

Abstract
Clostridium difficile infection (CDI) is a common and debilitating nosocomial infection with high morbidity and mortality. C. difficile mediates diarrhea and colitis by releasing two toxins, toxin A and toxin B. Since both toxins stimulate proinflammatory signaling pathways in human colonocytes and both are involved in the pathophysiology of CDI, neutralization of toxin A and B activities may represent an important therapeutic approach against CDI. Recent studies indicated that human monoclonal antibodies (MAbs) against toxins A and B reduce their cytotoxic and secretory activities and prevent CDI in hamsters. Moreover, anti-toxin A and anti-toxin B MAbs together with antibiotics also effectively reduced recurrent CDI in humans. However, whether these MAbs neutralize toxin A- and toxin B-associated immune responses in human colonic mucosa or human peripheral blood monocyte cells (PBMCs) has never been examined. We used fresh human colonic biopsy specimens and peripheral blood monocytes to evaluate the effects of these antibodies against toxin A- and B-associated cytokine release, proinflammatory signaling, and histologic damage. Incubation of anti-toxin A (MK3415) or anti-toxin B (MK6072) MAbs with human PBMCs significantly inhibited toxin A- and toxin B-mediated tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) expression. MK3415 and MK6072 also diminished toxin A- and toxin B-mediated NF-κB p65 phosphorylation in human monocytes, respectively, and significantly reduced toxin A- and B-induced TNF-α and IL-1β expression as well as histologic damage in human colonic explants. Our results underline the effectiveness of MK3415 and MK6072 in blocking C. difficile toxin A- and toxin B-mediated inflammatory responses and histologic damage.
INTRODUCTION
Clostridium difficile infection (CDI) is a common gastrointestinal nosocomial infection with increasing incidence and mortality over the past decade (1). The pathophysiology of CDI involves the release of two large exotoxins from pathogenic strains, toxin A and toxin B (2). Despite early reports indicating that toxin A is the primary toxin mediating the enterotoxic effects of C. difficile, studies in human colonic explants and human colonic xenografts indicate that both toxins can induce inflammation and cytotoxicity in human colon (3, 4). Studies in hamsters using isogenic toxin A and toxin B mutants of a virulent C. difficile strain confirmed that both toxins are important for the pathophysiology of CDI (5, 6).
The proinflammatory action of toxins A and B involves release of various cytokines and chemokines, prostaglandins, and additional inflammatory mediators from human colonocytes (7, 8) and monocytes (9, 10). The mechanisms involved in the toxin actions involve activation of established proinflammatory signaling pathways, including the global mediator of inflammation NF-κB (7) (11, 12), mitogen-activated protein (MAP) kinases (10, 13), as well as cyclooxygenase-2 (14).
Earlier evidence indicated that immune responses to C. difficile toxins are an important component for the pathogenesis of CDI (15–17). More recent studies confirmed the importance of antitoxin antibodies against toxin A and toxin B in primary and recurrent CDI in humans (18, 19) and animals (20, 21). Human monoclonal antibodies (MAbs) against toxin A reduced mortality in hamsters caused by C. difficile, but neutralization of both toxins provided better protection (22). Both monoclonal antibodies also neutralized the cytotoxicity of both toxins in human fibroblasts (22). Importantly, the results of a large multicenter phase II trial using human neutralizing MAbs against both toxins, together with standard antimicrobial therapy, in CDI patients showed a significant reduction of recurrent CDI (23).
Although the efficacy of human MAbs against toxins A and B in reducing relapsing CDI has been demonstrated (23), formal studies on the effect of these antibodies in the toxin A- and B-mediated innate immune responses in target human cells or human colonic mucosa in vitro have not been done. In this study, we addressed the hypothesis that neutralization of toxins A and B by human MAbs against toxin A (MK3415) or toxin B (MK6072) provided by Merck inhibits toxin A- and B-mediated innate immune responses in human colonic mucosa and human peripheral blood monocyte cells (PBMCs). Our results demonstrate that both MAbs can significantly reduce toxin A- and toxin B-mediated expression of the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) in monocytes and human colonic mucosal biopsy specimens and diminish epithelial tissue damage in human colonic tissues in response to these toxins.
