

Abstract
The widespread emergence of antifungal drug resistance poses a severe clinical problem. Though predicted to play a role in this phenomenon, the drug:H+ antiporters (DHA) of the major facilitator superfamily have largely escaped characterization in pathogenic yeasts. This work describes the first DHA from the pathogenic yeast Candida glabrata reported to be involved in antifungal drug resistance, the C. glabrata QDR2 (CgQDR2) gene (ORF CAGL0G08624g). The expression of CgQDR2 in C. glabrata was found to confer resistance to the antifungal drugs miconazole, tioconazole, clotrimazole, and ketoconazole. By use of a green fluorescent protein (GFP) fusion, the CgQdr2 protein was found to be targeted to the plasma membrane in C. glabrata. In agreement with these observations, CgQDR2 expression was found to decrease the intracellular accumulation of radiolabeled clotrimazole in C. glabrata and to play a role in the extrusion of this antifungal from preloaded cells. Interestingly, the functional heterologous expression of CgQDR2 in the model yeast Saccharomyces cerevisiae further confirmed the role of this gene as a multidrug resistance determinant: its expression was able to complement the susceptibility phenotype exhibited by its S. cerevisiae homologue, QDR2, in the presence of imidazoles and of the antimalarial and antiarrhythmic drug quinidine. In contrast to the findings reported for Qdr2, CgQdr2 expression does not contribute to the ability of yeast to grow under K+-limiting conditions. Interestingly, CgQDR2 transcript levels were seen to be upregulated in C. glabrata cells challenged with clotrimazole or quinidine. This upregulation was found to depend directly on the transcription factor CgPdr1, the major regulator of multidrug resistance in this pathogenic yeast, which has also been found to be a determinant of quinidine and clotrimazole resistance in C. glabrata.
INTRODUCTION
Multidrug resistance has emerged in most organisms and poses a severe clinical problem for the treatment of tumors and infectious diseases, having reached alarming proportions in recent years. Systemic fungal infections are a problem of increasing clinical significance for immunocompromised patients. Infections caused by Candida species are recognized as the fourth or fifth most common cause of nosocomial infections; Candida glabrata infections rank second in frequency, after those caused by Candida albicans (1). The extensive use of antifungal drugs both as treatment and as prophylaxis has led to a huge increase in the number of intrinsically resistant infections with fungal pathogens. The frequency of these infections and their relatively high fatality rates (up to 45% for Candida glabrata) are generally attributed to the capacity of these pathogenic yeasts to efficiently develop multiple drug resistance (MDR) (2, 3).
In pathogenic yeasts, as in many other organisms (4), clinical manifestation of antifungal drug resistance has been linked mostly to the activation of multidrug efflux pumps of the ATP-binding cassette (ABC) transporter superfamily and the major facilitator superfamily (MFS). Both ABC and MFS multidrug transporters are proposed to actively extrude or compartmentalize drugs and other xenobiotics, thus providing protection from these compounds (5–10). In Candida albicans clinical isolates, overexpression of the ABC multidrug transporters encoded by the CDR1 (11) and CDR2 (12) genes, which share a high degree of homology with Saccharomyces cerevisiae PDR5 (ScPDR5), has been found to confer resistance to azoles. In Candida glabrata, antifungal drug resistance has been shown to rely on the ABC drug efflux pumps C. glabrata CDR1 (CgCDR1) (another ScPDR5 homologue), CgPDH1/CgCDR2 (a ScPDR15 homologue), and CgSNQ2 (a ScSNQ2 homologue) (13, 14).
