

Abstract
New classes of antiparasitic drugs active against Trypanosoma brucei are needed to combat human African trypanosomiasis. Inhibitors of methionyl-tRNA synthetase (MetRS) have excellent potential to be developed for this purpose (S. Shibata, J. R. Gillespie, A. M. Kelley, A. J. Napuli, Z. Zhang, K. V. Kovzun, R. M. Pefley, J. Lam, F. H. Zucker, W. C. Van Voorhis, E. A. Merritt, W. G. Hol, C. L. Verlinde, E. Fan, and F. S. Buckner, Antimicrob. Agents Chemother. 55:1982–1989, 2011). In order to assess the potential for resistance to develop against this new class of inhibitors, T. brucei cultures were grown in the presence of MetRS inhibitors or comparison drugs. Resistance up to ∼50 times the baseline 50% inhibitory concentration (IC50) was induced against a MetRS inhibitor after ∼120 days. A similar level of resistance to the clinical drug eflornithine was induced after ∼50 days and for pentamidine after ∼80 days. Thus, resistance was induced more slowly against MetRS inhibitors than against clinically used drugs. The parasites resistant to the MetRS inhibitor were shown to overexpress MetRS mRNA by a factor of 35 over the parental strain. Southern analysis indicated that the MetRS gene was amplified in the genome by nearly 8-fold. When injected into mice, the MetRS inhibitor-resistant parasites caused a reduced level of infection, indicating that the changes associated with resistance attenuated their virulence. This finding and the fact that resistance to MetRS inhibitors developed relatively slowly are encouraging for further development of this class of compounds. Published studies on other antitrypanosomal drugs have primarily shown that alterations in membrane transporters were the mechanisms responsible for resistance. This is the first published report of induced drug resistance in the African trypanosome due to overexpression of the target enzyme.
INTRODUCTION
The protozoan Trypanosoma brucei causes human African trypanosomiasis (HAT) and impacts large regions of sub-Saharan Africa. Cases have fortunately declined in number in the past decade (1); however, historically HAT has been a cyclical disease, and it seems likely that future outbreaks can be anticipated (2). Vector control combined with case identification and treatment has been the main modality for controlling HAT. Vaccines have never been introduced into clinical use. Unfortunately, existing chemotherapeutics have major drawbacks that limit their application and effectiveness. All of the anti-HAT treatment regimens involve parenteral administration of drugs, which creates logistical and cost issues for the resource-limited settings in which this disease is most prevalent. On top of this, all drugs for treating HAT have risks of serious side effects, and treatment of late-stage HAT (when parasites have entered the central nervous system) is associated with high treatment failure rates. New drugs that could ideally be given by mouth and that could simultaneously treat early- and late-stage HAT would provide needed tools to aggressively combat this human scourge (2). Recently, hopes of delivering a new HAT drug ended in disappointment when the diamidine DB289 was discontinued in late clinical development due to toxicity affecting the kidneys (3). This occurrence underscores the importance of maintaining a robust pipeline of candidate drugs for diseases like HAT due to the inevitably high attrition rate in drug development.
The discovery and validation of new drug targets is central to the process of developing new candidate drugs for HAT. Aminoacyl-tRNA synthetases (aaRS) are a class of enzymes in T. brucei that, in several cases, have excellent potential to be exploited as drug targets. These enzymes are responsible for specifically charging tRNAs with their cognate amino acids. The enzymatic reaction of aaRS consists of the following steps: recognition of a specific amino acid and ATP, the formation of an aminoacyl-adenylate, the recognition of a specific tRNA, and the transfer of the aminoacyl group to the 3′ end of the tRNA. Due to their central role in protein synthesis, it is not surprising that aaRS enzymes are essential for cell growth, including that of trypanosomes (4–6). T. brucei contains 23 annotated tRNA synthetase genes in its genome, with redundancy for 3 amino acids (7). As a general rule, the T. brucei tRNA synthetases operate in both the cytoplasm and the mitochondrion of the cell (7). This is different from mammals, which generally encode separate complements of tRNA synthetases for the cytoplasm and the mitochondria (8). Comparisons of the ATP-amino acid binding pocket of the single T. brucei methionyl-tRNA synthetase (MetRS) to the corresponding pockets of the human mitochondrial and cytoplasmic MetRS enzymes have enabled us to determine that there are substantial differences that increase the likelihood of being able to discover small-molecule inhibitors that are selective for the trypanosome over the mammalian homologs (6, 9).
Two classes of MetRS inhibitors have been discovered with selective activity on T. brucei over mammalian cells (6, 10). A class of aminoquinolones (Fig. 1, compound 1312) have low nanomolar activity on T. brucei MetRS and T. brucei cells but suffer from poor oral bioavailability, thus limiting their potential development as oral drug candidates (6). A similar class of compounds with a urea core (Fig. 1, compound 1433) were somewhat less active on T. brucei (50% inhibitory concentration [IC50], 220 nM) but demonstrated dramatically improved permeability properties compared to the aminoquinolones (10). The urea-based compounds are serving as leads for further optimization in the HAT drug discovery program.
