

Abstract
Pseudomonas aeruginosa is a leading cause of hospital-acquired infections and is resistant to many antibiotics. Among its primary mechanisms of resistance is expression of a chromosomally encoded AmpC β-lactamase that inactivates β-lactams. The mechanisms leading to AmpC expression in P. aeruginosa remain incompletely understood but are intricately linked to cell wall metabolism. To better understand the roles of peptidoglycan-active enzymes in AmpC expression—and consequent β-lactam resistance—a phenotypic screen of P. aeruginosa mutants lacking such enzymes was performed. Mutants lacking one of four lytic transglycosylases (LTs) or the nonessential penicillin-binding protein PBP4 (dacB) had altered β-lactam resistance. mltF and slt mutants with reduced β-lactam resistance were designated WIMPs (wall-impaired mutant phenotypes), while highly resistant dacB, sltB1, and mltB mutants were designated HARMs (high-level AmpC resistant mutants). Double mutants lacking dacB and sltB1 had extreme piperacillin resistance (>256 μg/ml) compared to either of the single knockouts (64 μg/ml for a dacB mutant and 12 μg/ml for an sltB1 mutant). Inactivation of ampC reverted these mutants to wild-type susceptibility, confirming that AmpC expression underlies resistance. dacB mutants had constitutively elevated AmpC expression, but the LT mutants had wild-type levels of AmpC in the absence of antibiotic exposure. These data suggest that there are at least two different pathways leading to AmpC expression in P. aeruginosa and that their simultaneous activation leads to extreme β-lactam resistance.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen, notable for its intrinsic antibiotic resistance. P. aeruginosa causes 10 to 15% of hospital-acquired infections worldwide (1), infecting the immunocompromised, burn victims, and cystic fibrosis patients. The emergence of antibiotic resistance in P. aeruginosa (and other Gram-negative bacteria) is of increasing concern as available treatment options become exhausted (2). A clearer understanding of the mechanisms leading to antibiotic resistance is necessary to identify avenues for remediation and treatment (3).
One of the primary mechanisms leading to β-lactam resistance in P. aeruginosa is the inducible expression of a chromosomally encoded AmpC β-lactamase. AmpC degrades many β-lactam antibiotics, which otherwise inhibit enzymes responsible for synthesis and maintenance of the peptidoglycan (PG) layer (4), a mesh-like polymer that protects the cell from high internal turgor pressures and provides cell shape. It is composed of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) sugars that are cross-linked through stem peptides attached to MurNAc residues (5).
Synthesis and turnover of PG require the concerted activities of penicillin-binding proteins (PBPs), N-acetylmuramoyl-l-alanine amidases, endopeptidases, carboxypeptidases, and lytic transglycosylases (LTs) (6). High-molecular-weight PBPs have transglycosylase and transpeptidase activities, responsible for linking GlcNAc and MurNAc sugars and cross-linking stem peptides, respectively. Low-molecular-weight PBPs have endopeptidase and carboxypeptidase activities and control the extent of cross-linking (5). Cell growth and division require insertion of new material at sites created when LTs cleave the β-1,4 glycosidic linkage between GlcNAc and MurNAc residues, releasing anhydromuropeptides (anhMPs) (7). Under normal conditions, free anhMPs enter the cytoplasm through the inner membrane permease, AmpG, and are efficiently processed by the β-N-acetylglucosaminidase, NagZ, and the N-acetylmuramoyl-l-alanine amidase, AmpD, for recycling back into the PG biosynthesis pathway. Perturbation of PG turnover—by either mutation or β-lactam treatment—causes increased cytoplasmic accumulation of anhMPs that bind to the AmpC transcriptional regulator, AmpR, inducing AmpC expression (8).
Recently, mutation of the nonessential dacB gene encoding the d,d-carboxypeptidase PBP4 was found to elicit AmpC expression in the absence of β-lactams (9). dacB mutations were identified in β-lactam-resistant clinical isolates of P. aeruginosa, suggesting that loss of PBP4 function is a medically relevant resistance mechanism (10). In Aeromonas spp., induction of AmpC expression requires a specific anhMP, anhydromuropentapeptide (11). These observations suggest that loss of specific enzyme activities, rather than general inhibition of PG turnover, leads to AmpC induction. However, whether loss of PBP4 activity is the only trigger is not yet clear.
In this study, we screened P. aeruginosa strains deficient in one or more PG-active enzymes to identify factors that contribute to β-lactam resistance. We confirmed that loss of PBP4—but not other PBPs—increases β-lactam resistance and AmpC activity, even in the absence of antibiotic induction. We further showed that the loss of either of two LTs—SltB1 or MltB—increases resistance, although unlike in PBP4 mutants, basal AmpC activity in sltB1 or mltB mutants remains at wild-type levels. The results show that PG-active enzymes with seemingly redundant activities have unique effects on β-lactam resistance.
MATERIALS AND METHODS
Bacterial strains, plasmids, and mutants.