The role of the yeast multidrug resistance transporters of the MFS, however, has received much less attention in the context of antifungal drug resistance. For S. cerevisiae, only after the release of the genome sequence (15) did this family of 23 predicted drug:H+ antiporters (DHA), of which 22 were completely unknown at the time, receive full attention. During the past 15 years, the majority of these S. cerevisiae transporters have been characterized as multidrug resistance transporters (9). Still, very little is known to date about this family of transporters in pathogenic yeasts. The C. albicans drug:H+ antiporter Mdr1, a close homologue of the S. cerevisiae Flr1 gene, is the single member of this family that has been linked to the clinical manifestation of azole drug resistance (16). Besides Mdr1, only Flu1, from C. albicans, and CgFlr1, from C. glabrata, have been characterized; they have been found to confer fluconazole (17) and benomyl (18) resistance, respectively. More recently, a strong association between the expression of the DHA Mdr1 from Candida dubliniensis and fluconazole resistance was also reported (19). In C. glabrata, there are 15 predicted DHA, of which 10 belong to the DHA1 family, predicted to have 12 transmembrane spans, and 5 belong to the DHA2 family, predicted to have 14 transmembrane segments (20). It is expected that these transporters, which remain uncharacterized so far, may play a significant role in multidrug resistance in C. glabrata, with important implications for the development of intrinsic or acquired drug resistance in clinical isolates.
In this study, functional analysis of the C. glabrata open reading frame (ORF) CAGL0G08624g (the CgQDR2 gene) was undertaken, with emphasis on its role in antifungal drug resistance. This ORF encodes a putative drug:H+ antiporter that belongs to the MFS and shares a high degree of homology with the Saccharomyces cerevisiae QDR2 gene (20). S. cerevisiae QDR2 (21–23) has been found to confer resistance to the antimalarial/antiarrhythmic drug quinidine, the antifungal drug ketoconazole, and the herbicide barban. In addition, QDR2 has been implicated in the resistance of yeast to the anticancer drugs cisplatin and bleomycin (21) and to toxic concentrations of the polyamines spermine, spermidine, and putrescine (24). It has also been found to play a role in the maintenance of intracellular K+ concentrations and to become crucial for the survival of yeast cells in the presence of limiting K+ concentrations, either in K+-depleted growth medium or under quinidine stress (22). Surprisingly, the levels of QDR2 transcripts have been found to remain constant when yeast is exposed to the chemical stresses to which QDR2 confers resistance; its regulation is apparently independent of the pleiotropic drug resistance (PDR) network in S. cerevisiae. In this study, the role and regulation of the CgQDR2 gene in antifungal drug resistance were examined, and CgQdr2 was found to confer resistance to the antifungal drugs clotrimazole, tioconazole, miconazole, and ketoconazole. The subcellular localization of this transporter was assessed, and its action in the extrusion of [3H]clotrimazole from preloaded Candida glabrata cells was verified. Finally, the transcriptional regulation of CgQDR2 under chemical stress was examined, and its expression was seen to be upregulated under quinidine- and clotrimazole-induced stress in a manner dependent on the transcription factor CgPdr1, the major regulator of multidrug resistance in this yeast.
MATERIALS AND METHODS
Strains, plasmids, and growth media.
Saccharomyces cerevisiae strain BY4741 (MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0) and its single deletion mutant BY4741_Δqdr2 were obtained from the Euroscarf collection. Candida glabrata strain CBS138, whose genome sequence was released in 2004, and KUE100 (25) were used in this study. Other C. glabrata strains, including L5U1 (cgura3Δ0 cgleu2Δ0), kindly provided by John Bennett (18) of the National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, and 66032 and 66032_Δcgpdr1, kindly provided by Thomas Edlind (26) of the Department of Microbiology and Immunology, Drexel University, College of Medicine, Philadelphia, PA, were also used. Plasmid pGREG576 was obtained from the Drag&Drop collection (27).