Fig 1.

Compound structures. Shown are the structures of methionyl-tRNA synthetase inhibitors (1433 and 1312) and the clinical antitrypanosomal drugs eflornithine and pentamidine.
Antimicrobial resistance has undermined or threatened the utility of many important classes of drugs used to treat infectious diseases (11). Drug resistance has impacted HAT, as well, with examples of melarsoprol-resistant parasites isolated from patients with relapsed HAT following melarsoprol chemotherapy (12). Cross-resistance between different drug classes is also recognized as a problem in T. brucei (13). With the ongoing efforts described above to exploit a new drug target in T. brucei (i.e., MetRS), it is prudent to assess the potential for drug resistance to develop against inhibitors of this target. There is general concern that enzyme targets are particularly vulnerable to resistance due to the potential for mutations to arise in the target's genetic sequence. A dramatic example of this situation occurs in Plasmodium falciparum with the rapid development of resistance to atovaquone mediated by mutations in the target enzyme, cytochrome b (14). The high frequency of induction of resistance mutations makes atovaquone ineffective as monotherapy and forced its development as a combination treatment with proguanil, known as Malarone (15). Recent research with tRNA synthetase inhibitors for bacterial infections has indicated a problem, specifically with resistant bacteria developing in patients treated with an inhibitor of bacterial leucyl-tRNA synthetase, GSK2251052 (http://www.sec.gov/Archives/edgar/data/1411158/000110465912068165/a12-23396_1ex99d1.htm). Mutations in the target enzyme were reportedly responsible for the observed clinical resistance.
Here, the potential for resistance to develop in T. brucei against two separate MetRS inhibitors (Fig. 1) is investigated. Resistant parasites were generated by continuous in vitro culture with incrementally increasing concentrations of inhibitors. The rate of induction of resistance was observed to be lower than the rates against other T. brucei drugs, eflornithine and pentamidine. Mutations in the gene sequence of MetRS were not observed, but dramatic amplification of the MetRS gene in the genome and at the transcript level revealed that overexpression of the target was the mechanism of resistance. The resistant parasites produced lower levels of parasitemia and mortality in mice than the parental strain, indicating that the genetic changes affected the fitness of the parasites. The studies indicate that resistance to MetRS inhibitors can be induced but that it occurs relatively slowly and at a cost to the virulence of the parasites. Future studies will be necessary to assess the potential for T. brucei resistance to develop against MetRS inhibitors when the compounds are administered to infected experimental animals.
MATERIALS AND METHODS
Chemicals.
The synthesis methods of 1312 and 1433 were previously described (6, 10). Eflornithine and pentamidine were purchased from Sigma-Aldrich.
Culture methods and growth inhibition assays. (i) Culture methods.
Wild-type T. brucei brucei (bloodstream form, strain 427, from K. Stuart, Seattle BioMed, Seattle, WA) were cultured in HMI-9 medium containing 10% fetal bovine serum, penicillin, and streptomycin at 37°C with 5% CO2 (29). Selection of resistant T. brucei cells was conducted in a volume of 5 ml in T-25 flasks. Wild-type T. brucei cells cultured without drug pressure were maintained during the study as a control. Eflornithine-, pentamidine-, 1312-, and 1433-resistant parasites were selected in increasing concentrations of drug starting at the IC25, except for pentamidine, which was started at the IC12.5. When T. brucei cells were grown to mid-log phase (range, 1 × 106 to 3 × 106 cells/ml), they were passed at a 1:10 dilution into a new flask containing the same amount of drug they had previously grown in and another flask containing double the amount of drug they had previously grown in. When parasites grew in the higher concentration, this was marked as a step up on the graph (Fig. 2). This process was repeated until the resistant parasites were growing in 32 times the IC50, except for 1312-resistant parasites, which did not grow successfully in concentrations exceeding 4 times the IC50. Resistant and wild-type parasites were then cloned by limiting dilution.
Fig 2.

In vitro induction of resistance in T. brucei. Compound concentrations were increased over time as tolerated by the cultures (see Materials and Methods). The asterisks indicate when the volume for cultures with 1312 was increased from 5 ml to 50 ml.
(ii) Growth inhibition assays.
Three clones from each group and the population were tested in the T. brucei growth inhibition assay as previously described (10). Cross-resistance was also tested for each clone by assaying the clone with the other compounds (1433, 1312, eflornithine, and/or pentamidine) that the clone was not cultured in.
(iii) Determination of in vitro growth rates of resistant T. brucei.
At time zero, two 1433-resistant and two wild-type clones (103 cells/ml) with and without 1.9 μM (∼10 times the baseline IC50) 1433 were cultured in 5 ml in T-25 flasks. Total parasites were quantified at designated time points using the PerkinElmer ATPlite kit as previously described (16).