MATERIALS AND METHODS
C. difficile culture and toxin purification.
C. difficile strain VPI 10463 (ATCC 43255 stock strain) was cultured in Difco cooked meat medium (catalog no. 226730; BD, Fisher Scientific) at 37°C under anaerobic conditions, and toxins A and B were purified to homogeneity as previously reported (3). The cytotoxicity of toxins A and B was determined by cell rounding, as previously described (24).
Human colonic explants and human primary monocyte cell culture.
Colonic explants and blood were obtained after informed consent in accordance with procedures established by the Cedars-Sinai Institutional Review Board (number 3358). PBMCs were isolated as we described previously (25). Human primary monocytes were isolated and cultured in RPMI 1640 medium (Invitrogen) containing 10% fetal calf serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen) as described previously (25). Monocyte preparations were >90% pure, as determined by esterase stain (Sigma-Aldrich, St. Louis, MO).
Mouse macrophage cell culture.
Mouse cultured RAW264.7 macrophages were cultured in Dulbecco modified Eagle medium with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen), as previously described (25).
Merck MK3415 and MK6072 neutralizing antibodies.
Anti-toxin A MK3415 and anti-toxin B MK6072 neutralizing antibodies were provided by Merck and used at the indicated concentrations. Details on the generation, characterization, and in vitro and in vivo neutralizing activities of these monoclonal antibodies were described by Babcock et al. (22). MK3415 was formerly called MDX006, and MK6072 was formerly called MDX1388. Control human IgG antibody was obtained from Southern Biotech (catalog no. 0150.01).
TNF-α and IL-1β ELISAs.
The levels of proinflammatory mediators human TNF-α (DY210, R&D Systems) and human IL-1β (DY201; R&D Systems) were measured by enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's instructions.
Phospho-NF-κB p65 and phospho-ERK ELISAs.
The treated cells were lysed in radioimmunoprecipitation assay buffer with protease inhibitor cocktail. Equal amounts of proteins (∼40 μg) were used for detection of phospho-p65 (kit number 7834; Cell Signaling, Danvers, MA) and phospho-extracellular signal-regulated kinase (phospho-ERK; kit number 7246; Cell Signaling) using ELISAs, according to the manufacturer's instructions.
Western blot analyses.
Cells were lysed in 1× blue loading buffer (catalog no. 7722; Cell Signaling, Danvers, MA). Equal amounts of cell extracts were fractioned by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and proteins were transferred onto nitrocellulose membranes (400 mA for 2 h at 4°C; Bio-Rad, Hercules, CA). Membranes were blocked in 5% nonfat milk in TBST (50 mM Tris, pH 7.5, 0.15 M NaCl, 0.05% Tween 20) and then incubated with antibodies (catalog no. 4370 for phospho-pERK and catalog no. 2128 for β-tubulin; Cell Signaling). Horseradish peroxidase-labeled antibody labeling was detected by chemiluminescence using an LAS4000 luminescent image analyzer (Fujifilm, Tokyo, Japan).
Quantitative real-time reverse transcription-PCR (RT-PCR).
Total RNA was isolated by an RNeasy kit (Qiagen, CA) and reverse transcribed into cDNA by a SuperScript III kit (catalog no. 11752; Invitrogen, Carlsbad, CA). Quantitative PCRs were run in an ABI Prism 7700 Fast sequence detector system as previously described (26). The levels of mRNA were determined by using cataloged primers (Invitrogen) for human TNF-α (Hs00174128_m1), IL-1β (Hs01555410_m1), and 18S (Hs99999901_s1). Results were expressed as the relative fold difference.
Histology scoring.
Epithelial cell damage in response to C. difficile toxins in human colonic explants was graded using a scoring system as previously published (27), and a score of 0 to 3 was assigned. Images from hematoxylin-eosin (H&E)-stained tissue sections were recorded at ×100 magnification.
Statistical analyses.
Quantitative results are expressed with error bars as mean ± standard error of the mean. Results were analyzed using the Prism professional statistics software program (GraphPad, San Diego, CA). Student's t tests with Mann-Whitney posttests were used for intergroup comparisons.
RESULTS
MK3415 and MK6072 inhibit toxin A- and toxin-B induced TNF-α expression in human PBMCs.