Cells were batch-cultured at 30°C, with orbital agitation (250 rpm), in YPD growth medium, composed of 20 g/liter glucose (Merck), 20 g/liter yeast extract (Difco), and 10 g/liter peptone (Difco). For some of the experiments, minimal medium, obtained by supplementation of basal medium (BM) [1.7 g/liter yeast nitrogen base without amino acids or NH4+ (Difco), 20 g/liter glucose (Merck), and 2.65 g/liter (NH4)2SO4 (Merck)] with different amino acids, was used. Wild-type S. cerevisiae BY4741 and strains derived from it were grown in MM4 medium, obtained by supplementation of BM with 20 mg/liter methionine, 20 mg/liter histidine, 60 mg/liter leucine, and 20 mg/liter uracil (all from Sigma). C. glabrata strains derived from CBS138, KUE100, and 66032 or L5U1 were cultured in unsupplemented BM or in BM supplemented with 20 mg/liter uracil and 60 mg/liter leucine (uracil only for L5U1, and uracil and leucine for 66032). Ammonium phosphate-derived KNA medium was also used to test BY4741 or KUE100 and the derived Δqdr2 or Δcgqdr2 deletion mutants under conditions of K+ limitation. KNA basal medium contains, per liter, a mixture of 0.492 g MgSO4·7H2O (Merck), 0.02 g CaCl2 (anhydrous) (Panreac), 1.056 g (NH4)2HPO4 (Merck), 3.96 g (NH4)2SO4, 20 g glucose, 2 mg niacin, 2 mg pyridoxine, 2 mg thiamine, 2 mg pantothenate, 0.02 mg biotin, and the desired concentration of KCl (all from Sigma). This medium was supplemented with 20 mg/liter methionine, 20 mg/liter histidine, 60 mg/liter leucine, and 20 mg/liter uracil (all from Sigma). To maintain selective pressure over the recombinant strains, uracil was added to this medium only to grow the host yeast cells. Solid media contained 20 g/liter agar (Iberagar) besides the ingredients listed above.
Cloning of the CgQDR2 gene (ORF CAGL0G08624g).
The pGREG576 plasmid from the Drag&Drop collection (27) was used to clone and express the C. glabrata ORF CAGL0G08624g in S. cerevisiae, as described previously for other heterologous genes (28). pGREG576 was acquired from Euroscarf and contains a galactose-inducible promoter (GAL1), the yeast selectable marker URA3, and the GFP gene, encoding a green fluorescent protein (GFPS65T), which allows monitoring of the expression and subcellular localization of the cloned fusion protein. CAGL0G08624g DNA was generated by PCR using genomic DNA extracted from the sequenced C. glabrata strain CBS138 and the following specific primers: 5′-GAATTCGATATCAAGCTTATCGATACCGTCGACAATGATGGAGGATCAGCAATCACTG-3′ and 5′-GCGTGACATAACTAATTACATGACTCGAGGTCGACTTATCTGCTTTCTTCGGC-3′. The designed primers contain, besides a region with homology to the first 24 and last 18 nucleotides of the CAGL0G08624g coding region (italicized), nucleotide sequences with homology to the cloning-site flanking regions of the pGREG576 vector (underlined). The amplified fragment was cotransformed into the parental S. cerevisiae strain BY4741 with the pGREG576 vector, previously cut with the restriction enzyme SalI, to obtain the pGREG576_CgQDR2 plasmid. Since the GAL1 promoter allows only slight expression of downstream genes in C. glabrata, a new construct was produced in order to visualize the subcellular localization of the CgQDR2 gene in C. glabrata by fluorescence microscopy. The GAL1 promoter present in the pGREG576_CgQDR2 plasmid was replaced by the copper-induced MTII C. glabrata promoter, giving rise to the pGREG576_MTII_CgQDR2 plasmid. The MTII promoter DNA was generated by PCR using genomic DNA extracted from the sequenced C. glabrata strain CBS138 and the following specific primers: 5′-TTAACCCTCACTAAAGGGAACAAAAGCTGGAGCTCGATCCGAGAAGTCATCCCT-3′ and 5′-GAAAAGTTCTTCTCCTTTACTCATACTAGTGCGGCAGGCATTTCGAAGCTATAA-3′. The designed primers contain, besides a region with homology to the first and last 19 nucleotides of the first 500 bp of the MTII promoter region (italicized), nucleotide sequences with homology to the cloning-site flanking regions of the pGREG576 vector (underlined). The amplified fragment was cotransformed into the parental strain BY4741 with the pGREG576_CgQDR2 plasmid, previously cut with the SacI and NotI restriction enzymes to remove the GAL1 promoter, to generate the pGREG576_MTII_CgQDR2 plasmid. Recombinant plasmids pGREG576_CgQDR2 and pGREG576_MTII_CgQDR2 were obtained through homologous recombination in S. cerevisiae and were verified by DNA sequencing.