Mouse infections.
Three 6- to 8-week-old Swiss Webster (CFW) female mice (Charles River, Wilmington, MA) were used per group. Six groups of mice were used, including three different clones from the 1433-resistant and three clones from the wild-type bloodstream form strain 427. On day zero, each mouse was infected by intraperitoneal (i.p.) injection with 5 × 104 parasites of the 1433-resistant or wild-type clone. On day 2 postinfection, parasitemia monitoring began by microscopic examination of tail blood on wet mounts. On day 4, parasites from two different mice from 1433-resistant and wild-type groups that had high levels of parasitemia were each passed into three new CFW mice via i.p. injection at 2 × 104 parasites. Parasitemia was monitored for 2 weeks postinfection or until the mice became moribund and had to be euthanized. All animal procedures were performed at the University of Washington and approved by its Institutional Animal Care and Use Committee (IACUC).
Molecular biology methods. (i) Northern analysis.
RNA was isolated from three different clones from 1312-resistant, 1433-resistant, and wild-type parasites using a Qiagen RNeasy kit. Northern analysis was performed as previously described (6), except that 12 μg per lane of total RNA was used.
(ii) Southern analysis.
DNA was isolated from three different clones from 1312-resistant, 1433-resistant, and wild-type parasites using standard procedures (16). Each DNA sample (6 μg per digestion) was digested with two different sets of restriction digestion enzymes (NsiI/EagI and NsiI/SacII) purchased from New England BioLabs. Digestions were designed using the gene coding for T. brucei methionyl-tRNA synthetase (Tb927.10.1500) and were performed in a 30-μl total volume according to the manufacturer's instructions. Each digest was separated on a standard 1% Tris-borate-EDTA (TBE) gel. The membranes were then probed and blotted as described previously (6).
(iii) Sequencing analysis.
DNA was isolated from three different clones from 1312-resistant, 1433-resistant, and wild-type parasites as described above. The gene coding for T. brucei methionyl-tRNA synthetase (Tb927.10.1500) was amplified from each DNA sample using primers 1 and 2 (see Table S1 in the supplemental material). The PCR products were purified using the Qiagen QIAquick PCR Purification Kit. Sequencing primers 3 through 12 (see Table S1 in the supplemental material) were then added to the cleaned PCR product according to the sequencing company's (GeneWiz) instructions. Sequencing chromatogram analysis was performed in ContigExpress.
(iv) Statistical analyses.
Two-way comparisons were performed by unpaired t tests using Graphpad (San Diego, CA) Prism software.
RESULTS
Selection of drug-resistant bloodstream form T. brucei.
T. brucei brucei strain 427 parasites were grown in culture with incrementally increasing concentrations of drugs (Fig. 2). The concentration of the indicated drug (eflornithine, pentamidine, 1312, or 1433) was doubled whenever the cells grew out as a robust population. Resistance to 32 times the baseline IC50 occurred most rapidly with eflornithine, appearing by ∼50 days. The next fastest, 32×-pentamidine-resistant parasites, grew out at ∼80 days. The MetRS inhibitor compound 1433 reached a level of 32 times the baseline IC50 by ∼120 days. However, we were not able to induce a level of resistance to compound 1312 beyond 4 times the baseline IC50 even after 127 days of continuous culture. We even increased the culture volume from 5 ml to 50 ml beginning around day 100 to increase the probability that potentially rare events (e.g., mutations or gene duplications) might occur to give rise to resistance, but this did not result in populations with more than 4 times the baseline IC50.
Cross-resistance studies.
Resistant parasites were cloned by limiting dilution, and three separate clones were tested for susceptibility to the other drugs used in these experiments (Fig. 3). The eflornithine-resistant clones were resistant only to eflornithine and were not resistant to pentamidine, 1312, or 1433. Similarly, the pentamidine-resistant clones were resistant only to pentamidine. However, the 1433-resistant clones showed cross-resistance to 1312 of the same magnitude as the resistance to 1433 (approximately 50 times the IC50 for the parent population). The 1312-resistant clones were also cross-resistant to 1433 at a level of ∼5 times the IC50 for the parent population. Thus, the mechanism of resistance to the MetRS inhibitors appears to act similarly on the two structurally different compounds (1312 and 1433). Finally, neither the 1312-resistant clones nor the 1433-resistant clones were cross-resistant to either eflornithine or pentamidine.
Fig 3.

