

Abstract
S. aureus combats cell wall antibiotic stress by altered gene expression mediated by various environmental signal sensors. In this study, we examined the transcriptional regulation of trfA, a gene related to mecA of Bacillus subtilis encoding an adaptor protein implicated in multiple roles, notably, proteolysis and genetic competence. Despite strong sequence similarity to B. subtilis mecA, the function of S. aureus trfA remains largely unexplored; however, its deletion leads to almost complete loss of resistance to oxacillin and glycopeptide antibiotics in glycopeptide-intermediate S. aureus (GISA) derivatives of methicillin-susceptible or methicillin-resistant S. aureus (MRSA) clinical or laboratory isolates. Northern blot analysis and 5′ rapid amplification of cDNA ends (RACE) mapping revealed that trfA was expressed monocistronically by three promoters. Cell wall-active antibiotic exposure led to both increased trfA transcription and enhanced steady-state TrfA levels. trfA promoter regulation was not dependent upon the cell wall stress sentinel VraSR and other sensory stress systems, such as GraRS, WalkRK, Stk1/Stp1, and SigB. Notably, we discovered that the global oxidative-stress regulator Spx controlled trfA transcription. This finding was also confirmed using a strain with enhanced Spx levels resulting from a defect in yjbH, encoding a Spx-interacting protein governing Spx proteolytic degradation. A cohort of clinical GISA strains revealed significant steady-state upregulation of trfA compared to corresponding susceptible parental strains, further supporting a role for trfA in antibiotic resistance. These data provide strong evidence for a link between cell wall antibiotic stress and evoked responses mediated by an oxidative-stress sensor.
INTRODUCTION
Diseases caused by Staphylococcus aureus range from relatively benign soft tissue infections to life-threatening invasive illness (1, 2). Of particular concern are infections arising from encounters with strains with altered susceptibility to antibiotics, such as methicillin-resistant S. aureus (MRSA). Glycopeptide antibiotics (vancomycin and teicoplanin) are frequently considered the mainstay for therapy of MRSA infections. Recent studies suggest, however, that relatively minor increases in MIC levels of glycopeptides, even at the upper range of glycopeptide susceptibility, are correlated with higher rates of therapeutic failure (3–6). This troubling issue has prompted recent changes in glycopeptide susceptibility breakpoints and underscores the need for alternative pharmacotherapeutic agents.
High-level resistance to glycopeptides in S. aureus, termed VRSA (vancomycin-resistant S. aureus), arises from infrequent horizontal acquisition of Tn1546 encoding the multiprotein VanA complex from Enterococcus faecalis. Mechanistically, the Van complex enzymes alter the stem peptide of cell wall precursor molecules so that glycopeptides no longer bind efficiently. Worldwide, less than a dozen examples of VRSA strains have occurred since the first outbreak was reported (7, 8). In contrast to high-level resistance, clinical S. aureus isolates showing low-level glycopeptide resistance (MIC range, 4 to 8 μg/ml) have been reported since 1997 and are referred to as glycopeptide-intermediate S. aureus (GISA). Low-level glycopeptide resistance is much more prevalent, and mechanistically, it is thought to occur by stepwise acquisition of mutations that confer survival advantage in the face of drug encounters (2, 9, 10). A complete understanding of the mechanism of acquisition of low-level resistance is currently lacking, although genetic studies to date have identified mutations in genes such as graRS, tcaA, stp1, vraRS, yjbH, walKR, and trfAB that contribute to the acquisition or loss of the resistance phenotype (10–14). The two-component histidine kinase sensor genes graRS and vraRS, as well as the serine/threonine kinase stk1-stp phosphatase, are phosphosignaling systems controlling a large number of downstream genes, suggesting that the mechanism of low-level glycopeptide resistance is complex. This is perhaps not surprising in light of the fact that glycopeptides inhibit end-stage cell wall assembly steps occurring outside the plasma membrane, and thus, for topological reasons, initiating a response to drug encounters must involve transmembrane-signaling steps.
Previous studies of a unique set of clinical isolates in our laboratory led to the discovery of two adjacent genes linked with teicoplanin resistance (Teir) that we named trfA and trfB for teicoplanin-resistant factors A and B (14). Detailed analysis showed that individual or combined deletion of trfA and/or trfB led to the loss of glycopeptide or oxacillin resistance in an in vitro-selected teicoplanin-resistant derivative of ISP794, as well as in the clinical GISA strain NRS3 (14). A clear functional role of the trfA or trfB gene in S. aureus remains undefined. Conceptual translation of trfA indicates that its product most closely resembles the MecA adaptor protein of Listeria monocytogenes and Bacillus subtilis (14), whereas the conceptual translation of trfB shows strong similarity with YjbF of B. subtilis, also called CoiA in Streptococcus pneumoniae (14). Studies with both organisms suggest that YjbF/CoiA contributes to competence for genetic transformation (15).
Importantly, the MecA adaptor protein has no known functional relation to the S. aureus mecA encoding the PBP2′ enzyme, which confers the MRSA phenotype on strains acquiring any of several allotypes of the horizontally transmitted SCCmec element. In B. subtilis, the MecA adaptor has been extensively studied and plays a regulatory role in genetic competence development, motility, and autolysis (16, 17). Notably, B. subtilis MecA serves dual functions as an assembly factor/chaperone for the AAA+ Hsp100/Clp ATPase family member ClpC and as a substrate specificity factor for regulated proteolysis (18).
A few substrates bound by MecA in B. subtilis and fed to proteolytic machinery are ComK, CtsR, and MurAA, the enzyme controlling the first committed step in cell wall biosynthesis (19–22). By virtue of strong overall sequence similarity, S. aureus TrfA is most likely a MecA ortholog, although this awaits experimental confirmation. MecA-dependent control of regulated proteolysis, and especially MurAA turnover, naturally suggests a link between MecA/TrfA function and biological mechanisms that exist to combat cell wall-active antibiotics.
In order to further our understanding of pathways that lead to altered sensitivity to cell wall-active antibiotics in S. aureus, we report in this study the detailed transcriptional regulation of trfA. Our results surprisingly reveal that trfA is a previously unrecognized member of the cell wall stress regulon, and we present evidence that it is under the transcriptional control of the global thiol/oxidative-stress regulator Spx. These findings are discussed in light of the growing body of evidence linking the bactericidal activities of various antibiotics to the production of reactive oxygen species (ROS).
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table 1. The rsbU-defective NCTC8325 strain ISP794 (MIC = 1 μg/ml) and its teicoplanin derivative AR376 (MIC = 8 μg/ml) were described previously (10, 14). All S. aureus strains were grown in Mueller-Hinton broth (MHB) and E. coli strains in Luria-Bertani medium supplemented with 100 μg/ml of ampicillin or carbenicillin when required. All antibiotics were obtained from Sigma-Aldrich, except teicoplanin (Sanofi-Aventis) and vancomycin (Sandoz). Diamide(1,1′-azobis(N,N-dimethylformamide)) was obtained from Sigma-Aldrich.
Table 1.