Bacterial strains and plasmids used in this study are listed in Table 1. P. aeruginosa knockouts were created using an Flp-FRT recombination system as previously described (12). To create dacB, sltB1, and ampC knockouts, Escherichia coli SM10 strains carrying the pEX18Ap-dacB::GmFRT, sltB1::GmFRT, or ampC::GmFRT disruption construct were incubated with P. aeruginosa strain PAO1, and mating mixtures were plated on Pseudomonas isolation agar (PIA) containing 100 μg/ml gentamicin (Gm) to counterselect the donor. Gm-resistant PAO1 colonies were plated on modified Luria-Bertani agar (LBA) containing no salt, 5% sucrose, and 100 μg/ml Gm to select for double recombinants that lost the sacB-expressing pEX18Ap suicide vector. Recombinants were screened for carbenicillin (Cb) susceptibility and Gm resistance. For excision of the Gm resistance cassette, mutants were transformed with pFLP2 and selected on LBA containing 200 μg/ml Cb. The pFLP2 plasmid was then cured on LBA containing 5% sucrose, without NaCl. PCR using gene-specific primers was used to confirm gene disruption.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| PAO1 | P. aeruginosa PAO1 WTa | 41 |
| PAO1 ampC::FRT | WT with FRT scar at nucleotide 1070 of ampC (PA4110) | This study |
| PAO1 dacB::FRT | WT with FRT scar at nucleotide 168 of dacB (PBP4, PA3047) | 16 |
| PAO1 sltB1::FRT | WT with FRT scar at nucleotide 577 of sltB1 (PA4001) | This study |
| PAO1 mgtA::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 495 of mgtA (PA0378) | 41 |
| PAO1 pbpG::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 282 of pbpG (PBP7, PA0869) | 41 |
| PAO1 sltG::Tn | WT with transposon ISphoA/hah insertion at nucleotide 227 of sltG (PA1171) | 41 |
| PAO1 mltA::Tn | WT with transposon ISphoA/hah insertion at nucleotide 397 of mltA (PA1222) | 41 |
| PAO1 mltD::Tn | WT with transposon ISphoA/hah insertion at nucleotide 868 of mltD (PA1812) | 41 |
| PAO1 pbpC::Tn | WT with transposon ISphoA/hah insertion at nucleotide 1226 of pbpC (PBP3a, PA2272) | 41 |
| PAO1 mltF2::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 991 of mltF2 (PA2865) | 41 |
| PAO1 slt::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 1256 of slt (PA3020) | 41 |
| PAO1 mltF::Tn | WT with transposon ISphoA/hah insertion at nucleotide 730 of mltF (PA3764) | 41 |
| PAO1 sltH::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 415 of sltH (PA3992) | 41 |
| PAO1 dacC::Tn | WT with transposon ISlacZ/hah insertion at nucleotide 549 of dacC (PBP6, PA3999) | 41 |
| PAO1 mltB::Tn | WT with transposon ISphoA/hah insertion at nucleotide 358 of mltB (PA4444) | 41 |
| PAO1 ponA::Tn | WT with transposon ISphoA/hah insertion at nucleotide 1573 of ponA (PBP1a, PA5045) | 41 |
| PAO1 mrcB::Tn | WT with transposon ISphoA/hah insertion at nucleotide 973 of mrcB (PBP1b, PA4700) | 41 |
| PAO1 sltB1::FRT dacB::FRT | sltB1::FRT mutant with FRT scar at nucleotide 168 of dacB (PBP4) | This study |
| PAO1 mltB::Tn dacB::FRT | mltB::Tn mutant with FRT scar at nucleotide 168 of dacB (PBP4) | This study |
| PAO1 mltB::Tn sltB1::FRT | mltB::Tn mutant with FRT scar at nucleotide 577 of sltB1 | This study |
| PAO1 ampC::FRT dacB::FRT | ampC::FRT with FRT scar at nucleotide 168 of dacB (PBP4) | This study |
| PAO1 sltB1::FRT ampC::FRT | sltB1::FRT mutant with FRT scar at nucleotide 1070 of ampC | This study |
| PAO1 mltB::Tn ampC::FRT | mltB::Tn mutant with FRT scar at nucleotide 1070 of ampC | This study |
| PAO1 pBADGr-dacB | WT with pBADGr derivative containing dacB | This study |
| PAO1 pBADGr-sltB1 | WT with pBADGr derivative containing sltB1 | This study |
| PAO1 dacB::FRT + pBADGr-dacB | dacB::FRT mutant with pBADGr derivative containing dacB | This study |
| PAO1 sltB1::FRT + pBADGr-sltB1 | sltB1::FRT mutant with pBADGr derivative containing sltB1 | This study |
| PAO1 sltB1::FRT dacB::FRT + pBADGr-dacB | sltB1::FRT dacB::FRT mutant with pBADGr derivative containing dacB | This study |
| PAO1 mltB::Tn dacB::FRT + pBADGr-dacB | mltB::Tn dacB::FRT mutant with pBADGr derivative containing dacB | This study |
| PAO1 sltB1::FRT dacB::FRT + pBADGr-sltB1 | sltB1::FRT dacB::FRT mutant with pBADGr derivative containing sltB1 | This study |
| PAO1 mltB::Tn/sltB1::FRT + pBADGr-sltB1 | mltB::Tn sltB1::FRT mutant with pBADGr derivative containing sltB1 | This study |
| Plasmids | ||
| pPS856 | Source of FRT-flanked gentamicin cassette; Gmr | 12 |
| pEX18Ap | Suicide vector used for gene replacement; Apr | 12 |
| pFLP2 | Suicide vector encoding Flp recombinase; Apr | 12 |
| pBADGr | pMLBAD backbone with dhfr replaced with aacC1; Gmr | 42 |
| pEX18Ap-dacB::FRTGmFRT | Suicide vector containing dacB disrupted at position 168 with FRT-flanked gentamicin resistance cassette; Apr Gmr | 16 |
| pEX18Ap-sltB1::FRTGmFRT | Suicide vector containing sltB1 disrupted at position 577 with FRT-flanked gentamicin resistance cassette; Apr Gmr | This study |
| pEX18Ap-ampC::FRTGmFRT | Suicide vector containing ampC disrupted at position 1070 with FRT-flanked gentamicin resistance cassette; Apr Gmr | This study |
| pBADGr-dacB | pBADGr derivative containing dacB on an EcoRI fragment; Gmr | This study |
| pBADGr-sltB1 | pBADGr derivative containing sltB1 on an EcoRI fragment; Gmr | This study |
WT, wild type.