Cross-resistance. The resistant parasite populations or clones (i.e., C1, C2, and C3) were tested for susceptibility to other compounds to assess cross-resistance. The heights of the bars indicate the resistant IC50/wild-type IC50 (R/WT) ratios. The averages of the resistant/wild-type ratios of the clones were compared for statistical significance by unpaired t tests. (A) The bars for 1433 and 1312 are significantly higher (P < 0.0001) than the bars for eflornithine and pentamidine. (B) Again, the bars for 1433 and 1312 are significantly higher (P < 0.005) than the bars for eflornithine or pentamidine. The inset displays the data on a smaller y axis (0 to 8 instead of 0 to 200). (C) The pentamidine bars are significantly higher (P < 0.0001) than the bars for 1433, 1312, and eflornithine. (D) The bars for eflornithine are significantly higher (P < 0.002) than the bars for 1433, 1312, and pentamidine.
Fitness studies.
Further studies were done with the 1433-resistant clones to determine if the changes resulting in resistance were associated with altered in vitro growth rates or virulence in vivo. The in vitro growth rate of 1433-resistant parasites was, in fact, diminished compared to that of clones from the parent population (Fig. 4). At 72 h, the cell density of 1433-resistant parasites was approximately half of that observed with clones from the parent strain. The calculated difference in doubling time was 6.7 h (parent strain) to 9.1 h (1433-resistant cells). The growth rate of the 1433-resistant clones was essentially the same whether they were grown with or without 1433 (1.9 μM) in the culture medium.
Fig 4.

In vitro growth rates of resistant T. brucei. The asterisks indicate 1.9 μM compound 1433 was present. The growth of wild-type clones in the absence of drug (♢ and △) was significantly greater (P < 0.0025) than the growth of 1433-resistant clones in the absence (● and ◆) or presence (▼ and ■) of 1433 at 24, 48, and 72 h. The growth of wild-type clones in the presence of 1433 (○ and □) was significantly less (P < 0.001) than the growth of the other clones at 24, 48, and 72 h. The error bars indicate the standard errors of the mean.
The ability of 1433-resistant parasites to produce parasitemia in mice was next analyzed for three separate clones. Each clone was injected into three individual mice and compared to three clones from the parental strain; thus, 18 mice were infected and monitored daily for parasitemia. The parental clones behaved fairly consistently, with parasitemia rising to high levels (>2 million parasites/ml blood) by 4 days postinfection (see Fig. 7A). All of these mice were sacrificed at day 3 or day 4 postinfection due to overwhelming parasitemia. In contrast, 7 of the 9 mice receiving 1433-resistant clones had undetectable or low levels of parasitemia for at least the first 10 days postinfection (see Fig. 7B). Interestingly, 2 of the mice infected with 1433-resistant clones displayed parasitemia levels of 25 to 50 million T. brucei parasites per ml at day 4 postinfection. Due to concerns that these mice might die before the next observation point in the morning, they were sacrificed, and the parasites were passed into new recipient mice at an inoculum of 2 × 104 (see Fig. 7C and D). In these six mice, it can be seen that parasitemia occurred in 4 mice around day 7 or 8 but then diminished to nearly undetectable levels and remained low until the end of the observation period of 21 days (see Fig. 7D). In contrast, all the mice receiving the mouse-passaged parent clones (at 2 × 104) developed high parasitemia and required euthanasia when they became moribund (see Fig. 7C). In summary, the 1433-resistant clones had a moderately diminished growth rate in vitro and appeared to be attenuated, although not entirely, with respect to their ability to produce parasitemia in mice.
Fig 7.

Diminished/delayed parasitemia in mice caused by 1433-resistant T. brucei. (A) Control group: parasitemia of wild-type clones injected into mice at 5 × 104 i.p. (C, clone; M, mouse). All mice were sacrificed by day 4 due to high parasitemia. (B) Parasitemia of 1433-resistant clones in the first pass in mice (infection dose, 5 × 104 i.p.). (C) Parasitemia of wild-type clones in the second pass in mice (infection dose, 2 × 104 i.p.). (D) Parasitemia of 1433-resistant clones in the second pass in mice (infection dose, 2 × 104 i.p.). See Results for further explanation.
We were interested to see if the 1433-resistant clones that had been passaged through mice were still resistant to 1433. Specifically, parasites were collected after the second mouse passage, representing growth in the animal host for 8 total days with no exposure to 1433 or other drugs. IC50 curves showed that there was essentially no decrease in the level of resistance in parasite clones recovered from mice compared to their level of resistance prior to mouse infection (see Fig. S1 in the supplemental material).
Genetic analysis of the basis of resistance.
Experiments were performed to determine if overexpression or mutations of the drug target (MetRS) might be responsible for the observed resistance. Northern blotting was conducted to quantify MetRS mRNA levels (Fig. 5). Upregulation of the MetRS mRNA was observed in all three 1433-resistant clones and all three 1312-resistant clones that were analyzed. When normalized against β-tubulin expression, the magnitude of upregulation was approximately 35-fold for the 1433-resistant clones compared to the wild-type strain (P < 0.0001) and 8-fold for the 1312-resistant clones compared to the wild-type strain (P < 0.0001). The magnitude of mRNA overexpression is roughly the same as the increase in IC50s observed in the in vitro growth experiments.