Bacterial strains and plasmids used in this study
| Strain/plasmid | Revelant genotype | Characteristics | Source/reference |
|---|---|---|---|
| Strains | |||
| RN4220 | 8325-4; r− m+; restriction-defective laboratory strain | 72 | |
| ISP794 | 8325 pig 131; rsbU mutant | 73 | |
| AR852 | ISP794; rsbU+ | ISP794; (rsbUVW-sigB)+ tetL nearby | 24 |
| AR376 | ISP4-2-1 | GISA in vitro-derived strain | 14 |
| AR774 | vraS (G45R) | ISP794; vraS (G45R); Kanr nearby | 10 |
| AR826 | stp1 (Q12stop) | ISP794, stp1 (Q12stop); Kanr nearby | 10 |
| AR1079 | yjbH (K23stop) | ISP794, yjbH (K23stop); Eryr nearby | 10 |
| Δspx | 8325-4-derived strain | spx-deleted strain | 30 |
| spx+ | 8325-4 Δspx Pspx-spx::geh | spx-deleted strain chromosomally complemented with the intact copy of spx inserted into the geh locus | 30 |
| Δspx rsbU+ | 8325-4-derived strain | spx-deleted strain; (rsbUVW-sigB)+; tetL nearby | This study |
| AR612 | ISP4-2-1; ΔtrfA | ISP4-2-1; trfA::tetK | 14 |
| AR916 | ISPΔvraSR | ISP794; ΔvraSR::Kanr | 74 |
| Pair 1 non-GISA | Clinical MRSA glycopeptide-susceptible strain of pair 1 | 27 | |
| Pair 1 GISA | Clinical MRSA glycopeptide-resistant strain of pair 1 | 27 | |
| Pair 2 non-GISA | Clinical MRSA glycopeptide-susceptible strain of pair 2 | 27 | |
| Pair 2 GISA | Clinical MRSA glycopeptide-resistant strain of pair 2 | 27 | |
| Pair 3 non-GISA | Clinical MRSA glycopeptide-susceptible strain of pair 3 | 27 | |
| Pair 3 GISA | Clinical MRSA glycopeptide-resistant strain of pair 3 | 27 | |
| Pair 4 non-GISA | Clinical MRSA glycopeptide-susceptible strain of pair 4 | 27 | |
| Pair 4 GISA | Clinical MRSA glycopeptide-resistant strain of pair 4 | 27 | |
| Pair 5 non-GISA | Clinical MRSA glycopeptide-susceptible strain of pair 5 | 27 | |
| Pair 5 GISA | Clinical MRSA glycopeptide-resistant strain of pair 5 | 27 | |
| BL2 α(DE3) | E. coli | IPTG-inducible T7 RNA polymerase | New England Biolabs |
| Plasmids | |||
| pBluescript II KS(+) | Cloning vector; Ampr | ||
| pAM845 | pKS+ containing a Kpn-Pst fragment coding for TrfA | This study | |
| pTYB12 | N-terminal fusion IMPACT intein and chitin binding domain plasmid | New England Biolabs | |
| pAM873 | pTYB12 containing trfA cloned into NdeI-PstI restriction sites | This study |
Total RNA extraction.
Overnight bacterial cultures were diluted in MHB (1/100) and grown at 37°C with agitation to an optical density at 600 nm (OD600) of 0.6. When indicated, oxacillin (1 μg/ml), teicoplanin (10 μg/ml), vancomycin (10 μg/ml), d-cycloserine (10 μg/ml), ciprofloxacin (1 μg/ml), or diamide (5 mM) was added and incubated for an additional hour (for oxacillin), 10 min (for vancomycin, teicoplanin, and d-cycloserine), or 30 min (for ciprofloxacin and diamide). Bacteria were harvested, and RNA extraction was performed as previously described (14). The absence of contaminating DNA was always verified for every experiment by PCR using quantitative real-time PCR (qRT-PCR) probes in the absence of reverse transcription.
qRT-PCR.
The mRNA levels were determined by qRT-PCR using the one-step reverse transcriptase qPCR Master Mix Kit (Eurogentec, Seraing, Belgium) as described previously (23). Primers and probes for trfA, spx, and hu were designed using PrimerExpress software (version 1.5; Applied Biosystems) and obtained from Eurogentec (see Table S1 in the supplemental material). Primers and probes for 16S, vraR, and asp23 genes were previously described (23–25). Reverse transcription and PCR were performed using primers and probes at a concentration of 0.2 and 0.1 μM, respectively. For Hu gene detection, primers and probes were all used at a concentration of 0.1 μM. All mRNA levels were normalized on the basis of their 16S rRNA levels, which were assayed in each round of qRT-PCR as internal controls, as described previously (23).
Expression of recombinant TrfA protein.
The open reading frame of the trfA gene (N315 SA0857) was PCR amplified with primers indicated in Table S1 in the supplemental material and cloned in pBluescriptII KS(+). A sequence-verified trfA fragment was next subcloned into E. coli expression vector pTYB12 (New England Biolabs) using NdeI-PstI sites, generating plasmid pAM873. E. coli strain BL21 λ(DE3) (New England Biolabs) containing pTYB12-TrfA protein was grown in Luria-Bertani medium containing carbenicillin at 100 μg/ml to an OD600 of 0.7 and induced with 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for an additional 150 min at room temperature with vigorous shaking. Bacteria were harvested by low-speed centrifugation and resuspended in Laemmli buffer, and whole-cell extracts were used in Western blot analysis as described below.
Anti-TrfA antibody production.
Rabbit polyclonal antibodies were raised in specific-pathogen-free (SPF) New Zealand White rabbits against a 15-amino-acid synthetic peptide (FSREDLWTNRKRGEE, corresponding to amino acids 25 to 39 of SA0857) with MBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester)-conjugated keyhole limpet hemocyanin (KLH) carrier protein using the 87-day protocol of Eurogentec (Seraing, Belgium). Specific antiserum was further affinity purified against the immunizing peptide. The peptide was affinity coupled using AF-amino Toyopearl 650 M (TOSOH Bioscience GmbH, Germany) and an equal mixture of ACH (Na-[e-aminocaproyl]-dl-homoarginine hexylester) Sepharose and CNBr Sepharose. The antibody was eluted with 0.1 M glycine, pH 2.5. The specificity of the antibody was assessed by Western blot analysis using E. coli whole-cell extracts expressing inducible recombinant TrfA (see below and Fig. 3).
Fig 3.
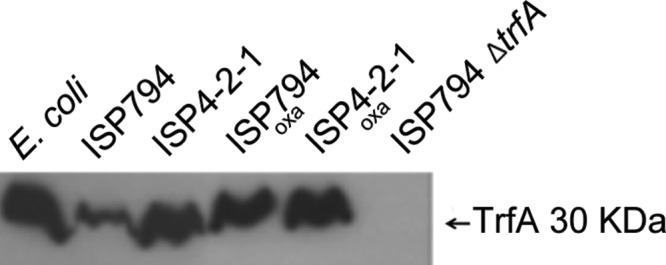
Western blot analysis of TrfA. Total soluble protein extracts (75 μg) from E. coli and S. aureus strains were loaded in SDS 15% acrylamide gels. TrfA protein (30 kDa) was detected using rabbit-polyclonal anti-peptide-TrfA antibodies (see Materials and Methods), and a typical Western blot is shown. As reference controls, an aliquot of a whole-cell extract of IPTG-induced S. aureus TrfA produced in E. coli (lane 1 from left) and an extract from an ISP794 control strain lacking trfA (lane 6) were included. Lanes 2 and 3, respectively, compare extracts derived from ISP794 or it isogenic Teir derivative. Lanes 4 and 5 show the results for ISP794 and ISP4-2-1, each exposed to 1 μg/ml of oxacillin (lane 4 and 5). The position of the 30-kDa protein marker is shown on the right.
Western blot analysis.
Western blot analyses of protein extracts from S. aureus were performed as follows. Overnight cultures of strains in MHB growing at 37°C with agitation were diluted (1/100) in MHB and grown at 37°C with agitation to an OD600 of 0.5. When indicated, oxacillin (1 μg/ml) was added, and bacteria were grown for an additional hour. After centrifugation, the cell pellets were washed and resuspended in 500 μl TE buffer (10 mM Tris, pH 7.5, 1 mM EDTA). Bacterial cells were disrupted by adding 500 μl of acid-washed glass beads (100 to 200 μm; Sigma) and using a FastPrep cell disrupter (MP Biomedicals). The cell debris was separated from soluble protein extracts by centrifugation at 14,000 rpm (10 min at 4°C). The supernatant was concentrated on Amicon spin columns (10-kDa cutoff; Milian, Geneva, Switzerland). Protein concentrations were determined by Bradford assay (Bio-Rad) using bovine serum albumin standards. Aliquots of proteins (75 μg) were loaded on 15% SDS-PAGE gels and blot transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad). After blocking using 5% low-fat milk in phosphate-buffered saline, TrfA was probed with anti-TrfA antibody at a 1/5,000 dilution, followed by incubation with a secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody at a 1/50,000 dilution. Chemiluminescence was detected using the Western Pico Super Signal reagent (Pierce).