For complementation, E. coli SM10 strains carrying pBADGr-dacB, pBADGr-sltB1, and pBADGr-ampC were conjugated with the P. aeruginosa strain of interest and mating mixtures were plated on PIA containing 100 μg/ml Gm to counterselect the donor.
Antibiotic susceptibility assays.
Antibiotic susceptibility assays were performed using Etest strips (bioMérieux, France). Overnight bacterial cultures were subcultured 1:50 in 5 ml Mueller-Hinton broth (MHB; Becton, Dickinson and Company, Mississauga, Ontario, Canada) and grown to logarithmic phase at 37°C, with shaking at 200 rpm. Cultures were standardized to an optical density at 600 nm (OD600) of 0.25 in MHB, and 100 μl was spread on Mueller-Hinton agar (MHA). Etest strips were overlaid and plates were incubated for 18 h at 37°C. MICs were determined as the concentration at which the zone of inhibition intersected the Etest strip. MICs were confirmed by three independent replicates, and differences 2-fold or greater in MICs compared to the control were considered significant.
AmpC β-lactamase activity assay.
AmpC activity was assessed using nitrocefin hydrolysis. Overnight bacterial cultures were subcultured 1:20 in 5 ml MHB and grown for 2 h at 37°C and 200 rpm. Cultures were split 1:1 in 5 ml MHB with or without 50 μg/ml cefoxitin (final concentration) and incubated for an additional 2 h at 37°C and 200 rpm. Following incubation, 1 ml culture was pelleted at 2,300 × g for 5 min, washed once with 1 ml 50 mM sodium phosphate (pH 7.0), and resuspended in 1 ml of the same buffer. Samples were placed on ice and then lysed by sonication with a microprobe (Misonix Sonicator 2000; Misonix Inc., Farmingdale, NY) using three 10-s pulses on power setting two with 10-s rests between pulses. Samples were centrifuged at 12,000 × g at 4°C for 5 min, and supernatants were collected. Supernatant protein concentration was determined using a Bradford assay (13) with bovine serum albumin as the standard (Bio-Rad Laboratories Ltd., Mississauga, Ontario, Canada). Nitrocefin hydrolysis assays were performed in 250-μl reaction mixtures of 50 mM sodium phosphate (pH 7.0) containing 10 μg total protein and 20 μg/ml nitrocefin (VWR International, Mississauga, Ontario, Canada). Nitrocefin hydrolysis was measured every min for 15 min at room temperature by absorbance at 486 nm. AmpC activity was calculated using a nitrocefin extinction coefficient of 20,500 M−1 cm−1. AmpC β-lactamase activity assays were independently performed three times, and statistical significance was assessed using a two-tailed Student t test. A P value of ≤0.05 was considered statistically significant.
RESULTS
Loss of PBP4, SltB1, or MltB increases β-lactam resistance, while loss of Slt or MltF decreases β-lactam resistance.
An antibiotic susceptibility screen of P. aeruginosa mutants lacking individual PG-active enzymes (see Table S1 in the supplemental material) revealed five strains with altered resistance profiles to the β-lactams piperacillin, cefotaxime, and ceftazidime but no changes in susceptibility to the carbapenem imipenem, the fluoroquinolone ciprofloxacin, or the aminoglycoside tobramycin (Table 2). Strains lacking low-molecular-weight PBP4 (dacB) or the LTs SltB1 and MltB had elevated MICs for β-lactam antibiotics (Table 2). Similar to a previously reported dacB mutant with 32-fold and ∼21-fold increases in resistance to piperacillin and ceftazidime, respectively (9), our dacB mutant had a 16-fold increase in resistance to piperacillin and an ∼21-fold increase in resistance to ceftazidime and cefotaxime compared to the wild type. The sltB1 and mltB mutants had 3- and 2-fold increases in piperacillin MICs, respectively, while the mltB mutant had in addition a 2-fold increase in ceftazidime resistance compared to the wild type (Table 2).
