

Abstract
Treatment of hepatitis C patients with direct-acting antiviral drugs involves the combination of multiple small-molecule inhibitors of distinctive mechanisms of action. ACH-806 (or GS-9132) is a novel, small-molecule inhibitor specific for hepatitis C virus (HCV). It inhibits viral RNA replication in HCV replicon cells and was active in genotype 1 HCV-infected patients in a proof-of-concept clinical trial (1). Here, we describe a potential mechanism of action (MoA) wherein ACH-806 alters viral replication complex (RC) composition and function. We found that ACH-806 did not affect HCV polyprotein translation and processing, the early events of the formation of HCV RC. Instead, ACH-806 triggered the formation of a homodimeric form of NS4A with a size of 14 kDa (p14) both in replicon cells and in Huh-7 cells where NS4A was expressed alone. p14 production was negatively regulated by NS3, and its appearance in turn was associated with reductions in NS3 and, especially, NS4A content in RCs due to their accelerated degradation. A previously described resistance substitution near the N terminus of NS3, where NS3 interacts with NS4A, attenuated the reduction of NS3 and NS4A conferred by ACH-806 treatment. Taken together, we show that the compositional changes in viral RCs are associated with the antiviral activity of ACH-806. Small molecules, including ACH-806, with this novel MoA hold promise for further development and provide unique tools for clarifying the functions of NS4A in HCV replication.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection is a major cause of liver diseases worldwide. It is estimated that 170 million individuals are infected with HCV (1–4). A significant portion of these infected people will develop liver diseases, including hepatitis, cirrhosis, and hepatocellular carcinoma (5). Treatment with pegylated alpha interferon (IFN-α) and ribavirin has a sustained virologic response or cure rate of ∼45% in genotype 1 HCV-infected patients (6, 7), and the addition of boceprevir or telaprevir, HCV NS3 protease inhibitors newly approved by the U.S. Food and Drug Administration, increases the cure rate to ∼70% (8). The new standard care of the triple combination, however, also leads to more toxic effects (9). Hence, development of new treatment regimens with higher efficacy, as well as better tolerability is urgently needed (10).
HCV, a member of the Flaviviridae family, is an enveloped virus with a positive-stranded RNA genome of 9.6 kb. The viral genome encodes a large polyprotein that is cleaved co- and/or posttranslationally into at least 10 mature viral proteins: structural proteins, including C, E1, E2, and p7, and nonstructural (NS) proteins, including NS2, NS3, NS4A, NS4B, NS5A, and NS5B. The functions of these viral proteins in the HCV life cycle have been extensively studied and mostly clarified (11). For example, NS5B has an RNA-dependent RNA polymerase activity, NS3 possesses a serine protease activity in its N-terminal domain and a helicase activity in the C-terminal domain, and NS4A is a cofactor of NS3 and activates NS3 protease function by forming a heterodimer (12–14). Several HCV nonstructural proteins such as NS3 protease, NS5B polymerase, and NS5A have been the prime targets for developing HCV direct-acting antiviral agents. Given the lack of a proofreading mechanism for HCV NS5B RNA-dependent RNA polymerase and the high-replication rate of HCV in patients, the emergence of resistant HCV variants is inevitable (15, 16) and has been observed in clinical trials of NS3 protease inhibitors, NS5A replication complex inhibitors, and NS5B polymerase inhibitors (17, 18). Therefore, combination therapies of antiviral agents that act via distinct mechanisms of action and lack cross-resistance will be necessary for sustained suppression of HCV replication.
ACH-806 (or GS-9132) is the result of discovery efforts aimed at the identification and characterization of small molecules that inhibit HCV replication via novel mechanisms. It was discovered through compound library screening, hit/lead identification, and lead optimization using HCV subgenomic replicon-containing cells (hereafter HCV replicon cells). ACH-806 has exhibited potent activity against genotype 1 HCV replication in vitro (19) and also showed antiviral activity in genotype 1 HCV-infected patients in a proof-of-concept clinical trial (1). Resistance substitutions that emerged under ACH-806 selection in replicon cells were mapped to the N-terminal region of NS3 and were not cross-resistant with NS3 protease inhibitors and NS5B polymerase inhibitors (19).
In HCV replicon cells, the mature nonstructural proteins, NS3, NS4A, NS4B, NS5A, and NS5B assemble on specialized intracellular membranes into replication complexes (RCs), where progeny viral RNA molecules are synthesized (11). NS4A is 54 amino acids (aa) in length and is the smallest nonstructural protein of HCV. It plays key roles in HCV replication by participating in RC assembly and regulating NS3 protease and helicase activities and NS5A phosphorylation (20–30). The central region of NS4A, aa 23 to 31, forms a complex with NS3 through extensive interactions with hydrophobic side chains on the two N-terminal β-strands of the NS3 protease domain (31–33). As a result, the positions of the catalytic triad of NS3 protease—His57, Ser139, and Asp81—are optimized for protease activity. Formation of the NS3-NS4A complex also enhances NS3 helicase activity, probably through interactions between the RNA helicase domain and the protease domain of NS3 protein. The N-terminal hydrophobic region of NS4A anchors the NS3-NS4A complex to host membranes as a component of RCs. The C-terminal acidic region of NS4A participates in the regulation of NS5A hyperphosphorylation and HCV replication.
In this study, we show that ACH-806 promotes the formation of a characteristic NS4A-containing product, p14, in HCV replicon cells, as well as in NS4A transfected cells. Further study demonstrated that p14 is a dimeric form of NS4A, and its production is negatively regulated by NS3. Associated with the formation of p14, ACH-806 causes reductions of NS3 and, especially, NS4A levels in RCs due to the accelerated degradation of both proteins. A previously described resistance substitution near the N terminus of NS3 attenuated both the decrease of NS3 and NS4A and the induction of p14, while conferring resistance to anti-HCV activity of ACH-806. Taken together, this study shows that the action of ACH-806 as an NS4A antagonist results in a series of viral RC compositional changes that probably contribute to impaired HCV RNA synthesis in treated replicon cells.
(This study was presented in part at the14th International Symposium on Hepatitis C Virus and Related Viruses, Glasgow, Scotland, United Kingdom, 2007.)
MATERIALS AND METHODS
Compounds.
ACH-806 (Fig. 1A), NS3 protease inhibitor telaprevir, nucleoside NS5B polymerase inhibitor (2′-C-methyladenosine), and non-nucleoside NS5B polymerase inhibitor 3-(1,1-dioxido-4H-benzo[e][1,2,4]thiadiazin-3-yl)-4-hydroxy-1-isopentylquinolin-2(1H)-one were synthesized by standard methods in organic chemistry and fully characterized by 1H-NMR high-pressure liquid chromatography and mass spectroscopy. A photo-affinity and tritium-labeled analog of ACH-806 (3H-ACH-119; specific activity, 103 Ci/mmol; see Fig. 8A) was synthesized at Amersham (now GE Healthcare UK).
Fig 1.
