicity, and increasing resistance to fluconazole. J. Med. Microbiol. 59:873–880 [DOI] [PubMed] [Google Scholar]
- 8. Chai YA, Wang Y, Khoo AL, Chan FY, Chow C, Kumarasinghe G, Singh K, Tambyah PA. 2007. Predominance of Candida tropicalis bloodstream infections in a Singapore teaching hospital. Med. Mycol. 45:435–439 [DOI] [PubMed] [Google Scholar]
- 9. Yang YL, Cheng MF, Wang CW, Wang AH, Cheng WT, Lo HJ, Hospitals T. 2010. The distribution of species and susceptibility of amphotericin B and fluconazole of yeast pathogens isolated from sterile sites in Taiwan. Med. Mycol. 48:328–334 [DOI] [PubMed] [Google Scholar]
- 10. Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cuenca-Estrella M, Gomez-Lopez A, Mellado E, Garcia-Effron G, Rodriguez-Tudela JL. 2004. In vitro activities of ravuconazole and four other antifungal agents against fluconazole-resistant or -susceptible clinical yeast isolates. Antimicrob. Agents Chemother. 48:3107–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franz R, Kelly SL, Lamb DC, Kelly DE, Ruhnke M, Morschhauser J. 1998. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob. Agents Chemother. 42:3065–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Warrilow AG, Martel CM, Parker JE, Melo N, Lamb DC, Nes WD, Kelly DE, Kelly SL. 2010. Azole binding properties of Candida albicans sterol 14-α demethylase (CaCYP51). Antimicrob. Agents Chemother. 54:4235–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 3:1639–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14α-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 42:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vale-Silva LA, Coste AT, Ischer F, Parker JE, Kelly SL, Pinto E, Sanglard D. 2012. Azole resistance by loss of function of the sterol Δ5,6-desaturase gene (ERG3) in Candida albicans does not necessarily decrease virulence. Antimicrob. Agents Chemother. 56:1960–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morio F, Pagniez F, Lacroix C, Miegeville M, Le Pape P. 2012. Amino acid substitutions in the Candida albicans sterol Δ5,6-desaturase (Erg3p) confer azole resistance: characterization of two novel mutants with impaired virulence. J. Antimicrob. Chemother. 67:2131–2138 [DOI] [PubMed] [Google Scholar]
- 18. Morschhauser J, Barker KS, Liu TT, Bla BWJ, Homayouni R, Rogers PD. 2007. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog. 3:e164. 10.1371/journal.ppat.0030164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanglard D, Ischer F, Monod M, Bille J. 1996. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob. Agents Chemother. 40:2300–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dunkel N, Blass J, Rogers PD, Morschhauser J. 2008. Mutations in the multidrug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 69:827–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain-of-function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268. 10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhauser J, Rogers PD. 2012. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 11:1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng LJ, Wan Z, Wang XH, Li RY, Liu W. 2010. Relationship between antifungal resistance of fluconazole-resistant Candida albicans and mutations in ERG11 gene. Chin. Med. J. 123:544–548 [PubMed] [Google Scholar]
- 24. Xiao L, Madison V, Chau AS, Loebenberg D, Palermo RE, McNicholas PM. 2004. Three-dimensional models of wild-type and mutated forms of cytochrome P450 14α-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob. Agents Chemother. 48:568–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geber A, Hitchcock CA, Swartz JE, Pullen FS, Marsden KE, Kwon-Chung KJ, Bennett JE. 1995. Deletion of the Candida glabrata ERG3 and ERG11 genes: effect on cell viability, cell growth, sterol composition, and antifungal susceptibility. Antimicrob. Agents Chemother. 39:2708–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Torelli R, Posteraro B, Ferrari S, La Sorda M, Fadda G, Sanglard D, Sanguinetti M. 2008. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 68:186–201 [DOI] [PubMed] [Google Scholar]
- 27. Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Rolley N, Kelly DE, Kelly SL. 2010. Identification and characterization of four azole-resistant erg3 mutants of Candida albicans. Antimicrob. Agents Chemother. 54:4527–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schubert S, Rogers PD, Morschhauser J. 2008. Gain-of-function mutations in the transcription factor MRR1 are responsible for overexpression of the MDR1 efflux pump in fluconazole-resistant Candida dubliniensis strains. Antimicrob. Agents Chemother. 52:4274–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eddouzi J, Lohberger A, Vogne C, Manai M, Sanglard D. Identification and antifungal susceptibility of a large collection of yeast strains isolated in Tunisian Hospitals. Med. Mycol., in press [DOI] [PubMed] [Google Scholar]