Table 2.
MICs for β-lactam, fluoroquinolone, and aminoglycoside antibiotics of various Pseudomonas aeruginosa strains
| Strain name or relevant genotype | MIC (μg/ml)a |
|||||
|---|---|---|---|---|---|---|
| PP | CT | TZ | IP | CI | TM | |
| PAO1 | 4 | 12 | 0.75 | 1 | 0.125 | 1.5 |
| dacB | 64 | >256 | 16 | 1 | 0.125 | 1 |
| sltB1 | 12 | 16 | 1 | 0.75 | 0.125 | 1.5 |
| mltB | 8 | 12 | 1.5 | 1.5 | 0.19 | 1 |
| slt | 1.5 | 6 | 0.19 | 1 | 0.125 | 1 |
| mltF | 2 | 6 | 0.38 | 1 | 0.125 | 1 |
| ampC | 3 | 4 | 1 | 0.25 | 0.5 | 1 |
| dacB ampC | 1 | 3 | 0.5 | 0.38 | 0.5 | 1 |
| sltB1 ampC | 1.5 | 2 | 0.5 | 0.25 | 0.75 | 1 |
| mltB ampC | 6 | 8 | 1 | 0.38 | 0.19 | 1 |
| sltB1 dacB | >256 | >256 | 24 | 1 | 0.125 | 1 |
| mltB dacB | 192 | >256 | 16 | 1.5 | 0.19 | 1 |
| mltB sltB1 | 24 | 64 | 2 | 1 | 0.125 | 1.5 |
| PAO1 + pBADGr-dacB | 3 | 4 | 0.75 | 1.5 | NA | NA |
| PAO1 + pBADGr-sltB1 | 3 | 4 | 0.75 | 1.5 | NA | NA |
| dacB + pBADGr-dacB | 3 | 4 | 0.75 | 1.5 | NA | NA |
| sltB1 + pBADGr-sltB1 | 3 | 4 | 0.75 | 1 | NA | NA |
| sltB1 dacB + pBADGr-dacB | 6 | 12 | 0.75 | 1.5 | NA | NA |
| mltB dacB + pBADGr-dacB | 4 | 8 | 0.75 | 1.5 | NA | NA |
| sltB1 dacB + pBADGr-sltB1 | 32 | 256 | 3 | 2 | NA | NA |
| mltB sltB1 + pBADGr-sltB1 | 6 | 8 | 0.75 | 1.5 | NA | NA |
Abbreviations: PP, piperacillin; CT, cefotaxime; TZ, ceftazidime; IP, imipenem; CI, ciprofloxacin; TM, tobramycin; NA, not applicable.
Strains lacking the LTs Slt or MltF had reduced resistance to β-lactam antibiotics (Table 2). These strains had ∼3- and 2-fold decreases in piperacillin MICs, respectively, compared to the wild type and 4- and 2-fold decreases in ceftazidime resistance, respectively. Cefotaxime resistance in both strains was decreased 2-fold compared to the wild type.
Growth curves showed no differences between the wild type and mutants over a 24-h period (see Fig. S1 in the supplemental material), ruling out growth rate-related changes in susceptibility. Since the loss of LTs has been reported to compromise membrane integrity (14), membrane permeability was assessed using the hydrophobic fluorescent probe 8-anilino-1-naphthylenesulfonic acid (ANS) (15, 16). ANS fluorescence of the mutant strains was similar to that of the wild-type control (data not shown), suggesting that the LT mutations did not affect permeability.
Increased β-lactam resistance is AmpC dependent.
To assess the role of AmpC in resistance of the selected mutants, ampC was disrupted in the dacB, sltB1, and mltB mutant backgrounds. β-Lactam susceptibility returned to wild-type levels in all three strains (Table 2). Resistance to imipenem was decreased ∼2.5-fold for all mutants lacking ampC, while susceptibilities to ciprofloxacin and tobramycin remained unchanged (Table 2).
To assess AmpC activity in mutant strains, nitrocefin hydrolysis assays were performed. Basal levels of AmpC activity were upregulated ∼50-fold in the dacB strain compared to the wild type, while those of the sltB1, mltB, slt, or mltF mutant strains were similar to that of the wild type (Fig. 1A). Induction by cefoxitin increased the AmpC activities of all strains, with an ∼60-fold increase in the wild-type strain compared to uninduced levels (Fig. 1B). The AmpC activities of sltB1, mltB, slt, or mltF mutant strains were also increased upon cefoxitin induction, but not to levels that were significantly different than those of the wild type. Upon cefoxitin induction, AmpC activity in the dacB strain increased ∼2.3-fold compared with uninduced levels and were ∼2-fold higher than the induced wild type (Fig. 1B).
Fig 1.