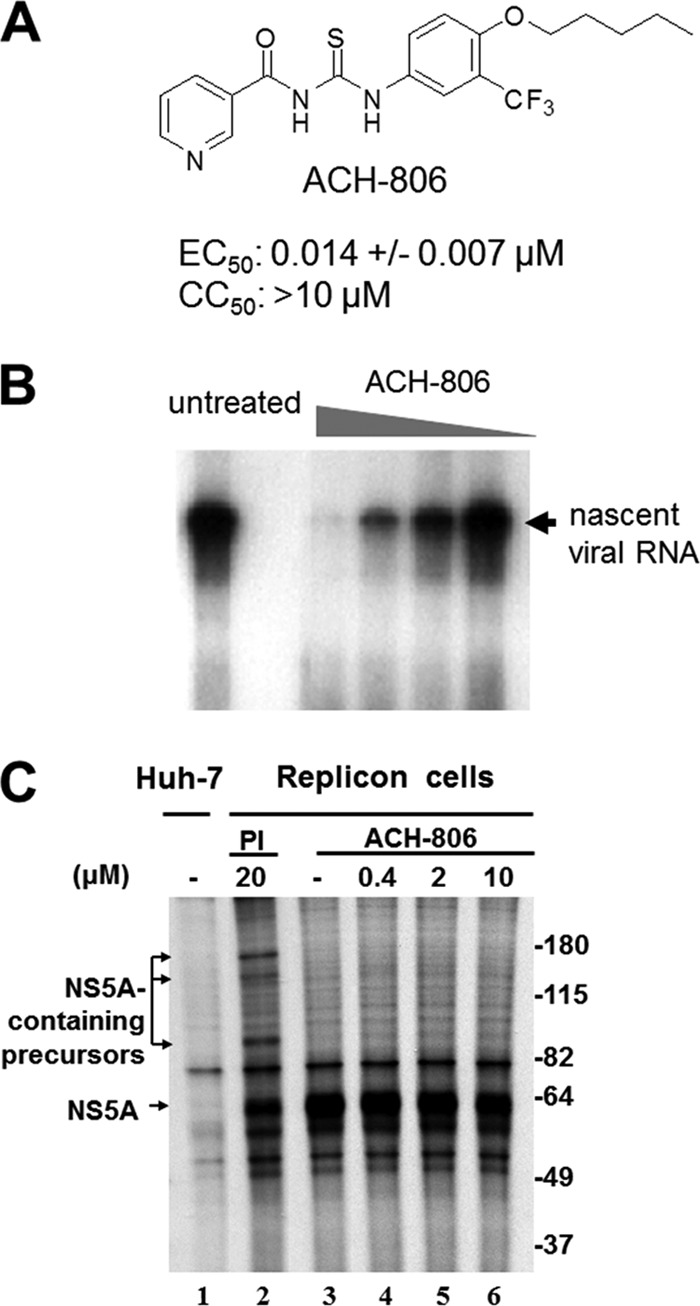
ACH-806 activity in HCV replicon cells. (A) ACH-806 structure. EC50 and CC50 values were determined in Huh-luc/neo cells (genotype 1b) using luciferase and MTS as endpoints after 3 days-exposure to the inhibitor. The mean values and standard deviations were derived from nine independent experiments. (B) Effect on the activity of RCs. RCs were prepared from Huh-9-13 cells that were treated with ACH-806 for 16 h at concentrations of 1.2, 0.24, 0.048, and 0.0096 μM before RC preparation. Isolated RCs were examined for their activity to synthesize nascent viral RNA in vitro in the absence of the inhibitor. The position of the major species of nascent viral RNA products is indicated on the right. (C) Effect on HCV polyprotein processing. Cells were subjected to simultaneous ACH-806 treatment and 35S metabolic labeling for 16 h, followed by anti-NS5A immunoprecipitation analysis. Molecular masses are indicated on the right in kilodaltons.
Fig 8.

Selective binding of an ACH-806 derivative to NS4A. (A) Compound structure. Tritiation sites for 3H-ACH-119 are indicated by asterisks (*). EC50, CC50, and SD values were determined in Huh-luc/neo cells using luciferase and MTS as endpoints after 3 days exposure to the inhibitor and were derived from three independent experiments. (B) In vitro 3H-ACH-119 binding. Concentrations (0.5 μM) of synthetic NS4A and recombinant full-length NS3 fused at the N terminus with NS4A (aa 23 to 31) were incubated with 40 nM 3H-ACH-119 in PBS at 30°C for 1 h. After UV irradiation, the proteins were subjected to a 4 to 20% SDS-PAGE, followed by fluorography and Coomassie blue staining. The positions of NS3 and NS4A are indicated. Molecular mass (MM) markers are indicated on the left in kilodaltons. (C) 3H-ACH-119 binds to synthetic NS4A in the presence of different concentrations of cold ACH-119, ACH-806, and ACH-281. (D) 3H-ACH-119 binding in cells. Huh-7 or Huh-9-13 cells were treated with 40 nM 3H-ACH-119 overnight, followed by photolysis and cell lysis (lanes 1 and 2). As controls, cells were treated with 40 nM ACH-119 (cold) and were metabolically labeled simultaneously with [35S]methionine-cysteine overnight, followed by cell lysis (lanes 3 and 4). All cell lysates were subjected to immunoprecipitation with anti-NS3 antibody (top three panels) and anti-NS4A antibody (bottom three panels), followed by SDS-PAGE and fluorography (lanes 3 and 4) or Western blot analysis (lanes 5 and 6). Positions of NS3 and NS4A are indicated on the left. Molecular mass (MM) markers are indicated on the right in kilodaltons. (E) Huh-9-13 and ACH-806 resistant cells were treated with 80 nM 3H-ACH-119 alone or together with 1 μM ACH-119 for 16 h. After UV irradiation, cell lysates were immunoprecipitated with anti-NS4A antibody. Equal amounts of immunoprecipitants were separated by 4 to 20% SDS-PAGE, followed by either fluorography or Western blot analysis. The positions of NS3 and NS4A are indicated on the left. Molecular masses are indicated on the right in kilodaltons.
Cells, plasmids, antibodies, and proteins.
Huh-7 cells, Huh-9-13 cells (genotype 1b replicon cells), and Huh-luc/neo cells (containing genotype 1b replicon with a firefly luciferase reporter gene) were licensed from ReBLikon (Schriesheim, Germany) (34). Plasmids expressing NS3, NS4A, NS4A-HA, NS4B-Flag, NS5A, and NS5B were constructed by cloning a PCR fragment amplified from pI377/NS3-3′ (a genotype 1b replicon-encoding plasmid) (34) into the pCL-neo mammalian expression vector (Promega, Madison, WI). Each PCR fragment was amplified with primers containing unique restriction enzyme sites and encoded one HCV nonstructural protein. A segment of sequence encoding the hemagglutinin (HA) or Flag tag was also included in the 3′ primer for NS4A-HA and the 5′ primer for NS4B-Flag, respectively. The PCR products were digested with the corresponding restriction enzymes and cloned into pCL-neo downstream of the cytomegalovirus immediate-early enhancer/promoter region. Full-length NS4A was synthesized at Synthetic Biomolecules, San Diego, CA. Full-length NS3 fused at the N terminus with cofactor NS4A was kindly provided by Gilead Sciences. Rabbit anti-NS3 (used for immunoprecipitation), rabbit anti-NS4B, rabbit anti-NS5A, and rabbit anti-NS5B antibodies were generous gifts from Ralf Bartenschlager and Volker Lohmann (Heidelberg, Germany). Mouse anti-NS3 (used for Western blotting) and mouse anti-NS4A antibodies were purchased from Virostat (Portland, ME) and Biodesign (Saco, ME), respectively. Anti-Flag antibody and secondary antibodies (goat anti-rabbit and goat anti-mouse) were purchased from Santa Cruz Biotechnology (San Diego, CA).
Measurement of anti-HCV activity and cytotoxicity of compounds in replicon cells.
Huh-luc/neo cells were seeded in 96-well plates at a density of 8,000 cells per well in a final volume of 200 μl of Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. One day after seeding, compounds were serially diluted in 100% dimethyl sulfoxide (DMSO) and added to cells at a 1:200 dilution, achieving a final concentration of 0.5% DMSO in a total volume of 200 μl. Cells were further incubated for 3 days (96 h post-seeding), and the inhibition of HCV replicon replication was quantified by measurement of luciferase activity using a commercial kit (Bright-Glo luciferase assay system; Promega, Madison, WI). Anti-HCV activity was expressed as the concentration that caused a reduction of luciferase activity (measured in relative luminescence units) by 50% (EC50) compared to the untreated controls. EC50s were calculated with a Microsoft Excel-based program. To measure each compound's cytotoxicity, the culture medium in each well was replaced by 100 μl of phenol red-free DMEM containing 10 μl of MTS (Promega, Madison, WI). After 2 to 4 h of incubation at 37°C, absorbance (i.e., the optical density at 490 nm) was measured, and concentrations that resulted in a 50% reduction of absorbance (CC50) compared to the untreated controls were calculated with a Microsoft Excel-based program.
Transient transfection.