As shown in a recent report, toxin A induces expression of the proinflammatory cytokine TNF-α in human PBMCs (25). In the current study, toxin A at 0.1 and 1 μg/ml significantly increased TNF-α protein and mRNA expression in monocytes in a concentration-dependent manner (Fig. 1A and B). While pretreatment of PBMCs with control IgG 30 min prior to toxin exposure did not significantly alter toxin A-induced TNF-α expression compared to that for the control (no antibody), pretreatment with the anti-toxin A antibody MK3415 significantly reduced toxin A-induced TNF-α expression at the protein and mRNA levels (Fig. 1A and B). Pretreatment with MK3415 dose-dependently inhibited TNF-α protein and mRNA expression in PBMCs in the presence of 0.1 μg/ml toxin A (Fig. 1C and D).
Fig 1.
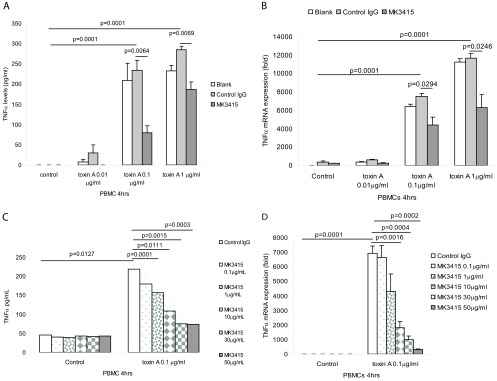
MK3415 attenuated toxin A-mediated TNF-α expression in PBMCs. (A and B) PBMCs were incubated with no antibody (blank), control IgG, or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 0 to 1 μg/ml for 4 h. (C and D) PBMCs were incubated with control IgG or MK3415 (0.1 to 50 mg/ml) for 30 min, followed by incubation with toxin A at 0.1 μg/ml for 4 h. Conditioned media were collected for TNF-α ELISA. TNF-α mRNA expression was detected by real-time RT-PCR. All experiments are representative of 3 independent experiments.
Similarly, toxin B at 0.1 to 10 μg/ml induced TNF-α protein and mRNA expression in human PBMCs (Fig. 2A and B). Toxin B at 1 μg/ml exerted the maximal response, while toxin B at 10 μg/ml stimulated modest TNF-α secretion (Fig. 2A). A similar lack of a dose-dependent response in macrophages in response to toxin B and toxin A was previously reported (28) and was probably related to cell death at high toxin doses. Pretreatment of PBMCs with control IgG did not significantly alter toxin B-induced TNF-α expression compared to that for the control, but pretreatment with the anti-toxin B antibody MK6072 significantly reduced toxin B-induced TNF-α mRNA and protein expression (Fig. 2A and B). MK6072 dose-dependently inhibited TNF-α protein and mRNA expression in PBMCs in the presence of 1 μg/ml toxin B (Fig. 2C and D).
Fig 2.
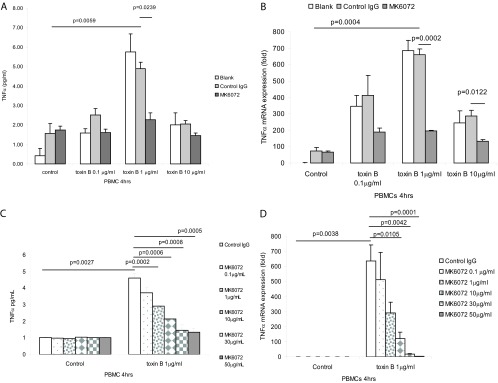
MK6072 attenuated toxin B-mediated TNF-α expression in PBMCs. (A and B) PBMCs were incubated with no antibody (blank), control IgG, or MK6072 (30 μg/ml) for 30 min, followed by incubation with toxin B at 0 to 10 μg/ml for 4 h. (C and D) PBMCs were incubated with control IgG or MK6072 (0.1 to 50 μg/ml) for 30 min, followed by incubation with toxin B at 1 μg/ml for 4 h. Conditioned media were collected for TNF-α ELISA. TNF-α mRNA expression was detected by real-time RT-PCR. All experiments are representative of 3 independent experiments.
MK3415 and MK6072 inhibit toxin A- and toxin B-induced IL-1β expression in human monocytes.
Previous experiments indicated that toxins A and B induce IL-1β expression in macrophages (29, 30). Consistent with these findings, toxin A significantly induced IL-1β protein secretion by human PBMCs in the conditioned medium, with a maximal response at 0.1 μg/ml (Fig. 3A). A further increase of the toxin A concentration reduced IL-1β protein expression, although mRNA expression continued to increase (Fig. 3A and B). There was no significant difference in IL-1β protein and mRNA expression between the control IgG group and the control (blank) group. Preincubation of PBMCs with the anti-toxin A antibody MK3415 significantly reduced toxin A-induced IL-1β protein and mRNA expression in these cells. MK3415 dose-dependently inhibited IL-1β protein and mRNA expression in PBMCs in the presence of 0.1 μg/ml toxin A (Fig. 3C and D).