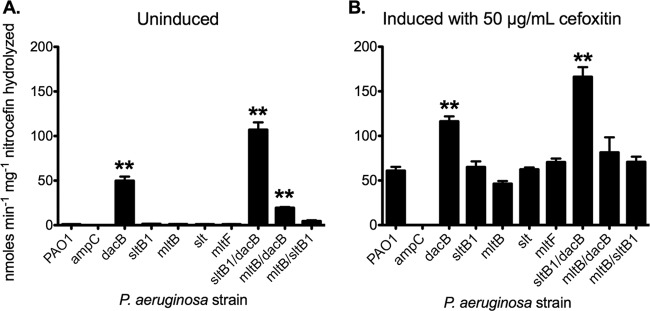
AmpC activity is increased in dacB mutants but not in LT mutants. Nitrocefin hydrolysis activity assays of P. aeruginosa mutants grown under basal conditions (A) and induced conditions (medium supplemented with 50 μg/ml cefoxitin) (B) were conducted. Only dacB mutants have increased basal AmpC activity, while the loss of LTs alone does not alter activity compared to that of the wild type. AmpC activity was measured in nmol min−1 mg−1 nitrocefin hydrolyzed. n = 3. Bars represent the means ± standard errors. **, P < 0.01.
β-Lactam resistance is additive in sltB1 dacB, mltB dacB, and mltB sltB1 double mutants.
The effect of combining mutations on resistance to β-lactams was determined using sltB1 dacB, mltB dacB, and mltB sltB1 double mutants. All had increased β-lactam resistance compared to the single mutants (Table 2). The MIC of the sltB1 dacB mutant for piperacillin was >256 μg/ml, versus 64 μg/ml for the dacB mutant, a >2.5-fold increase. The mltB dacB and mltB sltB1 mutants had 2-fold (to 192 μg/ml) and 1.5-fold (to 24 μg/ml) increases in piperacillin MICs, respectively, compared to the single mutants (Table 2). Resistance to ceftazidime also increased 1.5-fold (to 24 μg/ml) for the sltB1 dacB mutant. Cefotaxime resistance was already >256 μg/ml for the dacB mutant (the highest level that can be measured using the Etest assay) and was unchanged for the sltB1 dacB and mltB dacB double mutants. Resistance to cefotaxime increased >5-fold (to 64 μg/ml) for the mltB sltB1 mutant. There were no differences in susceptibility to imipenem, ciprofloxacin, or tobramycin for any of the double mutants (Table 2).
The sltB1 dacB strain had basal levels of AmpC activity >2-fold higher than that of the dacB mutant (P = 0.008), while the mltB sltB1 double mutant had AmpC activity ∼4.5-fold higher than either of its single-mutation parent strains (P = 0.034) (Fig. 1A). Interestingly, the mltB dacB double mutant had basal AmpC activity ∼20-fold above that of the mltB mutant but ∼2.5-fold below that of the dacB mutant (Fig. 1A). Upon cefoxitin induction, the sltB1 dacB double mutant had the highest level of AmpC activity, significantly higher than that of the dacB mutant (∼1.4-fold; P = 0.015) (Fig. 1B). The cefoxitin-induced AmpC activity of the mltB sltB1 double mutant was significantly higher than that of the mltB1 mutant (P = 0.022) but not significantly higher than that of the sltB1 mutant. Induced AmpC activities in the mltB dacB double mutant were not significantly different than in either of the single mutants (Fig. 1B).
dacB and sltB1 mutants were complemented with the cognate genes using the low-copy number, arabinose-inducible pBADGr plasmid (17). Initial experiments were performed using low-level expression from the leaky promoter in the absence of induction (17), to avoid the previously reported toxic effects of sltB1 overexpression (18). Complementation in trans restored β-lactam MICs to—and in several cases, below—that of the parent strain (Table 2). Therefore, multicopy expression of dacB or sltB1 in trans can increase susceptibility to β-lactams. Expression of the same constructs in the wild-type strain over a range of arabinose concentrations did not significantly change the MICs beyond that of the 0% arabinose control (data not shown).
SltB1 was previously reported to interact with PBP2 (19, 20); therefore, we asked whether increased β-lactam resistance in the sltB1 deletion mutant was due to loss of its LT activity or the potential disruption of a PBP2-containing complex due to absence of the SltB1 protein. To address the second possibility, the sltB1 gene was replaced at its chromosomal locus (to ensure native expression levels) with mutant versions expressing one of two putative active-site mutants, one replacing the proposed catalytic Glu135 (21) with Ala and the other replacing Ser189, responsible for orientation of PG in the active site (22), with Ala. Neither point mutation restored β-lactam susceptibility, suggesting that it is the loss of SltB1 activity, rather than disruption of PBP2 activity due to loss of the SltB1 protein, that underlies the resistant phenotype.
DISCUSSION
Peptidoglycan (PG) maintenance, turnover, and recycling are dynamic processes relying on the coordinated activities of several enzymes (6). Since PG degradation products can trigger AmpC induction and subsequent β-lactam resistance (8), we assessed whether the loss of PG-active enzymes altered antibiotic resistance in P. aeruginosa. We identified five strains with altered β-lactam resistance profiles. Three of these strains had increased resistance, and we termed them high-level AmpC resistant mutants (HARMs). HARMs lack PBP4 or the family 3 lytic transglycosylases (LTs) SltB1 and MltB. We also made the novel observation that loss of the family 1 LTs, Slt and MltF, increased β-lactam susceptibility compared with that of the wild type, and we termed those strains wall-impaired mutant phenotypes (WIMPs). Loss of other LTs (MltA, MltD, MltF2, SltG, and SltH; see Table S1 in the supplemental material) had no effect on resistance. To our knowledge, this is the first report that the loss of specific LTs alters antibiotic resistance in P. aeruginosa.