Northern blotting.
TrfA transcript analysis was essentially performed as previously described (24). Total RNA (6 μg) was separated in 1% agarose formaldehyde gels and blotted to nylon membranes (Hybond-N; Amersham). An [α-32P]UTP (Hartmann Analytics; FP-110; 15 TBq/mmol)-labeled trfA riboprobe was generated from pAM845 (Table 1; see Table S1 in the supplemental material). After plasmid linearization with Acc65I and gel purification, an [α-32P]UTP-labeled complementary antisense transcript was produced by in vitro transcription using T7 polymerase essentially as described previously (26). Unincorporated nucleotide was removed by passage over a microspin ProbeQuant G-50 column (GE Healthcare). The riboprobe mixture was treated with DNase I (Promega; RQ1) to eliminate the template DNA, extracted with phenol-chloroform-isoamyl alcohol (25:24:1), and precipitated with ethanol in the presence of 16 μg glycogen carrier. The pellet was washed with ice-cold 70% ethanol, dried, and resuspended in a minimal volume of TE. An aliquot was tested for probe purity on a 6% polyacrylamide, 8 M urea sequencing gel. The membrane was prehybridized with QuikHyb (Stratagene) buffer and incubated overnight with the trfA riboprobe at 65°C. Washes were done as follows: the first wash at 55°C with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% SDS for 15 min and a second wash with 1× SSC, 0.1% SDS for 10 min at 55°C, followed by one wash with 0.1× SSC, 0.1% SDS for 10 min at 55°C. The membrane was transferred to 3MM paper (Amersham Hyperfilms) without drying, sealed, and autoradiographed.
Mapping of the trfA transcriptional start site.
The 5′ ends of trfA transcripts were mapped using a Smarter RACE cDNA Amplification Kit from Clontech (catalog no. 634923). Total RNA was extracted from an oxacillin-induced culture of strain ISP794, conditions under which levels of trfA transcription were shown to be high in pilot experiments. Gene-specific cDNA with a Smarter IIA tail was generated using 1 μg of total RNA and a trfA PstI-specific primer (see Table S1 in the supplemental material), according to the manufacturer's protocol. Rapid amplification of cDNA ends (RACE) was next generated using UPM (Clontech kit) and trfA (see Table S1 in the supplemental material) nested primers, with the following specific PCR program: 5 cycles of 94°C for 30 s, 94°C for 30 s, and 72°C for 5 min; 5 cycles of 94°C for 30 s, 70°C for 30 s, and 72°C for 5 min; and, finally, 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 5 min. The PCR products were run in a 2% agarose gel, and each gel band was extracted and sequenced using a trfA nested primer. Primer-transcript junction sites revealed by direct sequencing permitted unambiguous assignment of the 5′ transcript ends.
RESULTS
Quantitative trfA transcription analysis in a cohort of clinical GISA strains.
Our previously published work revealed that trfA deletion significantly reduced glycopeptide and oxacillin resistance levels in both GISA clinical and laboratory-derived isolates (14). This result led us to the hypothesis that trfA plays an important role in modulating resistance levels to these antibiotics and, in addition, led us to predict that (i) trfA mRNA levels are altered upon addition of cell wall-active antibiotics and (ii) trfA steady-state mRNA levels were significantly higher in MRSA strains displaying stable reduced susceptibility to glycopeptides than in their susceptible counterparts. As a first step to test these possibilities, we addressed the hypothesis that steady-state trfA transcript levels were altered in a set of five isogenic clinical-strain pairs consisting of a pretherapy susceptible MRSA isolate and its corresponding posttherapy GISA derivative (27). We observed that trfA transcript levels analyzed by qRT-PCR were indeed significantly (P < 0.05) increased by 2- to 3-fold in four of the five GISA derivatives tested compared to their non-GISA parents (Fig. 1). Identical transcriptional trfA alterations are not expected in all GISA strains, since different genetic changes could drive emergence of the GISA phenotype. We conclude from these results that the GISA phenotype could be correlated with increased trfA transcription and that trfA levels could be considered a characteristic feature of some GISA strains.
Fig 1.
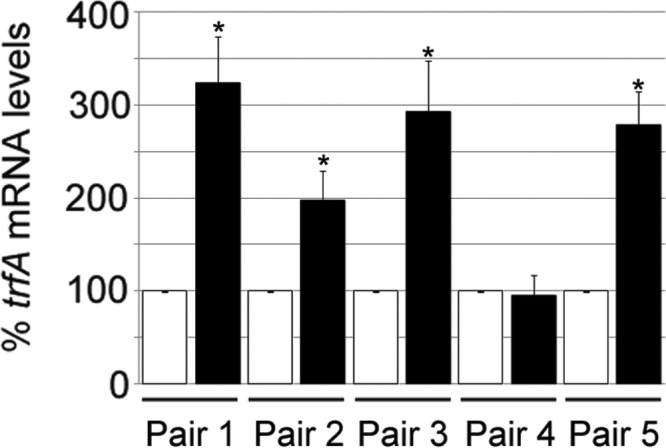
Analysis of trfA mRNA levels in glycopeptide-susceptible and GISA paired clinical strains. Steady-state levels of trfA transcripts were determined by qRT-PCR and normalized to 16S rRNA. All GISA mRNA levels (black bars) were compared to the corresponding glycopeptide-susceptible strains (white bars). The values represent the means and standard errors of the mean (SEM) of three independent experiments. *, results significantly different by Student's two-tailed t test (P < 0.05).
Cell wall-active antibiotics induce trfA gene transcription.
We next explored how trfA transcription was affected by exposure of cells to cell wall-active antibiotics. We chose to use the 8325-derived strain ISP794 for these studies because of the extensive use of the strain in our and other laboratories for genetic analysis of antibiotic resistance mechanisms. We observed a significant 3-fold induction (P < 0.01) of trfA following a 1-h exposure of strain ISP794 to 1 μg/ml oxacillin (Fig. 2). These conditions were chosen because oxacillin has been widely used to induce cell wall stress in a variety of S. aureus strains (28, 29). In contrast, we observed that control mRNA levels of a housekeeping gene encoding the nucleoid protein Hu were not significantly affected by oxacillin treatment (data not shown).
Fig 2.

Induction of trfA mRNA levels by cell wall-active antibiotics. Steady-state mRNA levels of trfA and hu (control) were determined by qRT-PCR and normalized to 16S rRNA. The mRNA levels of antibiotic-treated bacteria were compared to that of strain ISP794 in the absence of antibiotic addition. oxa, oxacillin; van, vancomycin; tec, teicoplanin; D-cyclo, d-cycloserine; Cip, ciprofloxacin. The values reported represent the means and SEM of at least three independent experiments. *, results significantly different by Student's two-tailed t test (P < 0.05). Note the absence of trfA induction by the non-cell-wall-targeting drug ciprofloxacin.