Fig 3.
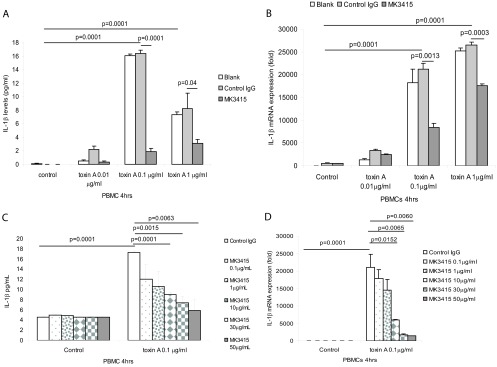
MK3415 attenuated toxin A-mediated IL-1β expression in PBMCs. (A and B) PBMCs were incubated with no antibody (blank), control IgG, or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 0 to 1 μg/ml for 4 h. (C and D) PBMCs were incubated with control IgG or MK3415 (0.1 to 50 μg/ml) for 30 min, followed by incubation with toxin A at 0.1 μg/ml for 4 h. Conditioned media were collected for IL-1β ELISA. IL-1β mRNA expression was detected by real-time RT-PCR. All experiments are representative of 3 independent experiments.
Toxin B at 1 μg/ml also induced maximal IL-1β protein and mRNA expression in PBMCs (Fig. 4A and B). Incubation of cells with anti-toxin B antibody MK6072 significantly inhibited toxin B-induced IL-1β protein and mRNA expression. MK6072 dose-dependently inhibited IL-1β protein and mRNA expression in PBMCs in the presence of 1 μg/ml toxin B (Fig. 4C and D).
Fig 4.
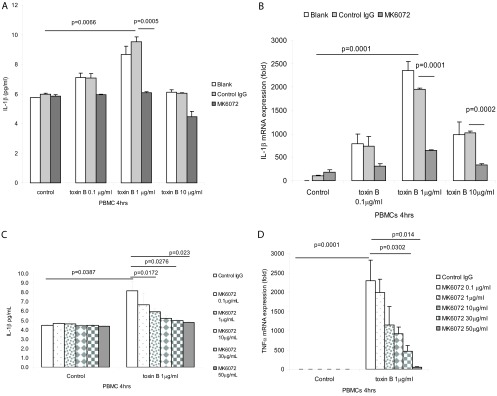
MK6072 attenuated toxin B-mediated IL-1β expression in PBMCs. (A and B) PBMCs were incubated with no antibody (blank), control IgG, or MK6072 (30 μg/ml) for 30 min, followed by incubation with toxin B at 0 to 10 μg/ml for 4 h. (C and D) PBMCs were incubated with control IgG or MK6072 (0.1 to 50 μg/ml) for 30 min, followed by incubation with toxin B at 1 μg/ml for 4 h. Conditioned media were collected for IL-1β ELISA. IL-1β mRNA expression was detected by real-time RT-PCR. All experiments are representative of 3 independent experiments.
MK3415 and MK6072 inhibit toxin A- and toxin B-induced NF-κB p65 phosphorylation in human monocytes.
Toxin A mediates TNF-α expression via NF-κB activation (6, 7). We previously reported that toxin A can induce NF-κB p65 subunit phosphorylation in human PBMCs (25). Here, we found that toxin A induces p65 phosphorylation in the same cells using an ELISA approach, as described in Materials and Methods. Addition of MK3415 inhibited toxin A (0.1 to 1 μg/ml)-induced p65 phosphorylation in PBMCs (Fig. 5A). MK3415 dose-dependently reduced toxin A-induced p65 phosphorylation in PBMCs (Fig. 5B).
Fig 5.