Transient transfection of plasmids expressing NS3, NS4A, NS4A-HA, Flag-NS4B, NS5A, and NS5B was performed using the FuGENE 6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's instructions. Briefly, Huh-7 cells were seeded in a six-well plate at a density of 3 × 105 cells per well. On the following day, ca. 1 to 2 μg of the plasmid was used for each transfection. At 4 h posttransfection, compounds were added, and the plates were further incubated for ∼16 h. Cells either were directly lysed in sample buffer (Laemmli sample buffer supplemented with 5% β-mercaptoethanol) before sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or were lysed in NBP lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% sodium deoxycholate, and 0.1% SDS, supplemented with protease inhibitors [Complete, Mini, EDTA-free; Roche]) before immunoprecipitation as described below.
Isolation of RCs from replicon cells.
RCs were prepared as membrane fractions as described previously with modifications (35). Huh-9-13 cells were seeded in a 35-mm dish and cultured to ca. 90% confluence, followed by incubation overnight in the medium containing various concentrations of compound. After treatment, the cells were washed once with phosphate-buffered saline (PBS) and frozen at −80°C overnight. The frozen plates were thawed on ice. The cells were lysed in 1 ml of hypotonic buffer (10 mM Tris-HCl [pH 7.8], 10 mM NaCl) and passed through a 21-gauge needle 30 times. Nuclei were removed by centrifugation in a microcentrifuge at 900 × g at 4°C for 6 min. RCs were collected from the supernatant by centrifugation in a microcentrifuge at ∼20,000 × g at 4°C for 20 min. The pellets were either resuspended in 6 μl of storage buffer (hypotonic buffer plus 15% glycerol) for activity evaluation or solubilized in sample buffer before SDS-PAGE as described below.
Measurement of the activity of RCs isolated from replicon cells.
The assay for HCV RNA biosynthesis by isolated RCs was performed according to the protocol described previously (35, 36).
Pulse-chase and continuous metabolic labeling.
Cells were seeded in six-well plates at a density of 3 × 105 cells per well. For pulse-chase labeling (see Fig. 7A), the medium was replaced on the second day with methionine- and cysteine-free medium to starve the cells for 2 h, and then the medium was replaced with 1 ml per well of fresh methionine- and cysteine-free medium containing 150 μCi of 35S-Express protein labeling mix (35S-labeled methionine and cysteine; Perkin-Elmer, Boston, MA) for 2.5 h. After the pulse-labeling, the cells were washed with complete medium to remove 35S-containing medium. After a 0.5-h culture in the complete medium, test compounds, including ACH-806, were added to the corresponding wells, and the cells were treated overnight before harvesting for immunoprecipitation analysis as described below. For continuous labeling (Fig. 1C), the 35S-Express protein labeling mix and test compound were added to wells simultaneously after a 2-h starvation of cells in methionine- and cysteine-free medium. The treatment and labeling lasted for 16 h before harvesting and immunoprecipitation analysis.
Fig 7.
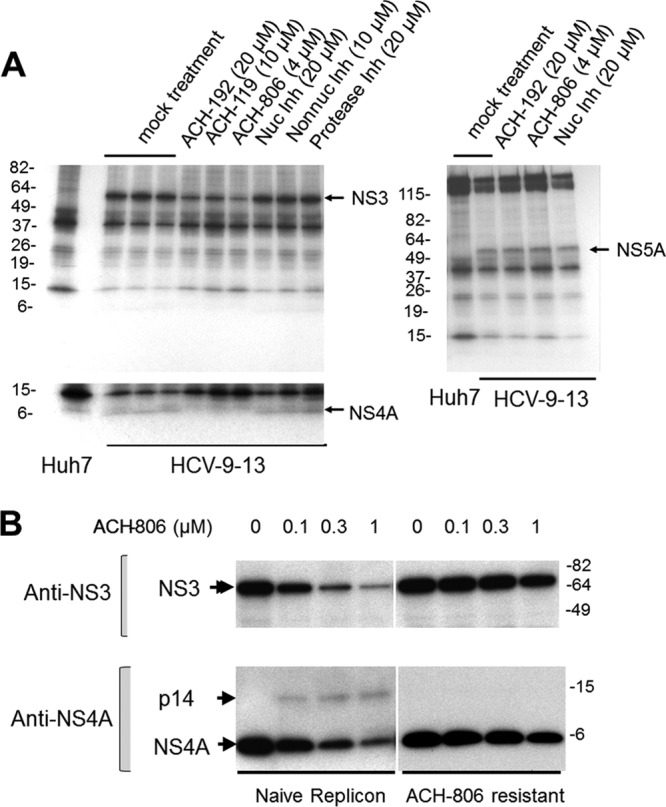
Effects of ACH-806 on NS3 and NS4A stability and on the amounts of p14, NS3, and NS4A in naive and resistant replicon cells. (A) Effect of ACH-806 on NS3 and NS4A stability. Huh-9-13 replicon cells were pulse-labeled with [35S]methionine-cysteine for 2.5 h, followed by a short chase (0.5 h) with complete medium. ACH-806 (4 μM), the two ACH-806 derivatives ACH-192 (20 μM) and ACH-119 (10 μM), NS5B nucleoside inhibitor (Nuc inh, 20 μM), NS5B non-nucleoside inhibitor (Non-nuc Inh, 10 μM), and protease inhibitor (20 μM) were added, and cells were further incubated overnight before the cells were lysed. Lysates were divided into two equal parts: each was subjected to immunoprecipitation with anti-NS5A (right panel) or anti-NS3 antibody (left panel), respectively, and followed by SDS-PAGE and fluorography. The lower left panel is a longer exposure of NS4A. Since the inhibitors were added after the pulse-labeling and chase periods, any differences in the amounts of pulse-labeled NS3, NS4A, and NS5A must represent differences in their degradation rates in response to inhibitor treatment. Molecular masses are shown on the left in kilodaltons. The positions of viral proteins are indicated on the right of each panel. (B) Effects of ACH-806 on the amount of p14, NS3, and NS4A in resistant cells. An ACH-806-resistant cell line that harbors a replicon carrying a C16S substitution in the N-terminal region of NS3 was chosen to compare the effects of ACH-806 on the amounts of p14, NS3, and NS4A to its parent (naive) replicon cell line Huh-9-13. After 16 h of treatment with ACH-806, cell lysates were subjected to SDS-PAGE and Western blotting with anti-NS3 and NS4A antibodies, respectively. The positions of viral proteins are indicated on the left. Molecular masses are indicated on the right in kilodaltons.
Photo-affinity labeling.
For in vitro photo-affinity labeling, certain amounts of protein were incubated in PBS with 40 nM 3H-ACH-119 at 30°C for 1 h. The solution was then irradiated from above with UV (λ = 254 nm) at a distance of 5 cm at room temperature for 7 min. Protein samples were subjected to SDS-PAGE as described below. To photo-affinity label protein in a cell culture, cells were seeded in six-well plates at a density of 3 × 105 cells per well. On the second day, the cells were incubated in the complete medium containing 40 nM or other concentrations of 3H-ACH-119 mentioned specifically in the text for 16 h. After the medium was removed, the confluent cell monolayer was irradiated similarly as described above for in vitro labeling. Cells were then harvested by addition of 0.6 ml of NBP lysis buffer per well, followed by immunoprecipitation, SDS-PAGE, and fluorography as described below.
Immunoprecipitation, SDS-PAGE, fluorography, and Western blot analyses.
For immunoprecipitation, cell lysates in the lysis buffer were incubated with protein A-beads that were prebound with an HCV antibody. After rotating on a rotator at 4°C overnight, the beads were washed to remove unbound proteins. The bound proteins were eluted in Laemmli sample buffer, boiled, and separated by SDS-PAGE. For direct visualization of labeled proteins, gels were treated with Enhancer (Amersham, Piscataway, NJ), dried, and exposed to X-ray film. For Western blot analysis, the proteins in the gel were transferred to a polyvinylidene difluoride (PVDF) membrane (Immun-Blot PVDF/Filter Paper Sandwich; Bio-Rad, Hercules, CA). Membranes were blocked and probed with a primary antibody specifically against an HCV nonstructural protein and then with a corresponding horseradish peroxide-conjugated secondary antibody. Lastly, the bound antibody was detected by chemiluminescence using an ECL Plus kit (Amersham). Membranes were then stripped and reprobed with different antibodies for sequential Western blot analysis of multiple HCV nonstructural proteins, where indicated.