Fig 5.
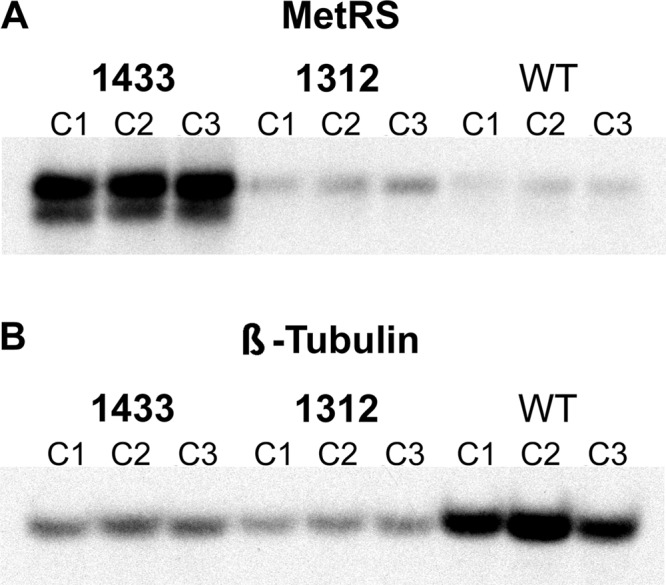
(A and B) Expression levels by Northern blotting. When normalized to the β-tubulin signal by densitometry, MetRS mRNA in the 1433-resistant parasites was upregulated by an average of 35-fold compared to wild-type parasites, and expression in the 1312-resistant parasites was upregulated 8-fold.
Southern blots were performed to assess for evidence of gene amplification. The blots showed increased signal in the 1433-resistant clones (8-fold) compared to the wild-type clones when normalized to tubulin (P < 0.001) (Fig. 6). The signal in the 1312-resistant clones was not significantly different (0.7-fold; P = 0.1) from the signal in the wild-type clones. The size of the bands (1.6 kb) was unchanged, indicating that portions of DNA that included the restriction sites of the flanking intergenic regions were being amplified.
Fig 6.
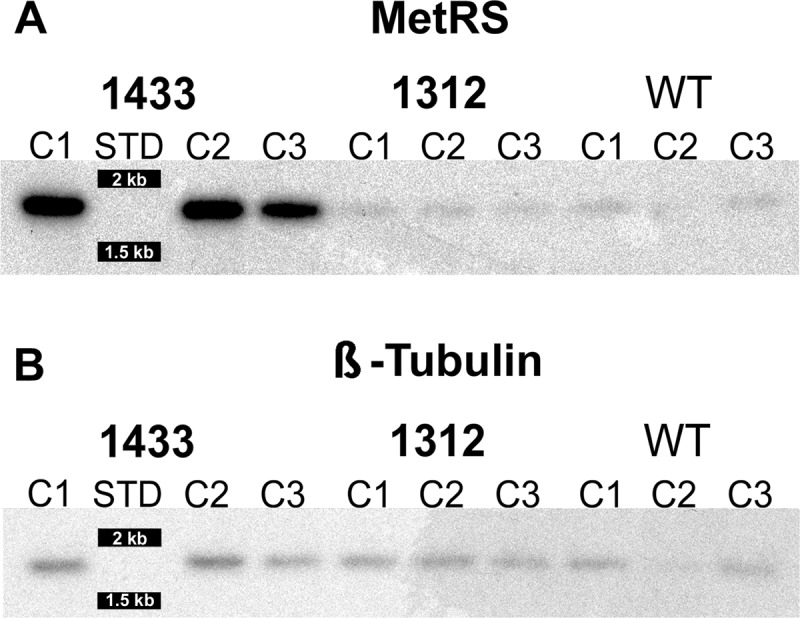
Southern blot. (A) Genomic DNA of wild-type, 1433-resistant, and 1312-resistant T. brucei digested with NsiI and EagI and hybridized with 32P-labeled probes to MetRS. The digest gives a predicted band size of 1,624 bases. (B) When normalized to the β-tubulin signal by densitometry, the signal for the MetRS gene in the 1433-resistant parasites was amplified 8-fold compared to wild-type parasites and 0.7-fold for 1312-resistant parasites.
The MetRS genes from the parental and 1433- and 1312-resistant clones (three each) were sequenced from PCR amplicons to look for mutations that might contribute to the resistance phenotype. First, for the parent strain, three single-nucleotide polymorphisms causing amino acid changes were detected in the open reading frame, indicating differences in the alleles of the MetRS gene. The same three polymorphisms were observed in the three 1312-resistant clones. In contrast, the three 1433-resistant clones had identical sequences with no detectable polymorphisms. This is consistent with one of the alleles being heavily amplified in the genome and thus overrepresented in the PCR sequences. No evidence for mutations in the open reading frame of the MetRS gene was detected that accounts for the resistant phenotype.