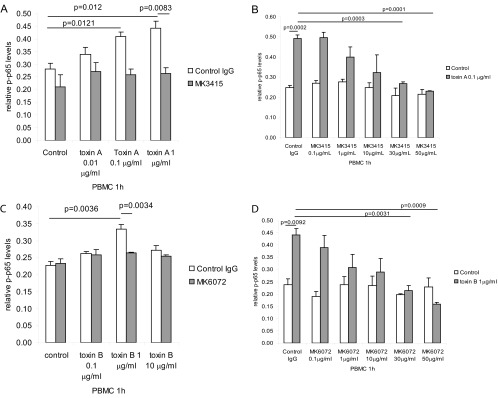
MK3415 and MK6072 inhibited toxin A- and toxin B-mediated NF-κB p65 phosphorylation in PBMCs. (A) PBMCs were incubated with control IgG or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 0 to 1 μg/ml for 1 h. (B) PBMCs were incubated with control IgG or MK3415 (0.1 to 50 mg/ml) for 30 min, followed by incubation with toxin A at 0.1 μg/ml for 1 h. (C) PBMCs were incubated with control IgG or MK6072 (30 μg/ml) for 30 min, followed by incubation with toxin B at 0 to 10 μg/ml for 1 h. (D) PBMCs were incubated with control IgG or MK6072 (0.1 to 50 mg/ml) for 30 min, followed by incubation with toxin B at 1 μg/ml for 1 h. Equal amounts of cellular protein were used for detection of phospho-p65 (p-p65) expression. All experiments are representative of 3 independent experiments.
Exposure of these cells to different toxin B concentrations induced p65 phosphorylation, with higher levels of induction occurring at 1 μg/ml (Fig. 5C). Incubation with MK6072 reduced toxin B-induced p65 phosphorylation in PBMCs (Fig. 5C). MK6072 dose-dependently inhibited p65 phosphorylation in PBMCs in the presence of 1 μg/ml toxin B (Fig. 5D).
Toxin A and toxin B also activate the MAP kinase ERK pathway in mouse dendritic cells and colonocytes (8, 31). ERK activation is necessary for proinflammatory cytokine CXCL2 and chemokine IL-8 expression in response to toxin A and toxin B. We found that both toxin A and toxin B activated ERK phosphorylation in mouse Raw264.7 macrophages, while addition of control IgG did not alter this response (see Fig. S1A in the supplemental material). Moreover, addition of the anti-toxin A MAb MK3415 blocked toxin A-associated ERK phosphorylation in these cells (see Fig. S1A and B in the supplemental material). In addition, the anti-toxin B antibody MK6072 reduced toxin B-induced ERK phosphorylation (see Fig. S1C and D in the supplemental material).
MK3415 and MK6072 significantly inhibit toxin A- and toxin B-induced TNF-α and IL-1β mRNA expression in human colonic explants.
Previous results indicated that toxins A and B can cause epithelial cell damage in human colonic explants (4). Here human colonic explants were divided into several pieces in Earle's balanced salt solution buffer and incubated with toxin A or B and the respective human MAbs overnight. Toxin A and toxin B dose-dependently increased TNF-α mRNA in human colonic explants (Fig. 6A and B). Incubation with MK3415 significantly inhibited TNF-α mRNA expression induced by 0.01 to 1 μg/ml toxin A (Fig. 6A), while MK6072 inhibited toxin B (10 μg/ml)-induced TNF-α mRNA in human colonic explants (Fig. 6B).
Fig 6.
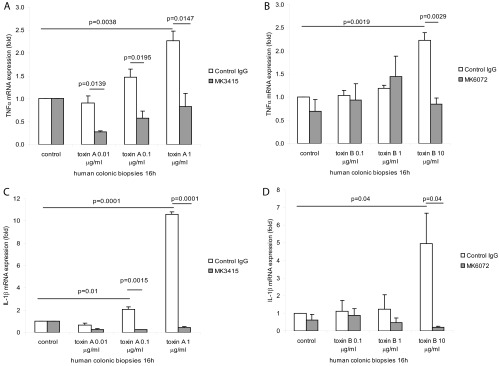
MK3415 and MK6072 attenuated toxin A- and toxin B-mediated TNF-α and IL-1β mRNA expression in human colonic explants. (A) Fresh human colonic explants were incubated with control IgG or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 0 to 1 μg/ml for 16 h. (B) Fresh human colonic explants were incubated with control IgG or MK6072 (30 μg/ml) for 30 min, followed by addition of toxin B at 0 to 10 μg/ml for 16 h. TNF-α mRNA expression was detected by real-time RT-PCR. (C) Fresh human colonic explants were incubated with control IgG or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 0 to 1 μg/ml for 16 h. (D) Explants were incubated with control IgG or MK6072 (30 μg/ml) for 30 min, followed by addition of toxin B at 0 to 10 μg/ml for 16 h. IL-1β mRNA expression was detected by real-time RT-PCR. All experiments are representative of 5 patient samples.