To extend these findings, we further tested the effects of other cell wall-active antibiotics: vancomycin, teicoplanin, and d-cycloserine. We observed that trfA transcription was significantly (P < 0.05) induced in every case compared to an untreated control, by 2-, 2.8-, and 2.4-fold following brief exposure to 10 μg/ml vancomycin, teicoplanin, and d-cycloserine, respectively. In contrast, no significant transcriptional alteration (exceeding ±25% of the untreated control) of the housekeeping control gene hu mRNA levels was observed with the same treated samples (data not shown). To address the question of whether trfA transcriptional induction was obtained exclusively with cell wall-active antibiotics, we exposed cells to ciprofloxacin, an inhibitor of DNA gyrase and type IV topoisomerase, and observed no induction of trfA transcription (Fig. 2). Collectively, we conclude from these results that trfA transcription is induced by four antibiotics encompassing three distinct classes and known to induce cell wall stress.
Steady-state TrfA protein levels rise in response to oxacillin challenge.
Affinity-purified rabbit polyclonal anti-TrfA antibody was prepared against an amino-terminal TrfA peptide (see Materials and Methods). The antibody specificity was first confirmed using recombinant S. aureus TrfA produced in Escherichia coli (Fig. 3, lane 1, and data not shown). Western blot analysis consistently detected a band apparently migrating at 30 kDa, consistent with the predicted 28.3-kDa TrfA molecular mass. TrfA migrating with the same apparent molecular mass was detected by Western blotting of whole-cell protein extracts derived from S. aureus Teis strain ISP794 and its Teir (GISA) derivative, ISP4-2-1 (Fig. 3, lanes 2 and 3). In contrast, no TrfA was detected in an S. aureus strain containing an internal disruption of trfA (Fig. 3, compare lanes 2, 3, and 6). Notably, we consistently observed higher steady-state TrfA levels in Teir ISP4-2-1 than in its Teis parent (Fig. 3, compare lanes 2 and 3). TrfA levels were also significantly increased in ISP794 exposed to oxacillin (under the same conditions as for Fig. 2) compared to the untreated control (Fig. 3, lanes 2 and 4). Oxacillin did not result in a significant increase in steady-state levels of TrfA in strain ISP4-2-1. It is worth mentioning that we consistently observed TrfA levels that were comparable between extracts from strain ISP4-2-1 and ISP794 treated with oxacillin.
Taken together, we conclude from these results that transcriptional induction of trfA by various stimuli is mirrored by comparable increased production of the TrfA protein.
Transcriptional analysis of trfA.
The induction of trfA by antibiotics targeting cell wall biosynthesis led us to examine trfA transcriptional regulation in detail. As a first step to dissect the regulatory pathways controlling trfA expression in S. aureus, trfA transcription was first examined by both Northern analysis and 5′-RACE mapping. Northern blots consistently detected three transcripts of approximately 0.9, 0.8, and 0.75 kb in both strains ISP794 and ISP4-2-1, using a strand-specific trfA riboprobe spanning only trfA coding sequence (Fig. 4A). Under the conditions of the assay, we consistently noted that the three transcripts were of comparable intensities, suggesting equivalent promoter usage. The transcripts detected were sufficient to encode full-length TrfA (predicted 239 amino acids) (Fig. 4B), and furthermore, Northern hybridization with a specific spx probe confirmed the presence of two monocistronic upstream spx transcripts of 0.6 and 0.5 kb, as previously described (30) (Fig. 4A). Characterization by 5′-RACE amplification of the 5′ ends of each trfA transcript of S. aureus allowed unambiguous identification of 3 distinct start sites, located at coordinates −178, −98, and −31 nucleotides upstream of the TrfA ATG translation initiation codon, respectively (Fig. 4C and D). Taking into consideration the length of the trfA coding sequence, but not 3′ untranslated regions, the three start sites produced calculated trfA transcripts of 898, 818, and 751 bp, corroborating the trfA monocistronic transcripts observed by Northern blotting.
Fig 4.

Transcriptional analysis of trfA. (A) Northern blot analysis of trfA and spx in strains ISP794 and ISP4-2-1, using 32P-radiolabeled RNA trfA- and spx-specific probes. The arrows indicate the trfA (0.7-kb, 0.8-kb, and 0.9-kb) and spx (0.6-kb and 0.5-kb) transcripts. Ethidium bromide-stained rRNA from the agarose gel prior to blot transfer is shown as a loading control. (B) Schematic representation of B. subtilis (B.s.) MecA and S. aureus (S.a.) TrfA/MecA proteins. Throughout the text, we refer to S. aureus TrfA to avoid confusion with S. aureus mecA, a gene unrelated to the MecA adaptor protein of B. subtilis and encoding an alternative penicillin binding protein responsible for the MRSA phenotype in the organism. The N-terminal and C-terminal protein regions are depicted in black and gray and show 57% and 31% protein identity, respectively. The linker region (white) is smaller in B. subtilis MecA. (C) Sequence of the trfA promoter region. The three different nucleotides corresponding to transcriptional start sites detected by 5′RACE are shown in boldface, and the rho-independent transcriptional terminator of spx is underlined. RBS, ribosome binding site. Asterisks mark each 10-nucleotide region. (D) Schematic diagram showing spx and trfA gene transcription organization. The arrows indicate spx or trfA transcripts produced from the corresponding promoters. Predicted rho-independent transcriptional terminators are shown.
Collectively, from these results, we conclude that trfA is transcribed monocistronically from three promoters. This finding contrasts with a previously published prediction placing trfA within a large multigene operon (31).
Searching for regulators of trfA.
Previous studies had established that many members of the cell wall stress regulon were under the control of the cell wall stress sentinel two-component system VraRS (29, 32). In light of the aforementioned induction of trfA by various cell wall-active antibiotics, the impact of VraRS on transcriptional regulation of trfA was tested in the presence or absence of oxacillin. We observed an identical pattern of trfA transcription by Northern blotting in a ΔvraSR disruption mutant of ISP794 compared with its isogenic wild-type parent when incubated in antibiotic-free medium (see Fig. S1A in the supplemental material). Importantly, incubation of ISP794 and its ΔvraSR mutant in an oxacillin-containing medium resulted in identical antibiotic-triggered upregulation of trfA transcription for both strains (see Fig. S1B in the supplemental material). We also detected no impact on trfA transcription by disruption of GraRS, another two-component regulator of cell wall antibiotic stress (data not shown). A third two-component phosphosignaling system (TCS), WalKR is essential in S. aureus, precluding direct examination of its genetic disruption upon trfA transcription. However, recent comprehensive mapping of WalRK-regulated genes and determination of a WalR consensus binding site failed to provide any evidence for its role in trfA regulation (33). Collectively, we conclude that none of the TCSs implicated in cell wall sensing play a detectable role in the transcriptional regulation of trfA.
The alternative sigma factor σB mediates many responses to diverse environmental stresses in S. aureus, so we next examined whether it played any role in trfA transcriptional regulation. Many laboratory strains derived from 8325 (including ISP794) show a defective σB stress response because of constitutive sequestration of σB by an anti-sigma factor resulting from a defective RsbU phosphatase (34).
We performed qRT-PCR using both ISP794 (rsbU mutant) and its corresponding rsbU+ restored derivative strain ISP794 (rsbU+) (see Fig. S1C in the supplemental material) (24). As expected, the restoration of rsbU+ in ISP794 strongly restored σB activity, since significant (P < 0.05) 7-fold-increased mRNA levels were observed for asp23, a gene known to be exclusively σB dependent (35). In contrast, no difference was observed for vraR mRNA levels known to be regulated in a σB-independent manner (36). We observed only a minor change in basal trfA transcription (<1.5-fold) in the rsbU+ restored strain compared to ISP794 (see Fig. S1C in the supplemental material). The σB regulon has been extensively studied in S. aureus, and consistent with our findings, no σB consensus promoter motif or altered trfA mRNA levels were reported by transcriptome analysis (36). Since the addition of various cell wall-active antibiotics results in a robust induction of trfA transcription in ISP794 in the absence of significant σB activity in this strain background (Fig. 2), we conclude from these experiments that trfA is not part of the σB regulon.
trfA transcription is modulated by stabilization of Spx, a global regulator of thiol/oxidative stress.