A recent study (23) of a P. aeruginosa strain PA14 transposon mutant library identified several genes whose disruption caused modest (∼2-fold) changes in ceftazidime MICs, and it was hypothesized that such changes could cause stepwise increases in resistance when combined. Such a phenomenon was previously described for combined mutations in the ampD genes (ampD, ampDh2, and ampDh3) in P. aeruginosa (24). Among the genes identified in the PA14 transposon mutant screen were those whose products are involved in cell wall and lipopolysaccharide biosynthesis; however, no LT genes were identified (23). In agreement with previous work (9) and our data, mutations in dacB—but importantly, not other PBP genes—increased resistance to ceftazidime in that study (23). Therefore, loss of particular sentinel protein activities and/or accumulation of certain PG metabolites—rather than a general perturbation of PG metabolism—triggers AmpC expression in this species. Although the activities of some PBPs and LTs appear to be redundant due to their nonessentiality (7, 18), our data suggest that they play distinct roles. Along these lines, studies with Salmonella showed that 2 of 9 LTs—MltC and MltE—modulated biofilm matrix formation (14), while the others had no effect. Interestingly, MltC and MltE are family 1 LTs, as are Slt and MltF from P. aeruginosa (14, 25).
Since LTs are responsible for generating the PG degradation products that are proposed to lead to AmpC induction, it may seem paradoxical that their absence can increase β-lactam resistance without inducing AmpC activity. LTs contribute to cell lysis following β-lactam exposure because their PG-lytic activities continue in the absence of the synthetic activities of PBPs, leading to loss of cell wall integrity. Thus, the loss of specific LTs—such as SltB1 and MltB in P. aeruginosa—may be protective when cells are treated with β-lactam antibiotics. In E. coli, the loss of LytM-containing PG hydrolases (26) or the combined loss of N-acetylmuramoyl-l-alanine amidases and LTs (27) delayed β-lactam-induced cell death. Similarly, pneumococci resistant to deoxycholate-induced cell lysis due to deficiencies in autolytic activity exhibited delayed β-lactam-induced cell death (28, 29). However, to our knowledge, this is the first report that the loss of a single LT causes such a phenotype.
In contrast, loss of Slt or MltF increased susceptibility to piperacillin and cephalosporin antibiotics, which does not appear to be due to a reduction in AmpC-inducing PG fragments, since these mutants had wild-type AmpC activities under basal and induced conditions (Fig. 1). We dubbed such mutants WIMPs (wall-impaired mutant phenotypes). Why does loss of these enzymes not increase resistance to β-lactams as was seen for SltB1 or MltB? Different LTs—with seemingly redundant activities—might be expressed at different times of the cell cycle, at different locations, or at different levels. It is also possible that Slt and MltF are members of protein complexes whose disruption alters antibiotic susceptibility. These data raise the question of whether loss of specific enzymes causes compensatory changes in the expression or activities of other LTs. While the loss of multiple hydrolases delayed β-lactam-mediated cell death in E. coli (26, 27), the loss of Slt70, MltA, or MltB reduced murein turnover, prevented AmpC induction, and sensitized cells to cefoxitin (30). Similarly, the combined loss of Slt and MltA to MltE in E. coli reduced β-lactam resistance by abolishing AmpC induction (31). These data support the idea that PG-active enzymes with similar activities can have specific and nonredundant physiological roles.
Slt and MltF are family 1 LTs, while SltB1 and MltB are family 3 LTs (25). Family designations for LTs are based on sequence motifs surrounding the catalytic regions; however, crystallographic studies with E. coli (32–34) and P. aeruginosa (20) showed that the catalytic domains of family 1 and family 3 LTs are structurally conserved despite their sequence dissimilarity, with a lysozyme fold and an essential catalytic Glu residue (35). One notable difference is the presence of an EF hand-type motif in the core domain of family 3 LTs. In SltB1, this motif contains a calcium-binding loop required for interaction with PBP2 and possibly other protein partners (20). Family 1 LTs have roles in conjugation and secretion systems, while family 3 LTs are involved in assembly of flagella and pili and in sporulation (36). How the loss of LTs from different families affects β-lactam resistance, however, has not previously been reported.
Loss of the predicted P. aeruginosa LTs MltA, MltD, MltF2, SltG, and SltH had no effect on antibiotic resistance, implying either some degree of redundancy between LTs or a lack of a phenotype under laboratory conditions. Loss of the PBP3 homolog, PBP3x, in P. aeruginosa caused no changes in cell morphology or growth rate under normal laboratory conditions; however, expression of PBP3x was limited to the stationary phase (37). It is likely that some LTs could have silent phenotypes under normal laboratory conditions. No functional or expression data for P. aeruginosa MltA, MltD, MltF2, SltG, or SltH are yet available.