Similarly, toxin A (0.1 to 1 μg/ml) and toxin B (10 μg/ml) induced IL-1β mRNA expression in human colonic explants (Fig. 6C and D). Incubation of the explants with MK3415 significantly inhibited the IL-1β mRNA expression induced by 0.1 to 1 μg/ml toxin A (Fig. 6C), while MK6072 inhibited toxin B (10 μg/ml)-induced IL-1β mRNA (Fig. 6D).
MK3415 and MK6072 attenuate toxin A- and toxin B-induced epithelial tissue damage in human colonic biopsy specimens.
Consistent with our previous findings (4), toxins A and B elicited dramatic epithelial tissue damage in fresh human colonic explants (Fig. 7A and C). MK3415 significantly decreased toxin A-mediated epithelial cell damage (Fig. 7B), while MK6072 inhibited epithelial tissue damage in response to toxin B (Fig. 7D).
Fig 7.

MK3415 and MK6072 attenuated toxin A- and toxin B-mediated epithelial damage in human colonic explants. (A) Fresh human colonic explants were incubated with control IgG or MK3415 (30 μg/ml) for 30 min, followed by incubation with toxin A at 1 μg/ml for 16 h. (C) Explants were incubated with control IgG or MK6072 (30 μg/ml) for 30 min, followed by addition of toxin B at 10 μg/ml for 16 h. (B and D) Epithelial tissue damage was evaluated semiquantitatively as described in Materials and Methods. All experiments are representative of 5 patient samples.
DISCUSSION
The Gram-positive anaerobe C. difficile mediates antibiotic-associated diarrhea and pseudomembranous colitis by releasing two large exotoxins, toxin A and toxin B (2, 32). Not only do these two toxins mediate intestinal inflammation and diarrhea in CDI patients, but also antitoxin antibodies may be involved in the pathogenesis of CDI (18, 19). In mice, concurrent administration of intravenous immunoglobulin reduced the lethality caused by C. difficile toxin-containing supernatants (33). Results from a recent phase 2 trial suggested no change in the length of hospital stay but a significant reduction of CDI recurrence when monoclonal antibodies against toxin A and toxin B were given together with standard antibiotic treatment (23).
The cellular and molecular mechanisms by which C. difficile toxins A and B mediate diarrhea and inflammation have been in good part elucidated by in vitro experiments and studies with relevant experimental models (2, 6). Studies also showed that pretreatment with antibodies against these toxins is able to neutralize the responses in vitro and in vivo (20, 34–36). Such studies included experiments with human MAbs against these toxins (22). The beneficial effects of human anti-toxin A and B MAbs in vivo and in vitro (22) led us to hypothesize that these MAbs may prevent the proinflammatory responses to these toxins in human colon and human PBMCs through modulation of important signaling pathways that regulate the mucosal inflammation activated by these toxins. We report here that human MAbs exert their anti-inflammatory activity against toxins A and B in native human colon and human PBMCs by neutralizing the toxins and thereby inhibiting their effects on activation of host MAP kinase and NF-κB signaling pathways. Most of our experiments with PBMCs demonstrated that 30 to 50 μg/ml of anti-toxin A antibody MK3415 and anti-toxin B antibody MK6072 is sufficient to block >90% of toxin A- and B-mediated TNF-α and IL-1β expression, while their 50% effective dose is approximately 10 to 30 μg/ml (Fig. 1 to 4).
Our data confirmed our earlier findings that native human colon is sensitive to both C. difficile toxins (4) and for the first time showed that toxins A and B can stimulate TNF-α and IL-1β gene transcription in human colonic explants. Interestingly, our results also showed that human MAbs against these toxins significantly reduced toxin A- and toxin B-mediated TNF-α and IL-1β expression in native human colon and human PBMCs. Confirming this protective toxin A- and B-induced innate immune response, we also showed that MK3415 and MK6072 also neutralized epithelial cell damage in human colonic explants in response to toxin A and B exposure, respectively.