RESULTS
ACH-806 does not inhibit HCV NS5B RNA polymerase and HCV polyprotein processing.
ACH-806 exhibits potent anti-HCV activities in both genotype 1a and 1b replicon-containing cells with EC50s between 0.01 and 0.05 μM, depending on the replicon cells and the endpoints (19). To study whether ACH-806 blocks de novo initiation of HCV RNA synthesis, RCs were prepared as membrane fractions from genotype 1b replicon cells and then treated with ACH-806 in vitro. Unlike some HCV NS5B non-nucleoside inhibitors that inhibit de novo HCV RNA synthesis in isolated RCs, ACH-806 failed to show any inhibitory effect at concentrations as high as 50 μM (data not shown), suggesting that allosteric sites on NS5B are probably not the target of ACH-806. On the other hand, in contrast to the result for ACH-806-treated RCs in vitro, RCs prepared from ACH-806-treated replicon cells showed a compound-dose-dependent reduction in HCV RNA synthesis with a 50% activity loss of RCs at a concentration as low as 50 nM (Fig. 1B). Therefore, although not directly inhibiting replication activity by the NS5B polymerase, ACH-806 treatment of replicon cells likely results in qualitative and/or quantitative changes in RC composition or function.
To address whether ACH-806 affects HCV polyprotein processing, HCV replicon cells were metabolically labeled with [35S]methionine and [35S]cysteine in the presence of ACH-806 or a protease inhibitor (PI [NS3 protease inhibitor]). Immunoprecipitation analysis with anti-NS5A antibody demonstrated that ACH-806 treatment at concentrations as high as 500-fold over its EC50 did not result in accumulation of NS5A-containing precursors in replicon cells (Fig. 1C). In contrast, a clear accumulation of NS5A-containing precursors was observed in the lysates of cells treated with PI at a concentration 20-fold above its EC50 (Fig. 1C). These results and other experiments described here support the claim that ACH-806 does not inhibit HCV polyprotein processing in replicon cells at concentrations where it displays its anti-HCV activity.
ACH-806 treatment results in compositional changes of HCV replication complexes.
To dissect the mechanism by which ACH-806 treatment of replicon cells compromises the function of RCs, we used Western blot analysis to examine the levels of each of the five HCV nonstructural proteins within RCs following ACH-806 treatment (Fig. 2A). No significant changes in the amounts of NS4B, NS5A, and NS5B were detected (Fig. 2A, third, fourth, and fifth panels). In contrast, dose-dependent reductions in the amounts of NS3 and, especially, NS4A were observed (Fig. 2A, first and second panels). Similar results about NS3 and NS4A in compound-treated replicon cells were shown in Fig. 7B. Besides reductions of NS3 and NS4A, a dose-dependent appearance of a new protein species was detected with anti-NS4A antibody (Fig. 2A, second panel). Significantly, therefore, ACH-806 treatment altered the composition of HCV RCs in replicon cells.
Fig 2.
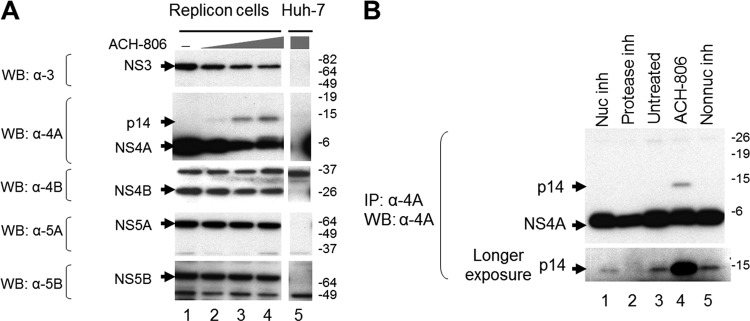
Alterations of viral protein composition of RCs in ACH-806-treated replicon cells. (A) Effect on each nonstructural protein. RCs were isolated from cells (Huh-9-13 or Huh-7) after exposure to ACH-806 (0.2, 1, and 5 μM for lanes 2, 3, and 4, respectively) overnight and were subjected to SDS-PAGE. After the transfer of proteins to a PVDF membrane, the membrane was probed sequentially with the indicated antibodies. Molecular masses are indicated on the right in kilodaltons. (B) Detection of p14. Cell lysates from Huh-9-13 cells, after exposure to ACH-806 (1 μM), NS3 protease inhibitor (Protease inh, 20 μM), NS5B nucleoside inhibitor (Nuc inh, 20 μM), and NS5B non-nucleoside inhibitor (Non-nuc inh, 20 μM) for 8 h, were subjected to immunoprecipitation, followed by a Western blot analysis with anti-NS4A antibody. The bottom panel is a longer exposure of the upper panel. Molecular masses are indicated on the right in kilodaltons.
The new protein species detected by anti-NS4A antibody migrated at ∼14 kDa and hence was designated as p14. The p14 species was not detected in RCs isolated from the untreated sample (Fig. 2A, lane 1, in the second panel). However, since the amount of viral protein loaded onto the gel was limited by large amounts of host proteins present in RC preparations, we could not exclude the possibility that p14 might be present at a low level in the control sample. To enrich p14, total cell lysates prepared from those replicon cells were subjected to an immunoprecipitation with anti-NS4A antibody before Western blot analysis (Fig. 2B). Indeed, p14 was present in the untreated sample (Fig. 2B, lane 3), and its level was unaffected by treatments with nucleoside and non-nucleoside NS5B polymerase inhibitors (lanes 1 and 5). Treatment with NS3 protease inhibitor, however, decreased the amount of p14 (lane 2). Consistent with Fig. 2A, treatment with ACH-806 greatly increased the amount of p14 (lane 4).
Other HCV nonstructural proteins are not required for the formation of p14.
Since all HCV nonstructural proteins expressed in the replicon cells used in the present study were derived from the processing of the single polyprotein, we wondered whether other HCV nonstructural proteins play a role in the formation of NS4A-related p14 in the presence of ACH-806. To address this question, we constructed five plasmids to express each of the HCV nonstructural proteins individually in Huh-7 cells. After individual transfections and subsequent ACH-806 or mock treatment, each nonstructural protein was examined in whole-cell lysates by Western blotting (Fig. 3). No significant changes in individual HCV protein expression were detected after ACH-806 treatment, except in NS4A-transfected cells, where a significant increase of p14 was observed with no associated change in the level of NS4A itself (Fig. 3, second panel). Recapitulation of the unique change of p14 accumulation in the compound-treated cells that expressed NS4A alone suggests that ACH-806 could trigger or accelerate p14 formation from NS4A protein and that the presence of other HCV nonstructural proteins and the assembly of HCV RCs are not required for the formation of p14.
Fig 3.
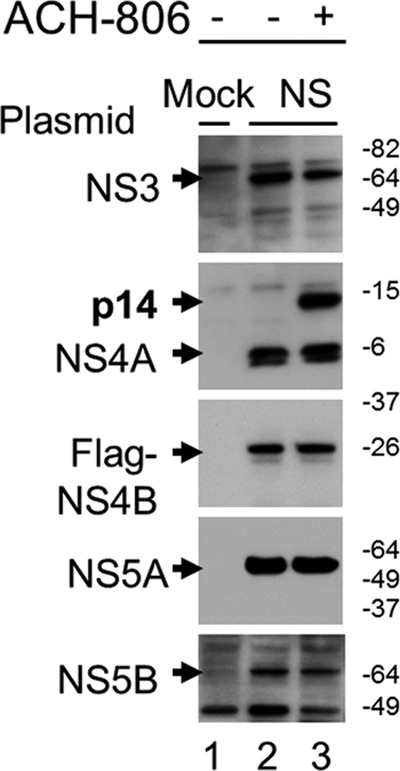
p14 formation in the absence of other HCV nonstructural proteins. Huh-7 cells were transfected with either a mock plasmid or plasmids encoding (from top to bottom) NS3, NS4A, NS4B with an N-terminal Flag, NS5A, and NS5B, respectively. Transfected cells were exposed overnight to ACH-806 (2 μM). Whole lysates were subjected to SDS-PAGE and Western blots with anti-NS3, anti-NS4A, anti-Flag, anti-NS5A, and anti-NS5B antibody (from top to bottom), respectively. Molecular masses are indicated on the right in kilodaltons.
p14 is a dimer of NS4A.