The strong constitutive upregulation of trfA transcription observed in the Teir strain ISP4-2-1 compared to Teis ISP794 (Fig. 4A) prompted us to examine in detail which of the three previously studied mutations discovered in ISP4-2-1 (10) could account for this observation. ISP4-2-1 harbors two nonsense mutations: one in stp1 (Q12stop), encoding a serine/threonine phosphatase, and one in yjbH (K23stop), encoding a negative regulator of the thiol/oxidative-stress global regulator Spx. The third mutation is a nonconservative missense mutation (G45R) in VraS, the sensor histidine kinase of the VraRS two-component system. In our previous study, we had reconstructed each mutation found in ISP4-2-1 and prepared all possible single, double, and triple mutations in the ISP794 genetic background (10).
The results of qRT-PCR analysis (Fig. 5) using the three single-mutation derivative strains of ISP794 shows that neither vraSG45R nor stp1Q12stop had any significant impact on trfA transcription. In contrast, we observed that yjbHK23stop significantly increased trfA transcription, by 4.2-fold compared with wild-type ISP794. Interestingly, this transcriptional upregulation of trfA was at least equivalent to the 3.5-fold increase observed in ISP4-2-1 compared with its parent, ISP794 (Fig. 5). We conclude that the loss of yjbH most likely fully accounts for the observed altered regulation of trfA transcription in strain ISP4-2-1.
Fig 5.

Analysis of vraS* (vraSG45R), stp1* (stp1Q12stop), yjbH* (yjbHK23stop), and spx on trfA mRNA levels. Strains harboring each of the three nucleotide changes detected in strain ISP4-2-1 compared to ISP794 (10) were used to determine which mutation(s) conferred enhanced trfA expression in strain ISP4-2-1. Steady-state levels of trfA transcripts were determined by qRT-PCR and normalized to 16S rRNA. Steady-state levels of trfA were also compared between the Δspx mutant and its spx+ restored derivative (Δspx-c), as well as between ISP794 treated with the thiol-specific oxidant diamide and untreated ISP794. The values represent the means and SEM of three independent experiments. *, results significantly different by Student's two-tailed t test (P < 0.05).
Disruption of yjbH in S. aureus, as well as in B. subtilis, is known to result in stabilization of Spx (10, 37, 38). Thiol/oxidative stress triggers a release of YjbH from Spx, permitting it to interact with the α-C-terminal domain (αCTD) of RNA polymerase to direct expression of Spx-regulated genes (38–40). In order to determine whether trfA was regulated by Spx, we performed qRT-PCR analysis on RNA obtained from a strain lacking spx or a derivative strain where spx had been restored by chromosomal insertion of cloned spx+ under the control of its own promoter (30). The data in Fig. 5 revealed a significant (3.1-fold) increase in trfA transcription in the complemented spx deletion strain compared to the spx deletion mutant. To further demonstrate Spx-dependent transcription of trfA, we hypothesized that induction conditions known to increase Spx protein levels via thiol stress (37, 41) would enhance transcription of the trfA gene. The addition of the thiol-specific oxidant diamide (5 mM) to strain ISP794 strongly induced trfA transcription by approximately 9-fold compared to the untreated control (Fig. 5). Taken together, these results led us to conclude that trfA is regulated by Spx.
Moreover, similar spx-dependent transcription of trfA in both rsbU mutant and rsbU+ strain backgrounds was observed (see Fig. S2 in the supplemental material). Restoration of the defective rsbU mutant gene present in the Δspx strain by phage transduction from donor strain AR852 (24) carrying the rsbU+VW-sigB operon tetracycline inserted nearby shows identical patterns of trfA expression in both backgrounds, with or without oxacillin administration.
Effect of oxacillin on spx expression.
Our finding that trfA transcription could be induced by a variety of antibiotics targeting various steps in cell wall biosynthesis led us to ask next whether cell wall antibiotic stress altered spx transcription. Using qRT-PCR, we measured spx transcription in oxacillin-treated compared to untreated bacteria, together with hu, encoding a nucleoid protein not known to be significantly altered by cell wall antibiotic stress (Fig. 6A). The results showed that neither spx nor hu transcription was detectably altered by the addition of oxacillin under conditions where trfA was otherwise strongly induced by this treatment (Fig. 2 and 5B). In contrast to these results, Western blot analysis performed with extracts from the same samples used for RNA extraction revealed strongly enhanced Spx protein levels in oxacillin-treated extracts compared to the untreated control (Fig. 6B). These data strongly suggest that posttranscriptional regulation of Spx accounts for the induction of trfA transcription following oxacillin exposure. Taken together with our findings with yjbH noted above, our results further suggest a model whereby exposure to cell wall-active antibiotics results in signals that disrupt the negative regulation of Spx by YjbH by an as-yet-unknown mechanism in S. aureus.
Fig 6.
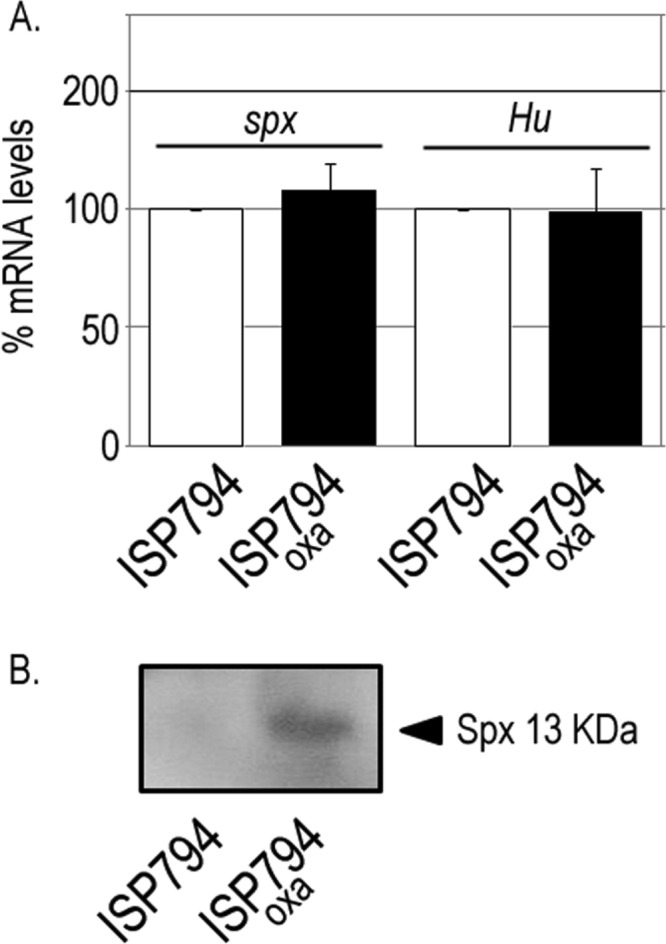
Effect of oxacillin on spx mRNA levels. (A) Steady-state levels of spx and hu transcripts were determined by qRT-PCR and normalized to 16S rRNA in strain ISP794 compared to oxacillin-treated (oxa) ISP794 cells. The values represent the means and SEM of three independent experiments. (B) Western blot analysis of total soluble protein extracts (75 μg) of S. aureus ISP794 or the same strain treated with oxacillin loaded in SDS-15% acrylamide gels. Spx protein (13 kDa) was detected using rabbit polyclonal anti-S. aureus Spx antibody as previously described (10).
DISCUSSION
The mechanisms underlying low-level glycopeptide resistance are multifactorial and still poorly understood. Since glycopeptides are considered to be among first-line drugs for the treatment of MRSA, there is considerable research devoted to understanding these mechanisms, as well as interest in identifying target genes or lead compounds that would restore sensitivity of MRSA strains to drugs such as β-lactams.