We show here that anti-toxin A and B MAbs MK3415 and MK6072, respectively, diminish toxin-induced NF-κB p65 subunit and ERK phosphorylation in monocytes/macrophages. Both NF-κB and ERK phosphorylation are required to elicit secretion of proinflammatory cytokines in response to toxin A and B in vivo and in vitro (7, 8, 12, 13, 31). Therefore, these two toxin A- and toxin B-neutralizing antibodies possibly prevented the toxins from triggering upstream signaling pathways and the resulting cytokine production and tissue damage.
In summary, the human anti-toxin A and anti-toxin B antibodies MK3415 and MK6072, respectively, significantly reduce C. difficile toxin A- and toxin B-mediated proinflammatory cytokine expression and colonic epithelial tissue damage in native human colon. This report provides further support for the clinical application of these two antibodies in CDI.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michelle Cheng, Ryan Ichikawa, Yuzu Kubota, Deanna Tran, and Irene Chang for assisting with our experiments. We also thank the Translational Pathology Core Laboratory, Department of Pathology, University of California at Los Angeles, for processing human colonic biopsy specimens for histologic assessment.
This work was supported by a research grant from Merck Pharmaceuticals (to C.P.). H.W.K. was supported by a Crohn's and Colitis Foundation of America Career Development Award (no. 2691) and NIH grant K01 DK084256 grant. S.H. was supported by a Crohn's and Colitis Foundation of America student research fellowship (no. 3831). Support was also provided by the Blinder Research Foundation for Crohn's Disease (to C.P.), the Eli and Edythe Broad Chair (to C.P.), and United States Public Health Service grant DK046763 (to D.Q.S. and S.R.T.). C.P.K. is supported by NIH grant RO1 AI095256, and X.C. is supported by a Career Development Award from the Crohn's and Colitis Foundation of America.
Footnotes
Published ahead of print 29 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02633-12.
REFERENCES
- 1. Kelly CP. 2012. Current strategies for management of initial Clostridium difficile infection. J. Hosp. Med. 7(Suppl 3):S5–S10 [DOI] [PubMed] [Google Scholar]
- 2. Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N. Engl. J. Med. 359:1932–1940 [DOI] [PubMed] [Google Scholar]
- 3. Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. 2003. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 125:413–420 [DOI] [PubMed] [Google Scholar]
- 4. Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT. 1995. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J. Clin. Invest. 95:2004–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713 [DOI] [PubMed] [Google Scholar]
- 7. He D, Sougioultzis S, Hagen S, Liu J, Keates S, Keates AC, Pothoulakis C, Lamont JT. 2002. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 122:1048–1057 [DOI] [PubMed] [Google Scholar]
- 8. Na X, Zhao D, Koon HW, Kim H, Husmark J, Moyer MP, Pothoulakis C, LaMont JT. 2005. Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes. Gastroenterology 128:1002–1011 [DOI] [PubMed] [Google Scholar]
- 9. Flegel WA, Muller F, Daubener W, Fischer HG, Hadding U, Northoff H. 1991. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect. Immun. 59:3659–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warny M, Keates AC, Keates S, Castagliuolo I, Zacks JK, Aboudola S, Qamar A, Pothoulakis C, LaMont JT, Kelly CP. 2000. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Invest. 105:1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jefferson KK, Smith MF, Jr, Bobak DA. 1999. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J. Immunol. 163:5183–5191 [PubMed] [Google Scholar]
- 12. Kim JM, Lee JY, Yoon YM, Oh YK, Youn J, Kim YJ. 2006. NF-kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand. J. Immunol. 63:453–460 [DOI] [PubMed] [Google Scholar]
- 13. Chen X, Fruehauf J, Goldsmith JD, Xu H, Katchar KK, Koon HW, Zhao D, Kokkotou EG, Pothoulakis C, Kelly CP. 2009. Saccharomyces boulardii inhibits EGF receptor signaling and intestinal tumor growth in Apc(min) mice. Gastroenterology 137:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H, Rhee SH, Kokkotou E, Na X, Savidge T, Moyer MP, Pothoulakis C, LaMont JT. 2005. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J. Biol. Chem. 280:21237–21245 [DOI] [PubMed] [Google Scholar]