Disruption of the CgQDR2 gene (ORF CAGL0G08624g).
The deletion of the CgQDR2 gene was carried out in the parental strain KUE100 by using the method described by Ueno et al. (29). The target gene CAGL0G08624g (CgQDR2) was replaced by a DNA cassette including the CgHIS3 gene through homologous recombination. The replacement cassettes were prepared by PCR using the following primers: 3′-CGTGATCAGCGGCCATGGCGCGAATATCAAAAGCAAATAATCTACTGAAACTCCCTATGATCTTTCAATA-5′and 3′-CCAGCCTCACGATGTAAATGGCTGGATGATGAGTTTATTTGACTTGTTCAAATTGTTTTTTTGTCAAAAG-5′. The pHIS906 plasmid including CgHIS3 was used as a template, and transformation was performed as described previously (25). The 3′ sequences of the primers, CCAGCCTCACGATG and CGTGATCAGCGGCC, were attached to the flanking regions of CgHIS3. The underlined 56-bp sequences were the flanking sequences of each gene. The recombination locus was verified by PCR using the following pairs of primers: 3′-GTTTTGTGGTTGCGGAAGCA-5′, which is downstream of CgQDR2, and 3′-AGAAAACCAGCCTCACGATG-5′ (ptet12F), which is specific for the cassette DNA. Appropriate PCR products were identified. To verify the gene deletion, PCR was performed using the following pairs of primers, which are assigned to the inside of the open reading frame of each gene: 3′-CAGTGATTGCTGATCCTCC-5′ and 3′-CTCGGGTGTATGCTTGGTC-5′. No PCR product was identified from the template DNA of the mutant, while clear PCR products were identified from the template DNA of the parental strain KUE100.
Assessment of the subcellular localization of CgQdr2.
The subcellular localization of the CgQdr2 protein was determined based on the observation of S. cerevisiae BY4741 or C. glabrata L5U1 cells transformed with the pGREG576-CgQDR2 or pGREG576-MTII-CgQDR2 plasmid, respectively. These cells express the CgQdr2_GFP fusion protein, whose localization may be determined using fluorescence microscopy. S. cerevisiae cell suspensions were prepared by cultivation in MM4-U medium, containing 0.5% glucose and 0.1% galactose, at 30°C, with orbital shaking (250 rpm), until a standard culture optical density at 600 nm (OD600) of 0.4 ± 0.04 was reached. At this point, cells were transferred to the same medium containing 0.1% glucose and 1% galactose to induce protein expression. C. glabrata cell suspensions were prepared in BM-U medium until a standard culture OD600 of 0.4 ± 0.04 was reached; they were then transferred to the same medium supplemented with 30 μM CuSO4 (Sigma), to induce protein overexpression. After 5 h of incubation, the distribution of the CgQdr2_GFP fusion protein in S. cerevisiae or in C. glabrata living cells was detected by fluorescence microscopy with a Zeiss Axioplan microscope (Carl Zeiss Microimaging) using excitation and emission wavelengths of 395 and 509 nm, respectively. Fluorescence images were captured using a cooled charge-coupled device (CCD) camera (CoolSnap fx; Roper Scientific Photometrics).
Drug susceptibility assays of S. cerevisiae.