Next, we conducted experiments to characterize the nature of p14. p14 was not affected by heating (95°C, 5 min) in 1× Laemmli sample buffer. Including a reducing agent β-mercaptoethanol in sample buffer could slightly reduce the level of p14 but the majority of p14 failed to be dissolved, suggesting disulfide bonds might not play a major role in the formation of p14. Urea treatment (8 M), however, completely dissociated p14 (Fig. 4A) no matter whether it was derived from NS4A-transfected cells (Fig. 4A, top panel) or replicon cells (Fig. 4A, bottom panel). These results provide evidence that p14 might be an aggregate in which NS4A is noncovalently linked to something, including NS4A itself (homodimer).
Fig 4.
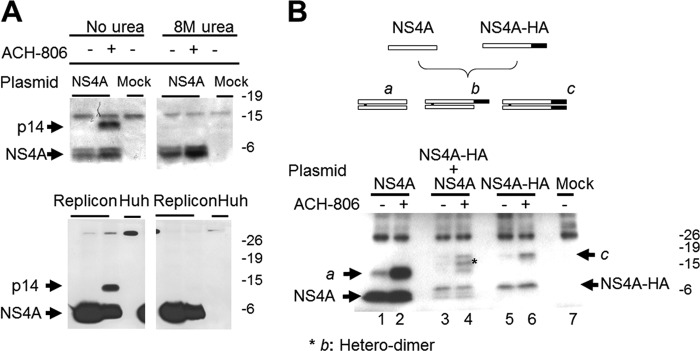
Nature of p14. (A) Effect of 8 M urea on p14. Huh-7 cells, transfected with either a mock plasmid or a plasmid encoding NS4A, or Huh-9-13 replicon cells were treated with ACH-806 (2 μM) overnight. Transfected Huh-7 cells (top panel) were lysed with sample buffer or sample buffer plus 8 M urea. For replicon cells (bottom panel), RCs were isolated and solubilized with sample buffer or sample buffer plus 8 M urea. All samples were subjected to Western blot analyses with anti-NS4A antibody. Molecular masses are indicated on the right in kilodaltons. (B) Dimeric form of NS4A. Two plasmids, one encoding NS4A and the other encoding NS4A-HA, were transfected alone or together in 1:1 ratio to Huh-7 cells. After overnight treatment with ACH-806 (2 μM), cells were lysed, and lysates were subjected to immunoprecipitation with anti-NS4A antibody, followed by a Western blot analysis with anti-NS4A antibody. The diagram above the Western blot illustrates the three expected dimer species if NS4A and NS4A-HA can form dimers. NS4A, open box; NS4A-HA, open box with a shaded box at its C terminus representing the HA tag; species a, NS4A homo-dimer; species b, NS4A and NS4A-HA heterodimer; species c, NS4A-HA homodimer. The asterisk on the Western blot indicates species b in the cotransfected cells. Molecular masses are indicated on the right in kilodaltons.
To test whether p14 is an NS4A homodimer, we cotransfected Huh-7 cells with plasmids encoding two forms of NS4A separable by size: native NS4A and NS4A with an HA epitope tag (NS4A-HA) (Fig. 4B, top). NS4A-HA is slightly larger than NS4A due to the presence of HA tag. Expression of NS4A alone (Fig. 4B, lanes 1 and 2) yielded p14 (labeled “a”), while expression of NS4A-HA alone (Fig. 4B, lanes 5 and 6) produced a species larger than p14 (labeled “c”). As expected, ACH-806 treatment significantly enhanced the levels of both p14 and the p14-like species (compare lane 1 to lane 2 and lane 5 to lane 6 in Fig. 4B). Notably, the coexpression of NS4A and NS4A-HA (Fig. 4B, lanes 3 and 4) resulted in an intermediately sized species, b, in addition to species a and c. Species a, b, and c all showed similar responses to ACH-806 treatment. Since species b was likely formed as a heterodimer of NS4A with NS4A-HA and species a and c therefore as homodimers, respectively, of NS4A and NS4A-HA, these results indicate that p14 is likely induced by ACH-806 treatment as a simple NS4A homodimer.
NS3 negatively regulates the formation of p14.
We noted a higher fraction of NS4A incorporated into p14 in cells expressing NS4A alone (compare lane 2 in the top panel with the bottom panel in Fig. 4A). Since NS4A interacts with NS3 in replicon cells to form an NS3-NS4A complex, we speculated that NS3 might play a negative role in the formation of p14. To answer this question, we coexpressed NS4A in Huh-7 cells with various amounts of NS3 and examined the amount of each protein with Western blot analysis after overnight ACH-806 treatment (Fig. 5A). The experiment revealed several interesting phenomena. First, NS4A levels were greatly increased when NS3 plasmid was included in a transfection. NS4A level achieved a plateau when 0.2 μg of NS3 plasmid was used. The enhanced detection of NS4A in a cotransfection was NS3-specific since coexpression with other HCV nonstructural proteins did not display this effect on NS4A (Fig. 5B). In an independent experiment (data not shown), coexpression of NS4A increased NS3 level similarly. These results are consistent with the previous findings that the formation of NS3-NS4A complexes in cells where NS3 and NS4A were coexpressed increased the stabilities of both proteins (14, 37). Second, as more NS3 protein was produced, the level of p14 was gradually decreased, although other conditions were kept the same. The result suggested that NS3 protein or NS3-NS4A complexes played a negative role in p14 formation. Third, ACH-806 treatment resulted in significant reductions of both NS3 and NS4A in the transfected cells. This finding was reminiscent of ACH-806-treated replicon cells in which the amounts of NS3 and NS4A were also both decreased (Fig. 2A). Together, these results indicate that ACH-806 might impair the interaction between NS3 and NS4A and thereby destabilize both proteins, while simultaneously increasing the incorporation of NS4A into p14.
Fig 5.
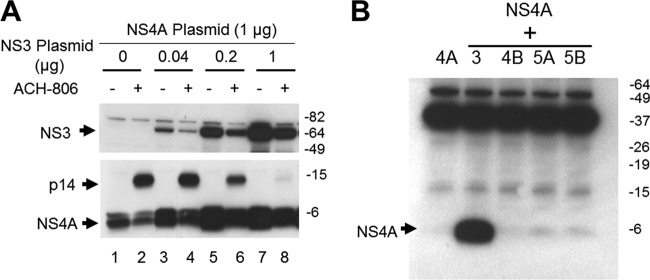
Impact of NS3 on p14 and NS4A. (A) Impact of NS3 on p14. Huh-7 cells were cotransfected with NS4A-expressing and NS3-expresssing plasmids in the indicated amounts (the total amount of plasmid used in each transfection was 2 μg, and an unrelated mock plasmid was used to make up any shortages in each transfection). After overnight treatment with ACH-806 (3 μM), the cells were lysed with sample buffer, and lysates were subjected to sequential Western blot analyses with anti-NS4A antibody and then anti-NS3 antibody. Molecular masses are indicated on the right in kilodaltons. (B) Impact of NS3 and other nonstructural proteins on NS4A. Huh-7 cells were cotransfected with NS4A and one of NS3-, NS4B-, NS5A-, and NS5B-expressing plasmids. Two days later, transfected cells were harvested, and cellular lysates were subjected to SDS-PAGE, followed by a Western blot analysis with NS4A antibody. Molecular masses are indicated on the right in kilodaltons.