DISCUSSION
A high level of resistance to the MetRS inhibitor 1433 was induced by long-term in vitro culture at sublethal doses of the compound. By approximately 120 days, the cultures grew in 1433 at concentrations 32 times greater than the IC50 for the parental strain. Following the same procedures, we were able to induce a similar level of resistance to eflornithine in a shorter period of ∼50 days. The result with eflornithine is very similar to results published by another group that generated T. brucei lines that were 40-fold less sensitive to eflornithine than the parental line (17). In those studies, resistance to eflornithine was due to the loss of a membrane transporter that mediates uptake of the drug. Specifically, the putative amino acid transporter (TbAAT6) was found to be deleted in two separate lines selected to be resistant to eflornithine. The target enzyme on which eflornithine acts, ornithine decarboxylase, was not altered in sequence or copy number. We did not investigate the genetic basis for eflornithine resistance in our experiments.
We also generated T. brucei lines that grew in pentamidine at 32 times the baseline IC50. The resistant parasites grew out in ∼80 days, somewhat faster than was observed with the MetRS inhibitors. Other investigators have induced high-level resistance to pentamidine in vitro and have shown that loss of surface transporters is responsible for the phenotype (18). In particular, loss of the P2 transporter (also involved with uptake of arsenical drugs in T. brucei) is partly responsible, as is loss of two other transporters named high-affinity pentamidine transporter 1 and low-affinity pentamidine transporter 1 (13). The mechanism of resistance to pentamidine in the lines generated in our studies was not investigated. The common theme of mechanisms of drug resistance in T. brucei, as shown with eflornithine, diamidines, and arsenicals, has been loss of surface transporters involved in drug uptake (19).
We did not see cross-resistance of the MetRS-resistant parasites with either eflornithine or pentamidine, indicating that the mechanism(s) mediating MetRS resistance did not significantly impact uptake or other aspects of the action of eflornithine or pentamidine. Similarly, the parasites with induced resistance to eflornithine or pentamidine were not resistant to MetRS inhibitors. Thus, if the surface transporters were altered in the eflornithine- or pentamidine-resistant cell lines in our studies, the changes had no apparent effect on the uptake or action of the MetRS inhibitors.
It is interesting that we were unable to induce the level of resistance with 1312 (only ∼4-times its IC50) that we were able to induce with 1433. We even attempted to increase the chances for higher levels of resistance to 1312 by increasing the culture size by a factor of 10 but were unsuccessful after 127 days. Note that the clones that were generated to be >32-fold resistant to 1433 were cross-resistant to 1312 by a similar magnitude (Fig. 3), so it is clearly possible to generate high-level resistance to 1312, but apparently not by inducing the resistance with 1312 itself. The most obvious differences between 1433 and 1312 are the respective IC50s of 220 nM and 4 nM against the parent strain, respectively. Enzyme assays have demonstrated that 1312 is a more potent inhibitor of T. brucei MetRS than is 1433 (10). It is possible that the apparently tighter binding of 1312 to MetRS prevents the in vitro selection for gene amplification events that allowed high-level resistance to develop against the weaker binder, 1433. The encouraging part of this observation is that we aim to develop analogs to 1433 with greater potency, and thus, we may not see the same high level of resistance to these future “urea” compounds that we saw with the current lead compound, 1433.
The 1433-resistant parasites have a moderately diminished growth rate in vitro (a doubling time of 9.1 h versus 6.7 h for the parent strain). An even greater effect was evident when mice were infected with the 1433-resistant clones (Fig. 7). Based on the finding that the resistance is due to gene amplification (discussed below), it seems likely that these changes add a burden to the cells that alters their in vitro and in vivo growth rates. This may be due to the increased energy demands on the cell to overexpress the target enzyme or to collateral damage to the genome that has deleterious effects on the growth rate and virulence.
Investigations into the mechanism of resistance to the MetRS inhibitors indicate that overexpression of the target gene is responsible for the phenotype. This was evident from the levels of MetRS mRNA observed on Northern blots (Fig. 5), as well as evidence for gene amplification on the Southern blots (Fig. 6). We assume that these changes translate into higher levels of expressed MetRS protein, although this was not directly measured. Two attempts were made to quantify the MetRS enzymatic activity of cell lysates from large cultures of T. brucei using a radiometric aminoacylation assay (20), but the method was not sufficiently sensitive to detect activity in any of the samples. We felt that the genetic evidence for gene amplification by Northern and Southern analyses was sufficiently compelling that the cost of preparing antibodies for Western blots was not justified.