The susceptibilities of the parental strain BY4741 to toxic concentrations of the selected drugs were compared to those of the deletion mutant BY4741_Δqdr2 by spot assays. The ability of CgQDR2 gene expression to increase wild-type resistance to the chemical stresses tested and to complement the susceptibility phenotype exhibited by the BY4741_Δqdr2 single deletion mutant was also examined, by using the pGREG576_CgQDR2 centromeric plasmid, in which CgQDR2 is expressed under the control of the GAL1 promoter.
For the S. cerevisiae cell suspensions used to inoculate the agar plates, mid-exponential-phase cells were first grown in basal MM4-U medium, containing 0.5% glucose and 0.1% galactose, until a culture OD600 of 0.4 ± 0.02 was reached and then diluted in sterile water to obtain suspensions with an OD600 of 0.05 ± 0.005. These cell suspensions and subsequent dilutions (1:5, 1:25) were applied as 4-μl spots to the surface of solid MM4-U medium, containing 0.1% glucose and 1% galactose, supplemented with growth-inhibitory concentrations of chemicals. The drugs and other xenobiotics tested included the following compounds, used in the specified concentration ranges: the azole antifungal drugs fluconazole (10 to 200 mg/liter), ketoconazole (10 to 50 mg/liter), clotrimazole (1 to 20 mg/liter), tioconazole (0.05 to 0.2 mg/liter), and miconazole (0.05 to 0.2 mg/liter), the polyene antifungal drug amphotericin B (0.05 to 0.5 mg/liter), the fluoropyrimidine 5-flucytosine (0.02 to 5 mg/liter), and the antimalarial/antiarrhythmic drug quinidine (3 to 5 mg/ml) (all from Sigma).
Drug susceptibility assays of C. glabrata.
The susceptibilities of the parental strain KUE100 to toxic concentrations of the selected drugs were compared to those of the deletion mutant KUE100_Δcgqdr2 by spot assays. Similarly, the susceptibilities of the parental strain 66032 to quinidine and clotrimazole were compared to those of the 66032_Δcgpdr1 deletion mutant. The ability of CgQDR2 gene expression to increase wild-type resistance to the chemical stresses tested was also examined with the URA3− strain L5U1 by using the pGREG576_CgQDR2 centromeric plasmid.
For the C. glabrata cell suspensions used to inoculate the agar plates, mid-exponential-phase cells of strain L5U1 harboring plasmids derived from pGREG576 were first grown in BM, containing 0.5% glucose and 0.1% galactose, without uracil until a culture OD600 of 0.4 ± 0.02 was reached and then diluted in sterile water to obtain suspensions with an OD600 of 0.05 ± 0.005. These cell suspensions and subsequent dilutions (1:5, 1:25) were applied as 4-μl spots to the surface of solid BM without uracil, containing 0.1% glucose and 1% galactose and supplemented with adequate concentrations of chemical stressors. The drugs tested included the following compounds, used in the specified concentration ranges: the azole antifungal drugs fluconazole (10 to 200 mg/liter), itraconazole (20 to 100 mg/liter), ketoconazole (10 to 50 mg/liter), clotrimazole (1 to 20 mg/liter), tioconazole (0.2 to 1 mg/liter), and miconazole (0.2 to 1 mg/liter), the polyene antifungal drug amphotericin B (0.1 to 0.5 mg/liter), the fluoropyrimidine 5-flucytosine (0.02 to 5 mg/liter), and the antimalarial/antiarrhythmic drug quinidine (3 to 9 mg/ml) (all from Sigma).
Clotrimazole MICs were determined in accordance with NCCLS (now CLSI) document M27-A2 (30) and European Committee on Antimicrobial Susceptibility Testing (EUCAST) Discussion Document E.Dis 7.1 (31) by using RPMI 1640 medium (Sigma). The antifungal concentrations ranged from 0.016 to 60 mg/liter.