In previous work from our laboratory using both GISA and MRSA strains, we discovered that S. aureus trfA played an important role in both glycopeptide and β-lactam resistance in the organism (14). Study of TrfA/MecA in other organisms, notably B. subtilis, suggests that it has numerous biological functions, including as an assembly chaperone for ClpC, as an adaptor protein for regulated proteolysis of bound substrates, and as a regulator of transcription factor function (42–45). Precisely how trfA contributes to drug resistance in S. aureus is unknown.
In the present study, we report that a panel of four cell wall-active antibiotics leads to induction of the S. aureus trfA promoter and that transcription is dependent upon Spx, a global regulator of oxidative-stress defense. We did not find evidence that linked trfA transcription to other known sensory systems related to cell wall stress or cell wall antibiotic resistance, such as VraRS or GraRS (32, 46), or the global alternative stress sigma factor SigB (36). Detailed transcriptome and/or binding site analysis also failed to reveal that trfA was controlled by WalKR/YycFG (33, 47) or detectably altered by disruption of the Stk1 kinase (48, 49). Our present findings, together with other published studies, indicate that encounters with cell wall-active antibiotics not only can trigger the induction of multiple sensory pathways that rely, for example, upon transmembrane phosphosignaling mechanisms, but also can trigger pathways that lead to posttranscriptional stabilization of Spx and concomitant changes in the expression of Spx-dependent genes. Figure 7 depicts a model summarizing these various sensory systems and their collective roles in mediating responses to antibiotic-induced cell wall damage.
Fig 7.

Model of proposed pathways regulating the evoked response to cell wall antibiotic encounter. Stk1/Stp, serine/threonine kinase-phosphatase sensor; RNAP, RNA polymerase. The dashed arrow denotes the presumptive pathway leading to Spx protein stabilization.
A central role for trfA as a modulator of multiple stress defenses is underscored by other published reports. A survey of global transcription profiling studies in S. aureus using various strains uncovered conditions showing that trfA is induced as part of the stringent response triggered by exposure to mupirocin (50), is upregulated following nitrosative stress and exposure to subinhibitory sodium nitrite (51), and responds to the proton ionophore carbonyl cyanide m-chloromethyl hydrazone (52). A microarray-based transcriptome study also revealed trfA transcriptional induction by both daptomycin (a calcium-dependent membrane-active lipopeptide antibiotic) and oxacillin, but it was not explored in detail (52).
In B. subtilis, supporting our results presented here, induction of a proteolytically stabilized Spx variant (Spx-DD) leads to upregulation of mecA and trfA (53), and recent work using chromatin immunoprecipitation methods revealed Spx occupancy of the trfA promoter under basal conditions that becomes strongly enhanced following exposure to diamide (54). Additional studies in B. subtilis have established that disulfide stress triggers the stringent response and that most major oxidative-stress genes were induced by disulfide stress (55). Loss of YjbH, a negative regulator and interacting partner of Spx, has been linked to reduced sensitivity to diamide (38) in B. subtilis, as well as nitrosative stress via altered susceptibility to sodium nitroprusside (56). In S. aureus, disruption of the corresponding YjbH ortholog results in pleiotropic effects that include altered sensitivity to β-lactam and glycopeptide antibiotics (10, 37, 57), as well as enhanced peptidoglycan cross-linking and overproduction of penicillin binding protein PBP4 (57).
The diversity of stress stimuli channeled through Spx comes primarily from studies in B. subtilis. The five spx promoters are controlled by four different sigma factors; at least two stress-sensitive repressors, PerR and YodB, binding to the RNA polymerase αCTD governed by a redox-sensitive CXXC switch; and protein levels modulated by ClpXP directed by the Spx partner protein and negative regulator YjbH (40, 58–60). The recently discovered YjbH-interacting protein YirB acts as an antiadaptor by inhibiting YjbH-mediated proteolysis of Spx (39). Many of these regulatory features are likely preserved in S. aureus (30, 38), with the exception of multiple sigma factor control, the fact that spx is bicistronic in B. subtilis but monocistronic in S. aureus, and the apparent lack of a protein with similarity to YirB (W. L. Kelley, unpublished observations).
Of the cell wall-active drugs tested in this study that lead to upregulation of trfA, three have sites of action outside the cell membrane (vancomycin, teicoplanin, and oxacillin), while one targets a cytosolic enzyme (d-cycloserine). The mechanism leading to spx-dependent upregulation of trfA in response to these various agents targeting cell wall biosynthesis must ultimately take into consideration how the various drug-induced stresses are sensed. Our observation that Spx protein levels were dramatically stabilized using oxacillin as a stimulus whereas spx transcription was unaffected strongly suggests that cell wall antibiotic stress acts on Spx primarily at the posttranscriptional level. Redox regulation of cysteine residues in YjbH has been proposed as a mechanism governing the proteolytic turnover of Spx (40, 57). In this scenario, oxidation of cysteines would have the dual effect of disrupting YjbH-Spx interaction, as well as possibly promoting Spx-αCTD interaction through oxidation of the Spx cysteine switch (58). The discovery of YirB in B. subtilis raises the additional possibility that competitor proteins can also disrupt YjbH-Spx interaction. In this regard, it is tempting to speculate that cell wall antibiotic stress in S. aureus results in the production of ROS, triggers induction of hypothetical YjbH antiadaptor protein expression, or modulates YjbH expression or turnover, resulting in altered YjbH-Spx stoichiometry. These mechanisms are not necessarily mutually exclusive. Evidence exists that S. aureus encounters with certain bactericidal antibiotics, including β-lactams and vancomycin, can trigger production of ROS, resulting in bacterial killing (61, 62). However, recent studies have challenged this model, and thus, the role of ROS production linked to antibiotic killing is controversial (63, 64).
A key question is what does a cell gain by stabilizing Spx in response to cell wall antibiotic stress and among the ensuing consequences driving trfA transcription? Among the genes included in the Spx regulon are those dedicated to oxidative-stress defense and redox homeostasis, which are clearly beneficial (41). In addition, Spx is thought to mediate both positive and negative regulation of genes that impact intermediary metabolism and has been proposed to exert a metabolic brake to attenuate growth and production of endogenous ROS until damage is repaired and the noxious stimulus is removed (41, 60). This notion is reminiscent of the SOS-response-mediated inhibition of cell division or the growth arrest mediated by PBP inhibition and the DpiAB TCS in E. coli in response to β-lactams (65). Finally, previous work from our laboratory (10) revealed that loss of spx resulted in a significant decrease in the frequency of emergence of low-level glycopeptide mutants, suggesting that Spx-dependent gene regulation impacts antibiotic resistance at many levels.
The understanding of glycopeptide resistance in S. aureus is far from complete. Mutations in numerous distinct genes, either individually or collectively, can contribute to altered susceptibilities. Since signaling systems often appear mutated, it is clear that effects mediated by genes under the control of these signaling systems will ultimately have an effect on drug resistance. Our present results add to this evolving story and, further, suggest a role for TrfA in some, but perhaps not all, pathways that govern glycopeptide and other cell wall-active antibiotic resistance mechanisms in S. aureus.
A role for TrfA as an adaptor and assembly factor for ClpC opens numerous possibilities for regulated proteolysis and cellular processes controlled by ClpCP (66–69). Global studies of ClpC reveal this network to be quite extensive (70, 71), and preliminary work shows that deletion of clpC in S. aureus closely mirrors the effect of trfA deletion with respect to glycopeptide resistance (A. Renzoni, unpublished data). Furthermore, cell wall antibiotic resistance is often correlated with changes in cell wall thickness, peptidoglycan cross-linking, or decreased autolysis (10). As an adaptor protein linked with proteolysis, TrfA could conceivably contribute to the regulation of any of these steps (17, 42, 43). Preliminary data indeed show that trfA deletion significantly affects cell wall thickness and morphology (Renzoni, unpublished). Clearly, identifying TrfA-interacting proteins and elucidating its role as a ClpCP adaptor will be of paramount importance in future studies.