ACH-806 impairs NS3-NS4A interaction.
Subsequently, we conducted a study to quantify the interaction between NS3 and NS4A in replicon cells to determine whether it was impaired by ACH-806 treatment (Fig. 6). After ACH-806 treatment, the cells were lysed. One-third of the lysates was directly used in a Western blot analysis by anti-NS3 antibody to compare the total NS3 levels in the compound-treated and untreated replicon cells. Based on the densities of NS3 in lanes 1 and 2 in Fig. 6, the total amount of NS3 in the ACH-806-treated sample was reduced by ∼6-fold (100/16). The remaining lysates (2/3) were subjected to coimmunoprecipitation with NS4A antibody, followed by a Western blot analysis with anti-NS3 antibody to measure the amount of NS3 in NS3-NS4A complex. Based on the densities of NS3 in lanes 3 and 4 in Fig. 6, ACH-806 treatment caused a reduction of NS4A-bound NS3 ∼29-fold (261/9). The more reduction of NS4A-bound-NS3 after ACH-806 treatment might be due to one or two of the following scenarios: ACH-806 caused more NS4A reduction, as shown in Fig. 2A, and therefore less NS3 was pulled down with anti-NS4A antibody. Here, a certain portion of NS3 in treated cells was free from the NS3-NS4A complex. Second, the binding between NS3 and NS4A became weaker in the presence of ACH-806. As a result, NS4A antibody failed to pull down all of the NS3 in the complex. Either the presence of “free” NS3 or the presence of loose NS3-NS4A complexes suggests impaired NS3-NS4A interaction.
Fig 6.
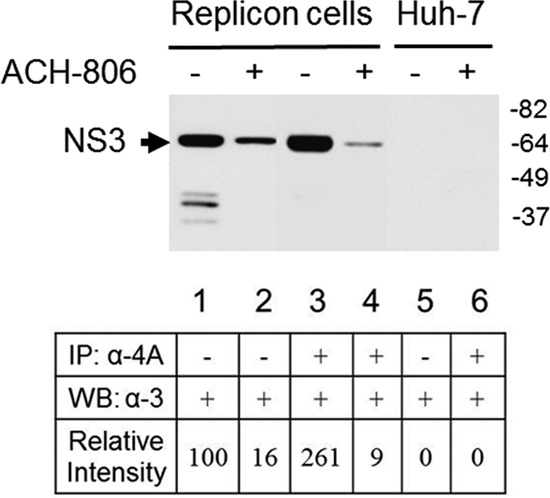
Impaired NS3-NS4A interaction in ACH-806-treated replicon cells. Cell lysates from Huh-9-13 replicon cells or Huh-7 cells after exposure to ACH-806 (5 μM) overnight were divided into two portions. The smaller portion was subjected directly to SDS-PAGE, followed by Western blotting with anti-NS3 antibody (lanes 1, 2, and 5); the larger portion was subjected to immunoprecipitation with anti-NS4A antibody, followed by SDS-PAGE and Western blotting with anti-NS3 antibody (lanes 3, 4, and 6). Molecular masses are indicated on the right in kilodaltons. The densities of NS3 in each lane were obtained by densitometry scanning. NS3 signal in lane 1 (untreated input) was given an arbitrary number 100, and the signals of the other lanes were normalized to lane 1. The numbers are shown at the bottom of each lane.
ACH-806 treatment leads to reduced stabilities of NS3 and NS4A.
To confirm that the reductions of both NS3 and NS4A in ACH-806-treated replicon cells were due to accelerated degradation, a pulse-chase experiment was executed to trace the decrease of labeled HCV proteins in replicon cells. Methionine- and cysteine-starved cells were pulse-labeled with 35S-labeled methionine and cysteine for 2.5 h. After washing with fresh culture medium, cultured cells were treated with HCV inhibitors, including ACH-806, for 16 h. As shown in Fig. 7A, the levels of labeled NS3 and NS4A immunoprecipitated by anti-NS3 antibody were apparently reduced after the treatment of ACH-806 and its two derivatives, ACH-192 and ACH-119. In contrast, HCV protease inhibitor, nucleoside inhibitor, and non-nucleoside inhibitor did not cause any reductions of pulse-labeled NS3 and its associated NS4A. The effect of ACH-806 was specific for NS3 and its bound NS4A because immunoprecipitated NS5A did not show a detectable change after these treatments. Since no inhibitor was present during labeling, reduction of a labeled protein, most likely, was due to its accelerated degradation.
To reinforce the studies described above and correlate the effects of ACH-806 on HCV NS3 and NS4A with its anti-HCV activities in replicon cells, a previously described ACH-806-resistant replicon cell line was utilized for a side-by-side comparison of the effects of ACH-806 on the amounts of p14, NS3, and NS4A between the ACH-806-resistant cell line and its parental cell line Huh-9-13. This ACH-806-resistant cell line harbors a replicon carrying a C16S substitution in the N-terminal region of NS3, where NS3 interacts with NS4A, and displays ∼10-fold less susceptibility to ACH-806 (19). As shown in Fig. 7B, ACH-806 induced significant decreases of NS3 and NS4A and promoted p14 formation in the parental replicon cells but not in the ACH-806-resistant replicon cells. This result linked together the following key facts of ACH-806: anti-HCV activity in a cell-based assay, the effects on the key viral proteins NS3 and NS4A, as well as on p14, and the corresponding substitution that conferred the resistant phenotype of replicons to this compound. Moreover, the location of the resistance substitution reinforces the importance of the NS3-NS4A interaction in the mechanism of ACH-806 action.
An ACH-806 derivative compound selectively binds to NS4A.
Although the evidence presented above demonstrated that ACH-806 acts on NS4A, it was uncertain whether there is a direct interaction between ACH-806 and NS4A. In order to investigate this, we synthesized a photo-affinity and tritium-labeled analog of ACH-806 (3H-ACH-119, Fig. 8A) and examined its binding to NS4A. First, we used a synthetic full-length NS4A and recombinant full-length NS3 fused at N terminus with cofactor domain of NS4A (aa 23 to 31) as potential binding targets. After a short incubation with 40 nM 3H-ACH-119 (30°C for 1 h), the protein solutions were irradiated with UV radiation and subjected to SDS-PAGE. The binding signal was detected through fluorography (Fig. 8B). Although the molar concentrations of NA4A and NS3-NS4A proteins used in this experiment were the same (0.5 μM), a much stronger signal was observed in NS4A (Fig. 8B). Moreover, this binding was competed for by both cold ACH-119 and ACH-806 in a dose-dependent manner but not by an anti-HCV inactive compound, ACH-281 (Fig. 8C).
To study compound binding in replicon cells, parental Huh-7 and replicon Huh-9-13 cells were treated with 3H-ACH-119, exposed to UV light to induce cross-linking, and then lysed with NBP buffer. As a control, another set of cell culture plates was metabolically labeled with [35S]methionine-cysteine and treated with unlabeled ACH-119 and lysed similarly. Cell lysates from both sets of cells were subjected to immunoprecipitation with anti-NS3 antibody (top three panels of Fig. 8D) and anti-4A antibody (bottom three panels of Fig. 8D), respectively. 3H-ACH-119-bound proteins, metabolic labeled proteins, and viral NS3 and NS4A in those immunoprecipitants were detected by fluorography (left and middle panels) and Western blotting (right panels), respectively. Although these antibodies precipitated many cellular proteins in addition to NS3 and NS4A, as shown by multiple bands in the sample labeled with [35S]methionine-cysteine, the dominant band was NS4A in the sample labeled with 3H-ACH-119. NS5A pulled down by anti-NS5A antibody, on the other hand, did not show any 3H-ACH-119 binding (data not shown). These data suggest that ACH-119 binds selectively to NS4A. We further compared the compound binding between Huh-9-13 cells and an ACH-806-resistant cell line. These cells were treated with 3H-ACH-119 alone or together with 1 μM cold ACH-119 for 16 h, followed by UV irradiation. Equivalent amounts of cell lysates were subjected to anti-NS4A immunoprecipitation and SDS-PAGE, followed by fluorography and Western blot analysis. As shown in Fig. 8E, although nearly equal levels of NS4A were detected in Western blots in two cell lines after treatment, more 3H-ACH-119-NS4A binding was present in Huh-9-13 cells, and this binding was attenuated by cold ACH-119. 3H-ACH-119-NS4A binding in ACH-806-resistant cells, however, was relatively weak and less affected by the presence of cold ACH-119. These data suggest that resistant substitution in NS3 (C16S), even not at NS4A, might play a negative role on ACH-119 binding through, probably, an altered NS3-NS4A interaction.