PG-active enzymes are spatially and temporally regulated, and disruption of specific enzymatic functions at a particular stage of the cell cycle may result in abnormal PG metabolism. Furthermore, PG-active enzymes can be organized into multienzyme complexes (19, 20, 38), and loss of one component might destabilize those complexes, affecting PG metabolism. SltB1 interacts with PBP2 (19, 20) but could have additional partners (20). Chromosomal expression of putative active-site mutants of SltB1 did not restore susceptibility, suggesting that increased β-lactam resistance in sltB1 mutants is due to the loss of SltB1 function, rather than disruption of protein interactions. Loss of PBP2 was previously reported to decrease P. aeruginosa β-lactam resistance (39), further suggesting that the phenotype of the sltB1 mutant is not indirectly due to loss of PBP2 activity.
Interestingly, loss of SltB1 conferred higher piperacillin and cephalosporin resistance than did loss of MltB, although these enzymes are proposed to produce the same PG degradation products (18). MltB binds PG with higher affinity than SltB1, but SltB1 mediates a higher rate of PG turnover (18). Thus, loss of sltB1 may more effectively limit PG degradation and protect against β-lactam challenge.
Increased β-lactam resistance in mltB and sltB1 strains is AmpC mediated (Table 2); however, our data suggest at least two different pathways to induction. AmpC activity in the dacB strain was significantly increased upon disruption of sltB1; however, AmpC activity in the sltB1 single mutant was similar to that in the wild type under both basal and induced conditions (Fig. 1B). Although AmpC expression in P. aeruginosa is controlled by AmpR, recent studies identified a role for the BlrAB (β-lactam resistance) two-component system (TCS)—formerly annotated as CreBC—in resistance (9, 40). Both β-lactam challenge and loss of PBP4 activate the Blr TCS; however, its exact role in β-lactam resistance is not yet known. Deletion of BlrAB was reported to restore β-lactam susceptibility in the PBP4 mutant, without affecting AmpC expression (9). Although BlrAB does not directly control AmpC production, it has an ancillary role in β-lactam resistance (11). Similarly, loss of SltB1 activity may be sensed through BlrAB (or another regulatory system), modulating β-lactam resistance without changing AmpC expression or activity.
A comprehensive understanding of the roles of PG-active enzymes in β-lactam resistance in P. aeruginosa is necessary to develop successful therapies. In addition to independently confirming that dacB mutations induce AmpC expression, we showed that simultaneous loss of specific LTs further increases resistance in the dacB background. More information on the specific roles of LTs, their spatial and temporal expression patterns, and their interaction partners is needed. LTs have been proposed as targets for the development of novel antibiotics (21, 36). However, specificity of inhibitors will be important, as depending on the enzymes targeted, LT inactivation could lead to increased β-lactam resistance.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Canada-UK Partnership on Antibiotic Resistance award 114045 from the Canadian Institutes of Health Research and by an award from the Natural Sciences and Engineering Research Council of Canada (NSERC). R.P.L. is the recipient of the Michael G. DeGroote Fellowship in Basic Biomedical Science and a postdoctoral fellowship from Cystic Fibrosis Canada. A.L.M. was the recipient of two NSERC undergraduate summer research awards.
Footnotes
Published ahead of print 22 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00268-13.
REFERENCES
- 1. Blanc DS, Francioli P, Zanetti G. 2007. Molecular epidemiology of Pseudomonas aeruginosa in the intensive care units—a review. Open Microbiol. J. 1:8–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giamarellou H, Poulakou G. 2009. Multidrug-resistant Gram-negative infections: what are the treatment options? Drugs 69:1879–1901 [DOI] [PubMed] [Google Scholar]
- 3. Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74:417–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mark BL, Vocadlo DJ, Oliver A. 2011. Providing beta-lactams a helping hand: targeting the AmpC beta-lactamase induction pathway. Future Microbiol. 6:1415–1427 [DOI] [PubMed] [Google Scholar]
- 5. Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32:259–286 [DOI] [PubMed] [Google Scholar]
- 6. Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell-wall recycling. Ann. N. Y. Acad. Sci. 1277:54–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee M, Hesek D, Llarrull LI, Lastochkin E, Pi H, Boggess B, Mobashery S. 2013. Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. J. Am. Chem. Soc. 135:3311–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J. 13:4684–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L, Haussler S, Oliver A. 2009. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353. 10.1371/journal.ppat.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moyá B, Beceiro A, Cabot G, Juan C, Zamorano L, Alberti S, Oliver A. 2012. Pan-beta-lactam resistance development in Pseudomonas aeruginosa clinical strains: molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 56:4771–4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. 2010. Induction of beta-lactamase production in Aeromonas hydrophila is responsive to beta-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335 [DOI] [PubMed] [Google Scholar]