The cell wall stress regulon/stimulon includes genes induced by certain antibiotics and subject to VraR-dependent regulation (29, 52). Although trfA is not formally part of the cell wall stress regulon by these criteria, trfA nevertheless is clearly upregulated in response to multiple cell wall-active antibiotics. Our study therefore highlights a previously unrecognized link between cell wall antibiotic stress and gene expression governed by an RNA polymerase-interacting factor responding to oxidative stress. These findings clearly reveal that the cell wall stress regulon is more complex than previously imagined.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Swiss National Science Foundation grants 310030-125109 (D.P.L.) and 3100A0-120428 (W.L.K.) and the Canton of Geneva.
We thank Pierre Vaudaux for critical comments and Dorte Frees and Hanne Ingmer (Royal Veterinary and Agricultural University, Denmark) for strains and helpful comments.
Footnotes
Published ahead of print 29 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00220-13.
REFERENCES
- 1. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 2. Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23:99–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gould IM, Cauda R, Esposito S, Gudiol F, Mazzei T, Garau J. 2011. Management of serious meticillin-resistant Staphylococcus aureus infections: what are the limits? Int. J. Antimicrob. Agents 37:202–209 [DOI] [PubMed] [Google Scholar]
- 4. Kullar R, Davis SL, Levine DP, Rybak MJ. 2011. Impact of vancomycin exposure on outcomes in patients with methicillin-resistant Staphylococcus aureus bacteremia: support for consensus guidelines suggested targets. Clin. Infect. Dis. 52:975–981 [DOI] [PubMed] [Google Scholar]
- 5. Tenover FC, Sinner SW, Segal RE, Huang V, Alexandre SS, McGowan JE, Jr, Weinstein MP. 2009. Characterisation of a Staphylococcus aureus strain with progressive loss of susceptibility to vancomycin and daptomycin during therapy. Int. J. Antimicrob. Agents 33:564–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lodise TP, Graves J, Evans A, Graffunder E, Helmecke M, Lomaestro BM, Stellrecht K. 2008. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob. Agents Chemother. 52:3315–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perichon B, Courvalin P. 2009. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:4580–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu W, Murray PR, Huskins WC, Jernigan JA, McDonald LC, Clark NC, Anderson KF, McDougal LK, Hageman JC, Olsen-Rasmussen M, Frace M, Alangaden GJ, Chenoweth C, Zervos MJ, Robinson-Dunn B, Schreckenberger PC, Reller LB, Rudrik JT, Patel JB. 2010. Dissemination of an Enterococcus Inc18-like vanA plasmid, associated with vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54:4314–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berger-Bachi B, McCallum N. 2006. State of the knowledge of bacterial resistance. Injury 37(Suppl. 2):S20–S25 [DOI] [PubMed] [Google Scholar]
- 10. Renzoni A, Andrey DO, Jousselin A, Barras C, Monod A, Vaudaux P, Lew D, Kelley WL. 2011. Whole genome sequencing and complete genetic analysis reveals novel pathways to glycopeptide resistance in Staphylococcus aureus. PLoS One 6:e21577. 10.1371/journal.pone.0021577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maki H, McCallum N, Bischoff M, Wada A, Berger-Bachi B. 2004. tcaA inactivation increases glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 48:1953–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuroda M, Kuwahara-Arai K, Hiramatsu K. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem. Biophys. Res. Commun. 269:485–490 [DOI] [PubMed] [Google Scholar]
- 13. Meehl M, Herbert S, Gotz F, Cheung A. 2007. Interaction of the GraRS two-component system with the VraFG ABC transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 51:2679–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Renzoni A, Kelley WL, Barras C, Monod A, Huggler E, Francois P, Schrenzel J, Studer R, Vaudaux P, Lew DP. 2009. Identification by genomic and genetic analysis of two new genes playing a key role in intermediate glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 53:903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kramer N, Hahn J, Dubnau D. 2007. Multiple interactions among the competence proteins of Bacillus subtilis. Mol. Microbiol. 65:454–464 [DOI] [PubMed] [Google Scholar]
- 16. Dubnau D, Roggiani M. 1990. Growth medium-independent genetic competence mutants of Bacillus subtilis. J. Bacteriol. 172:4048–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rashid MH, Tamakoshi A, Sekiguchi J. 1996. Effects of mecA and mecB (clpC) mutations on expression of sigD, which encodes an alternative sigma factor, and autolysin operons and on flagellin synthesis in Bacillus subtilis. J. Bacteriol. 178:4861–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, Bukau B, Turgay K. 2006. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 25:1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kock H, Gerth U, Hecker M. 2004. MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol. Microbiol. 51:1087–1102 [DOI] [PubMed] [Google Scholar]
- 20. Kruger E, Zuhlke D, Witt E, Ludwig H, Hecker M. 2001. Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J. 20:852–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakano S, Zheng G, Nakano MM, Zuber P. 2002. Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J. Bacteriol. 184:3664–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turgay K, Hamoen LW, Venema G, Dubnau D. 1997. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 11:119–128 [DOI] [PubMed] [Google Scholar]
- 23. Vaudaux P, Francois P, Bisognano C, Kelley WL, Lew DP, Schrenzel J, Proctor RA, McNamara PJ, Peters G, Von Eiff C. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect. Immun. 70:5428–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galbusera E, Renzoni A, Andrey DO, Monod A, Barras C, Tortora P, Polissi A, Kelley WL. 2011. Site-specific mutation of Staphylococcus aureus VraS reveals a crucial role for the VraR-VraS sensor in the emergence of glycopeptide resistance. Antimicrob. Agents Chemother. 55:1008–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Renzoni A, Francois P, Li D, Kelley WL, Lew DP, Vaudaux P, Schrenzel J. 2004. Modulation of fibronectin adhesins and other virulence factors in a teicoplanin-resistant derivative of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 48:2958–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelley WL, Georgopoulos C. 1997. Positive control of the two-component RcsC/B signal transduction network by DjlA: a member of the DnaJ family of molecular chaperones in Escherichia coli. Mol. Microbiol. 25:913–931 [DOI] [PubMed] [Google Scholar]
- 27. Uckay I, Bernard L, Buzzi M, Harbarth S, Francois P, Huggler E, Ferry T, Schrenzel J, Renzoni A, Vaudaux P, Lew DP. 2012. High prevalence of isolates with reduced glycopeptide susceptibility in persistent or recurrent bloodstream infections due to methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 56:1258–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yin S, Daum RS, Boyle-Vavra S. 2006. VraSR two-component regulatory system and its role in induction of pbp2 and vraSR expression by cell wall antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Utaida S, Dunman PM, Macapagal D, Murphy E, Projan SJ, Singh VK, Jayaswal RK, Wilkinson BJ. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719–2732 [DOI] [PubMed] [Google Scholar]
- 30. Pamp SJ, Frees D, Engelmann S, Hecker M, Ingmer H. 2006. Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. J. Bacteriol. 188:4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. ten Broeke-Smits NJ, Pronk TE, Jongerius I, Bruning O, Wittink FR, Breit TM, van Strijp JA, Fluit AC, Boel CH. 2010. Operon structure of Staphylococcus aureus. Nucleic Acids Res. 38:3263–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807–821 [DOI] [PubMed] [Google Scholar]