Gene amplification is a well-described phenomenon in trypanosomatids in response to evolutionary pressure (21, 22) and drug selection (23, 24). The responses to environmental pressures take place in a setting in which individual gene promoters are absent, and polycistronic transcription through tandem gene arrays is a means to increase gene dosage (21). Examples of genetic plasticity in trypanosomatids include expansion of linear chromosomes and amplification of extrachromosomal circular elements (25, 26). An example of the latter phenomenon is the amplification of P-glycoprotein genes on extrachromosomal circular elements in Leishmania that confer resistance to antimony drugs by efflux mechanisms (27). In the current study, the gene amplification appears to directly involve the specific target of action of the drug under study (as opposed to involving an efflux pump, for example). Additional studies will be necessary to characterize the nature of the gene amplification (e.g., episomal or tandem gene duplications) in the parasites selected in the current experiments and the implications for the stability of the drug-resistant phenotype.
We did not observe mutations to the MetRS gene in the 1433- or 1312-resistant clones. Whether this would occur during selection in an animal model remains an open question for future studies. The potential for resistance due to gene mutations is important because of recent reports of antibacterial tRNA synthetase inhibitors failing in phase II clinical trials due to the emergence of resistant bacteria (http://www.sec.gov/Archives/edgar/data/1411158/000110465912068165/a12-23396_1ex99d1.htm). This problem occurred with oxaborole compounds that target the editing site of the bacterial leucyl-tRNA synthetase (28), so entirely different molecular interactions are at play than with our inhibitors that target the amino acid binding site and an auxiliary pocket of the methionyl-tRNA synthetase (9). We speculate that mutations may be better tolerated in the editing site of a tRNA synthetase than in an amino acid binding site.
The limitations of these studies are related to the fact that drug resistance was induced in in vitro culture. The genetic changes that caused resistance had deleterious effects on the infectiousness of these parasites in mice, presumably due to the burden of the additional genetic baggage or to alterations of other genes necessary for normal virulence. Future studies may be necessary to address the question of whether resistance develops (and by what mechanism) when T. brucei is exposed to compounds in infected animals. Unfortunately, these studies were not feasible with the currently available MetRS inhibitors due to suboptimal pharmacokinetic properties in mice that make long-term administration at doses that substantially suppress T. brucei parasitemia technically impractical. If resistance could be induced in the animal model, it would be interesting to determine if a different resistance mechanism, such as gene mutations, was responsible, since there would be selection pressure against changes causing attenuation of virulence.
In conclusion, this research showed that resistance to MetRS inhibitors could be induced in cultured T. brucei exposed to incrementally increasing concentrations of compounds but that the rate of resistance development was considerably lower than the rate observed in parallel experiments with eflornithine and (slightly less so) with pentamidine, two drugs in clinical use for treating HAT. Furthermore, the mechanism of resistance by amplification of the target gene appeared to come at a fitness cost to the parasites, as evidenced by the diminished parasitemia that they produced in the mouse model. This information is encouraging in that the parasites did not show evidence of rapidly developing resistance in a manner that would allow them to remain fully virulent. As MetRS inhibitors are developed with improved pharmacokinetic properties, future studies can be conducted to assess the potential for resistance to occur in more clinically relevant animal models. The occurrence of mutations in the target enzyme remains a theoretical possibility but was not observed in these studies. Amplification of an enzyme that is targeted by a specific inhibitor has not been previously reported as a mechanism of resistance for T. brucei.
Supplementary Material
ACKNOWLEDGMENT
Support for this research was provided by National Institute of Allergy and Infectious Diseases grants AI067921 and AI084004.
Footnotes
Published ahead of print 15 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02578-12.
REFERENCES
- 1. Brun R, Blum J, Chappuis F, Burri C. 2010. Human African trypanosomiasis. Lancet 375:148–159 [DOI] [PubMed] [Google Scholar]
- 2. Simarro PP, Jannin J, Cattand P. 2008. Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med. 5:e55. 10.1371/journal.pmed.0050055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brun R, Don R, Jacobs RT, Wang MZ, Barrett MP. 2011. Development of novel drugs for human African trypanosomiasis. Future Microbiol. 6:677–691 [DOI] [PubMed] [Google Scholar]
- 4. Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391 [DOI] [PubMed] [Google Scholar]