DISCUSSION
HCV RNA replication occurs on viral replication complexes (RCs) that locate in a specialized cytoplasmic vesicular structure designated as the membranous web (38, 39) or lipid rafts (40). One of the RC components is the NS3-NS4A heterodimer in which the N-terminal transmembrane α-helix of NS4A and an amphipathic helix α0 helix at N terminus of NS3 coordinate for the attachment of the heterodimer to the membrane structure (41). Here, we have described mechanism of action studies conducted on a novel HCV inhibitor, ACH-806. We have shown that the RNA replication activity of RCs was lost upon treatment of replicon cells with ACH-806. In isolated RCs there were alterations in the composition of viral proteins (decreased amounts of NS3 and NS4A and an increased amount of a novel NS4A homodimer designated here as p14), as well as in the interactions among viral proteins (diminished interaction between NS3 and NS4A). Combined with the locations of resistance substitutions (in the N-terminal portion of NS3 where NS4A interacts with NS3) (19), we think that these alterations in RCs might contribute to the presence of defective RCs and diminished HCV RNA synthesis in ACH-806-treated replicon cells.
The most characteristic effect of ACH-806 on replicon cells was the induction of p14, a dimer form of NS4A. Although p14 was not reported previously, we found that it presents at very low levels in untreated replicon cells (Fig. 2B). We believe that, due to a tight association between NS3 and NS4A after NS3-NS4A translation and cleavage, most, if not all, NS4A is present as a form of NS3-NS4A complex in RCs. NS3, acting as a chaperone through forming a complex with NS4A, might preclude NS4A from dimerization or oligomerization. In the absence of NS3, on the other hand, the small hydrophobic NS4A may tend to fold incorrectly. Subsequently, the misfolded NS4A may be degraded and/or form dimeric aggregates, i.e., p14. This hypothesis is supported by the observations that NS4A was not stable when expressed alone and the formation of p14 in those NS4A-alone transfected cells was more marked compared to replicon cells (Fig. 5A). Coexpressing NS3 with NS4A, however, not only improved NS4A stability but also inhibited p14 production in a dose-dependent manner (Fig. 5A).
Another interesting observation about p14 is the nature of NS4A-NS4A interaction which, presumably, is strong because only urea at a concentration of 8 M separates it (Fig. 4A). Since p14 was efficiently generated in Huh-7 cells where NS4A was expressed, we think this might be a suitable starting point to study the nature of p14. Through systematic mutagenesis, for example, it could be possible to locate a key region(s) or amino acids in NS4A that is(are) critical or responsible for the NS4A-NS4A dimerization. This information will help us to understand how ACH-806 triggers p14 formation, as well as benefit clarification of the relationship between p14 formation and impaired NS3-NS4A interaction.
After ACH-806 treatment, the reduction of NS3 and, especially, NS4A in replicon cells (Fig. 2A) is the most significant change besides the reduced level of HCV RNA. We believe this phenomenon is most probably caused by accelerated degradation of these HCV nonstructural proteins since in in vitro and in cells we failed to find any evidence that ACH-806 inhibits protein translation. For example, there is no reduction of NS5A, a part of polyprotein downstream to NS3 and NS4A, in metabolically labeled replicon cells (Fig. 1C) in the presence of ACH-806. Although p14 and NS3-NS4A reduction are clearly shown in treated replicon cells, we, however, do not know the sequence/relations of these two events. Initially, two scenarios could be raised to interpret these data. First, ACH-806 might trigger a conformational change of NS4A, resulting in p14 aggregates and NS4A degradation. Without NS4A, NS3 is not stable in RCs. Second, ACH-806 leads an abnormal NS3-NS4A interaction that is followed by accelerated degradation of both NS3 and NS4A. p14 is formed during the process of degradation as a casual by-product. The following analysis of the effects of ACH-806 might argue against the second scenario. As shown extensively, NS3-NS4A interaction is required for NS4A-dependent cleavages of HCV polyprotein. Throughout the present study, we failed to find an apparent inhibitory effect of ACH-806 on those major cleavage events no matter whether the corresponding experiments were executed in an in vitro system (data not shown) or in replicon cells (Fig. 1C). This fact challenges the idea that ACH-806 impairs NS3-NS4A interaction in a universal manner. Rather, impaired NS3-NS4A interaction in the presence of ACH-806 is the result of some preceding effect(s) of this compound.
To summarize the discussions above, we propose a model to unify the effects of ACH-806 treatment on HCV RCs (Fig. 9). ACH-806 does not affect apparently polyprotein translation and processing, the early events of RCs formation. After individual nonstructural proteins are released from HCV polyprotein, they interact with each other and might adapt to serial conformational changes for the formation of RCs. ACH-806 initiates its action during or after RC assembly. First, ACH-806 could cause a conformational change of its primary target, NS4A. As a result, several events might happen simultaneously or sequentially: NS4A-NS4A dimerization between misfolded NS4A to form p14, impaired NS3-NS4A interaction due to the misfolding of NS4A, and NS4A degradation. Associated with NS4A reduction in RCs, NS3-NS4A interaction is further damaged. Finally, in the absence of its NS4A partner, NS3 degradation occurs. Due to the compositional change and abnormal nonstructural protein interactions in RCs, HCV RNA synthesis is causatively dysfunctional in replicon cells.
Fig 9.
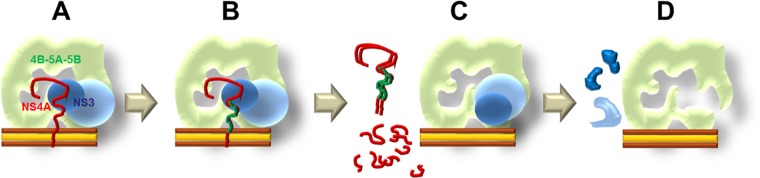
Model of mechanism of action of ACH-806. (A) In HCV replicon cells, RCs are formed on specialized cellular membrane (sandwiched brown color lines) for HCV RNA synthesis. NS4A (shown as a thick curved line in red) interacts with the protease domain of NS3 (shown as a small blue ball). The bigger whitish-blue ball represents the helicase domain of NS3 protein. The rest of the RC is shown as an intact, membrane-bound structure in mint-green color. (B) ACH-806 triggers a local conformational change(s) on NS4A (represented as a thick green line with a hypothetic position) that results in the formation of p14 (a dimer of NS4A) and degradation of NS4A shown in panel C. Without NS4A, NS3 in RCs (a “free form” shown in panel C) is not stable. The degradation of NS3 and NS4A leads to the rest RCs (shown in panel D) defective for HCV RNA replication.
Although NS4A is the smallest nonstructural protein of HCV, evidence suggests that NS4A plays multiple roles in HCV life cycle and pathogenesis. Besides acting as a cofactor of NS3, NS4A forms a complex with other HCV proteins (20, 21, 26, 30), which suggests a role in assisting the formation of RCs. Together with NS3 or NS4B, or by itself, NS4A modulates NS5A phosphorylation (20, 24, 27, 29). Overexpression of NS4A inhibits host and viral protein translation (22, 23) and causes mitochondrial damage (28). Selected substitutions in the NS4A C-terminal region reduce HCV replication fitness (27). Hence, considering the diverse functions of NS4A, ACH-806 can impact multiple aspects of HCV life cycle by targeting NS4A. Indeed, in addition to the discoveries described here, we noted other sporadic effects of ACH-806 on NS4A. For example, we detected in replicon cells that NS4B interacts with the residual NS4A with an enhanced affinity. Since it is not clear how to relate these effects of ACH-806 with its inhibitory effect on HCV RNA replication, we would like to leave these results for further studies. On the other hand, these results and data presented above suggest that ACH-806 and its derivatives are useful tools in investigating the role of NS4A in HCV replication.