- 15. Viscidi R, Laughon BE, Yolken R, Bo-Linn P, Moench T, Ryder RW, Bartlett JG. 1983. Serum antibody response to toxins A and B of Clostridium difficile. J. Infect. Dis. 148:93–100 [DOI] [PubMed] [Google Scholar]
- 16. Aronsson B, Granstrom M, Mollby R, Nord CE. 1985. Serum antibody response to Clostridium difficile toxins in patients with Clostridium difficile diarrhoea. Infection 13:97–101 [DOI] [PubMed] [Google Scholar]
- 17. Johnson S, Gerding DN, Janoff EN. 1992. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J. Infect. Dis. 166:1287–1294 [DOI] [PubMed] [Google Scholar]
- 18. Kyne L, Warny M, Qamar A, Kelly CP. 2000. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N. Engl. J. Med. 342:390–397 [DOI] [PubMed] [Google Scholar]
- 19. Kyne L, Warny M, Qamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357:189–193 [DOI] [PubMed] [Google Scholar]
- 20. Kelly CP, Pothoulakis C, Vavva F, Castagliuolo I, Bostwick EF, O'Keane JC, Keates S, LaMont JT. 1996. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob. Agents Chemother. 40:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kink JA, Williams JA. 1998. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 66:2018–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD., Jr 2006. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74:6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362:197–205 [DOI] [PubMed] [Google Scholar]
- 24. Pothoulakis C, LaMont JT, Eglow R, Gao N, Rubins JB, Theoharides TC, Dickey BF. 1991. Characterization of rabbit ileal receptors for Clostridium difficile toxin A. Evidence for a receptor-coupled G protein. J. Clin. Invest. 88:119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hing TC, Ho S, Shih DQ, Ichikawa R, Cheng M, Chen J, Chen X, Law I, Najarian R, Kelly CP, Gallo RL, Targan SR, Pothoulakis C, Koon HW. 3 July 2012. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut [Epub ahead of print.] 10.1136/gutjnl-2012-302180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koon HW, Shih D, Karagiannides I, Zhao D, Fazelbhoy Z, Hing T, Xu H, Lu B, Gerard N, Pothoulakis C. 2010. Substance P modulates colitis-associated fibrosis. Am. J. Pathol. 177:2300–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pothoulakis C, Castagliuolo I, LaMont JT, Jaffer A, O'Keane JC, Snider RM, Leeman SE. 1994. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 91:947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Linevsky JK, Pothoulakis C, Keates S, Warny M, Keates AC, Lamont JT, Kelly CP. 1997. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am. J. Physiol. 273:G1333–G1340 [DOI] [PubMed] [Google Scholar]
- 29. Warny M, Kelly CP. 1999. Monocytic cell necrosis is mediated by potassium depletion and caspase-like proteases. Am. J. Physiol. 276:C717–C724 [DOI] [PubMed] [Google Scholar]
- 30. Rocha MF, Maia ME, Bezerra LR, Lyerly DM, Guerrant RL, Ribeiro RA, Lima AA. 1997. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1beta, tumor necrosis factor alpha, and leukotrienes. Infect. Immun. 65:2740–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JY, Kim H, Cha MY, Park HG, Kim YJ, Kim IY, Kim JM. 2009. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J. Mol. Med. (Berl.) 87:169–180 [DOI] [PubMed] [Google Scholar]
- 32. Gerding DN, Johnson S. 2011. Clostridium difficile infection in 2010: advances in pathogenesis, diagnosis and management of CDI. Nat. Rev. Gastroenterol. Hepatol. 8:67–68 [DOI] [PubMed] [Google Scholar]
- 33. Saito T, Kimura S, Tateda K, Mori N, Hosono N, Hayakawa K, Akasaka Y, Ishii T, Sumiyama Y, Kusachi S, Nagao J, Yamaguchi K. 2011. Evidence of intravenous immunoglobulin as a critical supportive therapy against Clostridium difficile toxin-mediated lethality in mice. J. Antimicrob. Chemother. 66:1096–1099 [DOI] [PubMed] [Google Scholar]
- 34. Salcedo J, Keates S, Pothoulakis C, Warny M, Castagliuolo I, LaMont JT, Kelly CP. 1997. Intravenous immunoglobulin therapy for severe Clostridium difficile colitis. Gut 41:366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warny M, Fatimi A, Bostwick EF, Laine DC, Lebel F, LaMont JT, Pothoulakis C, Kelly CP. 1999. Bovine immunoglobulin concentrate—Clostridium difficile retains C difficile toxin neutralising activity after passage through the human stomach and small intestine. Gut 44:212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kelly CP, Kyne L. 2011. The host immune response to Clostridium difficile. J. Med. Microbiol. 60:1070–1079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.