- 5. Charriere F, Helgadottir S, Horn EK, Soll D, Schneider A. 2006. Dual targeting of a single tRNA(Trp) requires two different tryptophanyl-tRNA synthetases in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 103:6847–6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, Merritt EA, Hol WG, Verlinde CL, Fan E, Buckner FS. 2011. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei infection in mice. Antimicrob. Agents Chemother. 55:1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Charriere F, O'Donoghue P, Helgadottir S, Maréchal-Drouard L, Cristodero M, Horn EK, Söll D, Schneider A. 2009. Dual targeting of a tRNA ASP requires two different aspartyl-tRNA synthetases in Trypanosoma brucei. J. Biol. Chem. 284:16210–16217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duchene AM, Pujol C, Marechal-Drouard L. 2009. Import of tRNAs and aminoacyl-tRNA synthetases into mitochondria. Curr. Genet. 55:1–18 [DOI] [PubMed] [Google Scholar]
- 9. Koh CY, Kim JE, Shibata S, Ranade RM, Yu M, Liu J, Gillespie JR, Buckner FS, Verlinde CL, Fan E, Hol WG. 2012. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 20:1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shibata S, Gillespie JR, Ranade RM, Koh CY, Kim JE, Laydbak JU, Zucker FH, Hol WG, Verlinde CL, Buckner FS, Fan E. 2012. Urea-based inhibitors of Trypanosoma brucei methionyl-tRNA synthetase: selectivity and in vivo characterization. J. Med. Chem. 55:6342–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright GD, Poinar H. 2012. Antibiotic resistance is ancient: implications for drug discovery. Trends Microbiol. 20:157–159 [DOI] [PubMed] [Google Scholar]
- 12. Matovu E, Geiser F, Schneider V, Mäser P, Enyaru JC, Kaminsky R, Gallati S, Seebeck T. 2001. Genetic variants of the TbAT1 adenosine transporter from African trypanosomes in relapse infections following melarsoprol therapy. Mol. Biochem. Parasitol. 117:73–81 [DOI] [PubMed] [Google Scholar]
- 13. de Koning HP. 2008. Ever-increasing complexities of diamidine and arsenical crossresistance in African trypanosomes. Trends Parasitol. 24:345–349 [DOI] [PubMed] [Google Scholar]
- 14. Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob. Agents Chemother. 44:2100–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Looareesuwan S, Chulay JD, Canfield CJ, Hutchinson DB. 1999. Malarone (atovaquone and proguanil hydrochloride): a review of its clinical development for treatment of malaria. Malarone Clinical Trials Study Group. Am. J. Trop. Med. Hyg. 60:533–541 [DOI] [PubMed] [Google Scholar]
- 16. Ojo KK, Gillespie JR, Riechers AJ, Napuli AJ, Verlinde CL, Buckner FS, Gelb MH, Domostoj MM, Wells SJ, Scheer A, Wells TN, Van Voorhis WC. 2008. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob. Agents Chemother. 52:3710–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vincent IM, Creek D, Watson DG, Kamleh MA, Woods DJ, Wong PE, Burchmore RJ, Barrett MP. 2010. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 6:e1001204. 10.1371/journal.ppat.1001204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bridges DJ, Gould MK, Nerima B, Maser P, Burchmore RJ, De Koning HP. 2007. Loss of the high-affinity pentamidine transporter is responsible for high levels of cross-resistance between arsenical and diamidine drugs in African trypanosomes. Mol. Pharmacol. 71:1098–1108 [DOI] [PubMed] [Google Scholar]
- 19. Barrett MP, Vincent IM, Burchmore RJ, Kazibwe AJ, Matovu E. 2011. Drug resistance in human African trypanosomiasis. Future Microbiol. 6:1037–1047 [DOI] [PubMed] [Google Scholar]
- 20. Rubio MA, Ragone FL, Gaston KW, Ibba M, Alfonzo JD. 2006. C to U editing stimulates A to I editing in the anticodon loop of a cytoplasmic threonyl tRNA in Trypanosoma brucei. J. Biol. Chem. 281:115–120 [DOI] [PubMed] [Google Scholar]
- 21. Jackson AP. 2007. Tandem gene arrays in Trypanosoma brucei: comparative phylogenomic analysis of duplicate sequence variation. BMC Evol. Biol. 7:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arner E, Kindlund E, Nilsson D, Farzana F, Ferella M, Tammi MT, Andersson B. 2007. Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants. BMC Genomics 8:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Segovia M. 1994. Leishmania gene amplification: a mechanism of drug resistance. Ann. Trop. Med. Parasitol. 88:123–130 [DOI] [PubMed] [Google Scholar]
- 24. Ouellette M, Borst P. 1991. Drug resistance and P-glycoprotein gene amplification in the protozoan parasite Leishmania. Res. Microbiol. 142:737–746 [DOI] [PubMed] [Google Scholar]
- 25. Jamnadass RH, Pelle R, Pandit P, Ricard B, Murphy NB. 2006. Trypanosoma brucei: composition, organisation, plasticity, and differential transcription of NlaIII repeat elements in drug-resistant and sensitive isolates. Exp. Parasitol. 113:244–255 [DOI] [PubMed] [Google Scholar]
- 26. Ouellette M, Fase-Fowler F, Borst P. 1990. The amplified H circle of methotrexate-resistant Leishmania tarentolae contains a novel P-glycoprotein gene. EMBO J. 9:1027–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ouellette M, Papadopoulou B. 1993. Mechanisms of drug resistance in Leishmania. Parasitol. Today 9:150–153 [DOI] [PubMed] [Google Scholar]
- 28. Sutcliffe JA. 2011. Antibiotics in development targeting protein synthesis. Ann. N. Y. Acad. Sci. 1241:122–152 [DOI] [PubMed] [Google Scholar]
- 29. Hirumi H, Hirumi K. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985–989 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.