- 30. Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karababa M, Coste AT, Rognon B, Bille J, Sanglard D. 2004. Comparison of gene expression profiles of Candida albicans azole-resistant clinical isolates and laboratory strains exposed to drugs inducing multidrug transporters. Antimicrob. Agents Chemother. 48:3064–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng Q, Sanglard D, Vanhanen S, Liu H, Bombelli P, Smith A, Slabas A. 2005. Candida yeast long-chain fatty alcohol oxidase is a c-type haemoprotein and plays an important role in long chain fatty acid metabolism. Biochim. Biophys. Acta 1735:192–203 [DOI] [PubMed] [Google Scholar]
- 33. Coste A, Turner V, Ischer F, Morschhauser J, Forche A, Selmecki A, Berman J, Bille J, Sanglard D. 2006. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172:2139–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. 2003. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob. Agents Chemother. 47:2404–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alcazar-Fuoli L, Mellado E, Cuenca-Estrella M, Sanglard D. 2011. Probing the role of point mutations in the cyp51A gene from Aspergillus fumigatus in the model yeast Saccharomyces cerevisiae. Med. Mycol. 49:276–284 [DOI] [PubMed] [Google Scholar]
- 36. Subcommittee on Antifungal Susceptibility Testing of the ESCMID European Committee for Antimicrobial Susceptibility Testing 2008. EUCAST definitive document EDef 7.1: method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts. Clin. Microbiol. Infect. 14:398–405 [DOI] [PubMed] [Google Scholar]
- 37. Vandeputte P, Pradervand S, Ischer F, Coste AT, Ferrari S, Harshman K, Sanglard D. 2012. Identification and functional characterization of Rca1, a transcription factor involved in both antifungal susceptibility and host response in Candida albicans. Eukaryot. Cell 11:916–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kelly SL, Lamb DC, Corran AJ, Baldwin BC, Kelly DE. 1995. Mode of action and resistance to azole antifungals associated with the formation of 14α-methylergosta-8,24(28)-dien-3β,6a-diol. Biochem. Biophys. Res. Commun. 207:910–915 [DOI] [PubMed] [Google Scholar]
- 39. Hiller D, Sanglard D, Morschhäuser J. 2006. Overexpression of the MDR1 gene is sufficient to confer increased resistance to toxic compounds in Candida albicans. Antimicrob. Agents Chemother. 50:1365–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol. Mol. Biol. Rev. 70:583–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morio F, Loge C, Besse B, Hennequin C, Pape PL. 2010. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn. Microbiol. Infect. Dis. 66:373–384 [DOI] [PubMed] [Google Scholar]
- 42. Sanglard D, Ischer F, Monod M, Bille J. 1997. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 143(Pt 2):405–416 [DOI] [PubMed] [Google Scholar]
- 43. Lees N, Skaggs B, Kirsch D, Bard M. 1995. Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae: a review. Lipids 30:221–226 [DOI] [PubMed] [Google Scholar]
- 44. Bard M, Lees N, Turi T, Craft D, Cofrin L, Barbuch R, Koegel C, Loper J. 1993. Sterol synthesis and viability of ERG11 (cytochrome P450 lanosterol demethylase) mutations in Saccharomyces cerevisiae and Candida albicans. Lipids 28:963–967 [DOI] [PubMed] [Google Scholar]
- 45. Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2010. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14α-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob. Agents Chemother. 54:3578–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mogavero S, Tavanti A, Senesi S, Rogers PD, Morschhauser J. 2011. Differential requirement of the transcription factor Mcm1 for activation of the Candida albicans multidrug efflux pump MDR1 by its regulators Mrr1 and Cap1. Antimicrob. Agents Chemother. 55:2061–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Silver PM, Oliver BG, White TC. 2004. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3:1391–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob. Agents Chemother. 49:1745–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanglard D, Coste A, Ferrari S. 2009. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 9:1029–1050 [DOI] [PubMed] [Google Scholar]
- 50. Alvarez-Rueda N, Fleury A, Morio F, Pagniez F, Gastinel L, Le Pape P. 2011. Amino acid substitutions at the major insertion loop of Candida albicans sterol 14α-demethylase are involved in fluconazole resistance. PLoS One 6:e21239. 10.1371/journal.pone.0021239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang C, Dong D, Yu B, Cai G, Wang X, Ji Y, Peng Y. 2013. Mechanisms of azole resistance in 52 clinical isolates of Candida tropicalis in China. J. Antimicrob. Chemother. 68:778–785 [DOI] [PubMed] [Google Scholar]
- 52. Vandeputte P, Larcher G, Berges T, Renier G, Chabasse D, Bouchara JP. 2005. Mechanisms of azole resistance in a clinical isolate of Candida tropicalis. Antimicrob. Agents Chemother. 49:4608–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barchiesi F, Calabrese D, Sanglard D, Falconi Di Francesco L, Caselli F, Giannini D, Giacometti A, Gavaudan S, Scalise G. 2000. Experimental induction of fluconazole resistance in Candida tropicalis ATCC 750. Antimicrob. Agents Chemother. 44:1578–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hull CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2012. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob. Agents Chemother. 56:4223–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]