Finally, subsequent to the completion of preclinical pharmacology and drug safety studies, a phase I trial was conducted in patients chronically infected with genotype 1 HCV to investigate the safety, tolerability, and antiviral efficacy of ACH-806. In the present study, after orally administering 300 mg of ACH-806 twice daily, a mean decrease in plasma HCV RNA of 0.91 log10 was observed after 5 days of monotherapy versus an increase of 0.05 log10 for placebo-treated patients (1). Further development of ACH-806 was discontinued due to reversible nephrotoxicity. However, the antiviral activity of ACH-806 demonstrated in this proof-of-concept clinical trial and the novel mechanism of ACH-806 revealed in this study warrant continued discovery and development efforts for this novel class of inhibitors in the treatment of HCV patients.
ACKNOWLEDGMENTS
We thank Ralf Bartenschlager and Volker Lohmann for the generous gift of anti-NS3, anti-NS4B, anti-NS5A, and anti-NS5B antibodies. We also thank Steven Podos for help in preparing the manuscript.
This study was supported by Achillion Pharmaceuticals and Gilead Sciences.
Footnotes
Published ahead of print 29 April 2013
REFERENCES
- 1. Pottage JC, Lawitz E, Mazur D, Wyles D, Vargas H, Ghalib R, Gugliotti R, Donohue M, Robison H. 2007. Short-term antiviral activity and safety of ACH-806 (GS-9132), an NS4A antagonist, in HCV genotype 1-infected individuals. J. Hepatol. 46(Suppl 1):A783 [Google Scholar]
- 2. Wasley A, Alter MJ. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis. 20:1–16 [DOI] [PubMed] [Google Scholar]
- 3. Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567.4 [DOI] [PubMed] [Google Scholar]
- 4. Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52 [DOI] [PubMed] [Google Scholar]
- 5. Seeff LB. 1997. Natural history of hepatitis C. Hepatology 26:21S–28S [DOI] [PubMed] [Google Scholar]
- 6. Zeuzem S. 2008. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat. Clin. Pract Gastroenterol. Hepatol. 5:610–622 [DOI] [PubMed] [Google Scholar]
- 7. Feld JJ, Hoofnagle JH. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972 [DOI] [PubMed] [Google Scholar]
- 8. Pawlotsky JM. 2012. New antiviral agents for hepatitis C. F1000 Biol. Rep. 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pearlman BL. 2012. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect. Dis. 12:717–728 [DOI] [PubMed] [Google Scholar]
- 10. Lemon SM, McKeating JA, Pietschmann T, Frick DN, Glenn JS, Tellinghuisen TL, Symons J, Furman PA. 2010. Development of novel therapies for hepatitis C. Antivir. Res. 86:79–92 [DOI] [PubMed] [Google Scholar]
- 11. Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 12. Bartenschlager R, Lohmann V, Wilkinson T, Koch JO. 1995. Complex formation between the NS3 serine-type proteinase of the hepatitis C virus and NS4A and its importance for polyprotein maturation. J. Virol. 69:7519–7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Failla C, Tomei L, De Francesco R. 1995. An amino-terminal domain of the hepatitis C virus NS3 protease is essential for interaction with NS4A. J. Virol. 69:1769–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin C, Thomson JA, Rice CM. 1995. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J. Virol. 69:4373–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, Gomez J. 1992. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 66:3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107 [DOI] [PubMed] [Google Scholar]
- 17. Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 18. Fridell RA, Wang C, Sun JH, O'Boyle DR, 2nd, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935 [DOI] [PubMed] [Google Scholar]
- 19. Yang W, Zhao Y, Fabrycki J, Hou X, Nie X, Sanchez A, Phadke A, Deshpande M, Agarwal A, Huang M. 2008. Selection of replicon variants resistant to ACH-806, a novel hepatitis C virus inhibitor with no cross-resistance to NS3 protease and NS5B polymerase inhibitors. Antimicrob. Agents Chemother. 52:2043–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Asabe SI, Tanji Y, Satoh S, Kaneko T, Kimura K, Shimotohno K. 1997. The N-terminal region of hepatitis C virus-encoded NS5A is important for NS4A-dependent phosphorylation. J. Virol. 71:790–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dimitrova M, Imbert I, Kieny MP, Schuster C. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 77:5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Florese RH, Nagano-Fujii M, Iwanaga Y, Hidajat R, Hotta H. 2002. Inhibition of protein synthesis by the nonstructural proteins NS4A and NS4B of hepatitis C virus. Virus Res. 90:119–131 [DOI] [PubMed] [Google Scholar]
- 23. Kato J, Kato N, Yoshida H, Ono-Nita SK, Shiratori Y, Omata M. 2002. Hepatitis C virus NS4A and NS4B proteins suppress translation in vivo. J. Med. Virol. 66:187–199 [DOI] [PubMed] [Google Scholar]
- 24. Koch JO, Bartenschlager R. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J. Virol. 73:7138–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kou YH, Chou SM, Wang YM, Chang YT, Huang SY, Jung MY, Huang YH, Chen MR, Chang MF, Chang SC. 2006. Hepatitis C virus NS4A inhibits cap-dependent and the viral IRES-mediated translation through interacting with eukaryotic elongation factor 1A. J. Biomed. Sci. 13:861–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin C, Wu JW, Hsiao K, Su MS. 1997. The hepatitis C virus NS4A protein: interactions with the NS4B and NS5A proteins. J. Virol. 71:6465–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindenbach BD, Pragai BM, Montserret R, Beran RK, Pyle AM, Penin F, Rice CM. 2007. The C terminus of hepatitis C virus NS4A encodes an electrostatic switch that regulates NS5A hyperphosphorylation and viral replication. J. Virol. 20:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nomura-Takigawa Y, Nagano-Fujii M, Deng L, Kitazawa S, Ishido S, Sada K, Hotta H. 2006. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondrion-mediated apoptosis. J. Gen. Virol. 87:1935–1945 [DOI] [PubMed] [Google Scholar]
- 29. Tanji Y, Hijikata M, Satoh S, Kaneko T, Shimotohno K. 1995. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J. Virol. 69:1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishido S, Fujita T, Hotta H. 1998. Complex formation of NS5B with NS3 and NS4A proteins of hepatitis C virus. Biochem. Biophys. Res. Commun. 244:35–40 [DOI] [PubMed] [Google Scholar]
- 31. Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O'Malley ET, Harbeson SL, Rice CM, Murcko MA, Caron PR, Thomson JA. 1996. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87:343–355 [DOI] [PubMed] [Google Scholar]
- 32. Yan Y, Li Y, Munshi S, Sardana V, Cole JL, Sardana M, Steinkuehler C, Tomei L, De Francesco R, Kuo LC, Chen Z. 1998. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: a 2.2 Å resolution structure in a hexagonal crystal form. Protein Sci. 7:837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Hostomska Z. 1996. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 87:331–342 [DOI] [PubMed] [Google Scholar]
- 34. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 35. Yang W, Huang M. 2009. Studying HCV RNA synthesis in vitro with replication complexes. Methods Mol. Biol. 510:177–184 [DOI] [PubMed] [Google Scholar]
- 36. Yang W, Sun Y, Phadke A, Deshpande M, Huang M. 2007. Hepatitis C virus (HCV) NS5B nonnucleoside inhibitors specifically block single-stranded viral RNA synthesis catalyzed by HCV replication complexes in vitro. Antimicrob. Agents Chemother. 51:338–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolk B, Sansonno D, Krausslich HG, Dammacco F, Rice CM, Blum HE, Moradpour D. 2000. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461 [DOI] [PubMed] [Google Scholar]
- 41. Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. U. S. A. 105:14545–14550 [DOI] [PMC free article] [PubMed] [Google Scholar]