- 12. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 13. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 14. Monteiro C, Fang X, Ahmad I, Gomelsky M, Romling U. 2011. Regulation of biofilm components in Salmonella enterica serovar Typhimurium by lytic transglycosylases involved in cell wall turnover. J. Bacteriol. 193:6443–6451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loh B, Grant C, Hancock RE. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamers RP, Cavallari JF, Burrows LL. 2013. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAbetaN) permeabilizes the outer membrane of Gram-negative bacteria. PLoS One 8:e60666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giltner CL, Habash M, Burrows LL. 2010. Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J. Mol. Biol. 398:444–461 [DOI] [PubMed] [Google Scholar]
- 18. Blackburn NT, Clarke AJ. 2002. Characterization of soluble and membrane-bound family 3 lytic transglycosylases from Pseudomonas aeruginosa. Biochemistry 41:1001–1013 [DOI] [PubMed] [Google Scholar]
- 19. Legaree BA, Clarke AJ. 2008. Interaction of penicillin-binding protein 2 with soluble lytic transglycosylase B1 in Pseudomonas aeruginosa. J. Bacteriol. 190:6922–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikolaidis I, Izore T, Job V, Thielens N, Breukink E, Dessen A. 2012. Calcium-dependent complex formation between PBP2 and lytic transglycosylase SltB1 of Pseudomonas aeruginosa. Microb. Drug Resist. 18:298–305 [DOI] [PubMed] [Google Scholar]
- 21. Reid CW, Blackburn NT, Clarke AJ. 2006. Role of arginine residues in the active site of the membrane-bound lytic transglycosylase B from Pseudomonas aeruginosa. Biochemistry 45:2129–2138 [DOI] [PubMed] [Google Scholar]
- 22. Reid CW, Legaree BA, Clarke AJ. 2007. Role of Ser216 in the mechanism of action of membrane-bound lytic transglycosylase B: further evidence for substrate-assisted catalysis. FEBS Lett. 581:4988–4992 [DOI] [PubMed] [Google Scholar]
- 23. Alvarez-Ortega C, Wiegand I, Olivares J, Hancock RE, Martinez JL. 2010. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob. Agents Chemother. 54:4159–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Juan C, Moya B, Perez JL, Oliver A. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob. Agents Chemother. 50:1780–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blackburn NT, Clarke AJ. 2001. Identification of four families of peptidoglycan lytic transglycosylases. J. Mol. Evol. 52:78–84 [DOI] [PubMed] [Google Scholar]
- 26. Uehara T, Dinh T, Bernhardt TG. 2009. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J. Bacteriol. 191:5094–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heidrich C, Ursinus A, Berger J, Schwarz H, Holtje JV. 2002. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J. Bacteriol. 184:6093–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomasz A, Albino A, Zanati E. 1970. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227:138–140 [DOI] [PubMed] [Google Scholar]
- 29. Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kraft AR, Prabhu J, Ursinus A, Holtje JV. 1999. Interference with murein turnover has no effect on growth but reduces beta-lactamase induction in Escherichia coli. J. Bacteriol. 181:7192–7198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korsak D, Liebscher S, Vollmer W. 2005. Susceptibility to antibiotics and beta-lactamase induction in murein hydrolase mutants of Escherichia coli. Antimicrob. Agents Chemother. 49:1404–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Asselt EJ, Dijkstra AJ, Kalk KH, Takacs B, Keck W, Dijkstra BW. 1999. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme-like catalytic domain with an EF-hand. Structure 7:1167–1180 [DOI] [PubMed] [Google Scholar]
- 33. van Asselt EJ, Thunnissen AM, Dijkstra BW. 1999. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J. Mol. Biol. 291:877–898 [DOI] [PubMed] [Google Scholar]
- 34. Artola-Recolons C, Carrasco-Lopez C, Llarrull LI, Kumarasiri M, Lastochkin E, Martinez de Ilarduya I, Meindl K, Uson I, Mobashery S, Hermoso JA. 2011. High-resolution crystal structure of MltE, an outer membrane-anchored endolytic peptidoglycan lytic transglycosylase from Escherichia coli. Biochemistry 50:2384–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Asselt EJ, Kalk KH, Dijkstra BW. 2000. Crystallographic studies of the interactions of Escherichia coli lytic transglycosylase Slt35 with peptidoglycan. Biochemistry 39:1924–1934 [DOI] [PubMed] [Google Scholar]
- 36. Scheurwater E, Reid CW, Clarke AJ. 2008. Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 40:586–591 [DOI] [PubMed] [Google Scholar]
- 37. Liao X, Hancock RE. 1997. Identification of a penicillin-binding protein 3 homolog, PBP3x, in Pseudomonas aeruginosa: gene cloning and growth phase-dependent expression. J. Bacteriol. 179:1490–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Heijenoort J. 2011. Peptidoglycan hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev. 75:636–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Legaree BA, Daniels K, Weadge JT, Cockburn D, Clarke AJ. 2007. Function of penicillin-binding protein 2 in viability and morphology of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 59:411–424 [DOI] [PubMed] [Google Scholar]
- 40. Balasubramanian D, Schneper L, Merighi M, Smith R, Narasimhan G, Lory S, Mathee K. 2012. The regulatory repertoire of Pseudomonas aeruginosa AmpC ss-lactamase regulator AmpR includes virulence genes. PLoS One 7:e34067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Asikyan ML, Kus JV, Burrows LL. 2008. Novel proteins that modulate type IV pilus retraction dynamics in Pseudomonas aeruginosa. J. Bacteriol. 190:7022–7034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.