- 33. Delaune A, Dubrac S, Blanchet C, Poupel O, Mader U, Hiron A, Leduc A, Fitting C, Nicolas P, Cavaillon JM, Adib-Conquy M, Msadek T. 2012. The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun. 80:3438–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giachino P, Engelmann S, Bischoff M. 2001. Sigma(B) activity depends on RsbU in Staphylococcus aureus. J. Bacteriol. 183:1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kullik I, Giachino P, Fuchs T. 1998. Deletion of the alternative sigma factor sigmaB in Staphylococcus aureus reveals its function as a global regulator of virulence genes. J. Bacteriol. 180:4814–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bachi B, Projan S. 2004. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 186:4085–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engman J, Rogstam A, Frees D, Ingmer H, von Wachenfeldt C. 2012. The YjbH adaptor protein enhances proteolysis of the transcriptional regulator Spx in Staphylococcus aureus. J. Bacteriol. 194:1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Larsson JT, Rogstam A, von Wachenfeldt C. 2007. YjbH is a novel negative effector of the disulphide stress regulator, Spx, in Bacillus subtilis. Mol. Microbiol. 66:669–684 [DOI] [PubMed] [Google Scholar]
- 39. Kommineni S, Garg SK, Chan CM, Zuber P. 2011. YjbH-enhanced proteolysis of Spx by ClpXP in Bacillus subtilis is inhibited by the small protein YirB (YuzO). J. Bacteriol. 193:2133–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garg SK, Kommineni S, Henslee L, Zhang Y, Zuber P. 2009. The YjbH protein of Bacillus subtilis enhances ClpXP-catalyzed proteolysis of Spx. J. Bacteriol. 191:1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakano S, Kuster-Schock E, Grossman AD, Zuber P. 2003. Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 100:13603–13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prepiak P, Defrancesco M, Spadavecchia S, Mirouze N, Albano M, Persuh M, Fujita M, Dubnau D. 2011. MecA dampens transitions to spore, biofilm exopolysaccharide and competence expression by two different mechanisms. Mol. Microbiol. 80:1014–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mei Z, Wang F, Qi Y, Zhou Z, Hu Q, Li H, Wu J, Shi Y. 2009. Molecular determinants of MecA as a degradation tag for the ClpCP protease. J. Biol. Chem. 284:34366–34375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang F, Mei Z, Qi Y, Yan C, Xiang S, Zhou Z, Hu Q, Wang J, Shi Y. 2009. Crystal structure of the MecA degradation tag. J. Biol. Chem. 284:34376–34381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Persuh M, Mandic-Mulec I, Dubnau D. 2002. A MecA paralog, YpbH, binds ClpC, affecting both competence and sporulation. J. Bacteriol. 184:2310–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Falord M, Mader U, Hiron A, Debarbouille M, Msadek T. 2011. Investigation of the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress response and cell wall signal transduction pathways. PLoS One 6:e21323. 10.1371/journal.pone.0021323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dubrac S, Bisicchia P, Devine KM, Msadek T. 2008. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70:1307–1322 [DOI] [PubMed] [Google Scholar]
- 48. Burnside K, Lembo A, de Los Reyes M, Iliuk A, Binhtran NT, Connelly JE, Lin WJ, Schmidt BZ, Richardson AR, Fang FC, Tao WA, Rajagopal L. 2010. Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 5:e11071. 10.1371/journal.pone.0011071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Donat S, Streker K, Schirmeister T, Rakette S, Stehle T, Liebeke M, Lalk M, Ohlsen K. 2009. Transcriptome and functional analysis of the eukaryotic-type serine/threonine kinase PknB in Staphylococcus aureus. J. Bacteriol. 191:4056–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 188:6739–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schlag S, Nerz C, Birkenstock TA, Altenberend F, Gotz F. 2007. Inhibition of staphylococcal biofilm formation by nitrite. J. Bacteriol. 189:7911–7919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Muthaiyan A, Silverman JA, Jayaswal RK, Wilkinson BJ. 2008. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob. Agents Chemother. 52:980–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakano S, Nakano MM, Zhang Y, Leelakriangsak M, Zuber P. 2003. A regulatory protein that interferes with activator-stimulated transcription in bacteria. Proc. Natl. Acad. Sci. U. S. A. 100:4233–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rochat T, Nicolas P, Delumeau O, Rabatinova A, Korelusova J, Leduc A, Bessieres P, Dervyn E, Krasny L, Noirot P. 2012. Genome-wide identification of genes directly regulated by the pleiotropic transcription factor Spx in Bacillus subtilis. Nucleic Acids Res. 40:9571–9583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leichert LI, Scharf C, Hecker M. 2003. Global characterization of disulfide stress in Bacillus subtilis. J. Bacteriol. 185:1967–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rogstam A, Larsson JT, Kjelgaard P, von Wachenfeldt C. 2007. Mechanisms of adaptation to nitrosative stress in Bacillus subtilis. J. Bacteriol. 189:3063–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gohring N, Fedtke I, Xia G, Jorge AM, Pinho MG, Bertsche U, Peschel A. 2011. New role of the disulfide stress effector YjbH in beta-lactam susceptibility of Staphylococcus aureus. Antimicrob. Agents Chemother. 55:5452–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakano S, Erwin KN, Ralle M, Zuber P. 2005. Redox-sensitive transcriptional control by a thiol/disulphide switch in the global regulator, Spx. Mol. Microbiol. 55:498–510 [DOI] [PubMed] [Google Scholar]
- 59. Reyes DY, Zuber P. 2008. Activation of transcription initiation by Spx: formation of transcription complex and identification of a cis-acting element required for transcriptional activation. Mol. Microbiol. 69:765–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zuber P. 2009. Management of oxidative stress in Bacillus. Annu. Rev. Microbiol. 63:575–597 [DOI] [PubMed] [Google Scholar]
- 61. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810 [DOI] [PubMed] [Google Scholar]
- 62. Kohanski MA, DePristo MA, Collins JJ. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 37:311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216 [DOI] [PubMed] [Google Scholar]
- 65. Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629–1631 [DOI] [PubMed] [Google Scholar]
- 66. Frees D, Savijoki K, Varmanen P, Ingmer H. 2007. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63:1285–1295 [DOI] [PubMed] [Google Scholar]
- 67. Donegan NP, Thompson ET, Fu Z, Cheung AL. 2010. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 192:1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chatterjee I, Neumayer D, Herrmann M. 2010. Senescence of staphylococci: using functional genomics to unravel the roles of ClpC ATPase during late stationary phase. Int. J. Med. Microbiol. 300:130–136 [DOI] [PubMed] [Google Scholar]
- 69. Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D. 2013. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J. Proteome Res. 12:547–558 [DOI] [PubMed] [Google Scholar]
- 70. Chatterjee I, Schmitt S, Batzilla CF, Engelmann S, Keller A, Ring MW, Kautenburger R, Ziebuhr W, Hecker M, Preissner KT, Bischoff M, Proctor RA, Beck HP, Lenhof HP, Somerville GA, Herrmann M. 2009. Staphylococcus aureus ClpC ATPase is a late growth phase effector of metabolism and persistence. Proteomics 9:1152–1176 [DOI] [PubMed] [Google Scholar]
- 71. Chatterjee I, Maisonneuve E, Ezraty B, Herrmann M, Dukan S. 2011. Staphylococcus aureus ClpC is involved in protection of carbon-metabolizing enzymes from carbonylation during stationary growth phase. Int. J. Med. Microbiol. 301:341–346 [DOI] [PubMed] [Google Scholar]
- 72. Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712 [DOI] [PubMed] [Google Scholar]
- 73. Stahl ML, Pattee PA. 1983. Confirmation of protoplast fusion-derived linkages in Staphylococcus aureus by transformation with protoplast DNA. J. Bacteriol. 154:406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jousselin A, Renzoni A, Andrey DO, Monod A, Lew DP, Kelley WL. 2012. The posttranslocational chaperone lipoprotein PrsA is involved in both glycopeptide and oxacillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56:3629–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.