[3H]clotrimazole transport assays.
[3H]clotrimazole transport assays were carried out as described previously (23). To estimate the accumulation of clotrimazole (intracellular/extracellular [3H]clotrimazole) from yeast cells, the parental strain KUE100 and the mutant strain KUE100_Δcgqdr2, or the parental strain L5U1 harboring plasmid pGREG576 or pGREG576_MTII_CgQDR2, were grown in BM to the mid-exponential phase and were harvested by filtration. Cells were washed and were resuspended in BM to obtain dense cell suspensions (OD600, 5.0 ± 0.2, equivalent to approximately 2.2 mg [dry weight] ml−1). After 5 min of incubation at 30°C with agitation (150 rpm), 0.1 μM [3H]clotrimazole (1 mCi/ml; American Radiolabeled Chemicals) and 30 mg/liter of unlabeled clotrimazole were added to the cell suspensions. Incubation proceeded for an additional 30 min. Alternatively, for [3H]clotrimazole efflux assays, yeast cells were resuspended in TM buffer (0.1 M morpholineethanesulfonic acid [MES; Sigma], 41 mM Tris [Sigma] adjusted to pH 4.5 with HCl) without glucose and were incubated for 30 min at 30°C with agitation (150 rpm) to deenergize the cell population. Then 0.1 μM [3H]clotrimazole was added to the cell suspensions, and the accumulation of [3H]clotrimazole in deenergized yeast cells was followed until equilibrium was reached, after 30 min. Afterwards, a pulse of 2% glucose was added to the cell suspension, and the efflux of [3H]clotrimazole, in the absence or presence of the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma), was followed for an additional 30 min until a new equilibrium was reached. In all cases, the intracellular accumulation of labeled clotrimazole was followed by filtering 200 μl of cell suspension, at adequate time intervals, through prewetted glass microfiber filters (GF/C; Whatman). The filters were washed with ice-cold TM buffer, and radioactivity was measured in a Beckman LS-5000 TD scintillation counter. The amount of extracellular [3H]clotrimazole was estimated by radioactivity assessment of 50 μl of the supernatant.
Nonspecific [3H]clotrimazole adsorption to the filters (less than 5% of the total radioactivity) was assessed and was subtracted from all the intracellular concentration readings. To calculate the intracellular concentration of labeled clotrimazole, the internal cell volume (Vi) of the exponential-phase cells, grown in the absence of drug and used for accumulation assays, was considered constant and equal to 2.5 μl (mg [dry weight])−1 (32).
Measurements of CgQDR2 expression.
The levels of CgQDR2 transcripts in C. glabrata 66032 and 66032_Δcgpdr1 cells were assessed by quantitative real-time PCR. Total-RNA samples were obtained from cell suspensions harvested under control conditions (mid-exponential-phase cells in the absence of drugs) or upon 1 h of exposure to 6 mg/ml quinidine or 30 mg/liter clotrimazole. cDNA for real-time reverse transcription-PCR (RT-PCR) experiments was synthesized from total-RNA samples by using the MultiScribe reverse transcriptase kit (Applied Biosystems) and the 7500 RT-PCR thermal cycler block (Applied Biosystems) according to the manufacturer's instructions. The quantity of cDNA for subsequent reactions was kept at ca. 10 ng. The subsequent RT-PCR step was carried out using SYBR Green reagents (Applied Biosystems). Primers for the amplification of CgQDR2 and CgACT1 cDNA were designed using Primer Express software (Applied Biosystems) and are 5′-TCACTGCATAGTTTCATATCGGACTA-3′ and 5′-CAACTTCAGATAGATCAGGACCATCA-3′ for CgQDR2 and 5′-AGAGCCGTCTTCCCTTCCAT-3′ and 5′-TTGACCCATACCGACCATGA-3′ for CgACT1. The RT-PCR was carried out using a thermal cycler block (7500 real-time PCR system; Applied Biosystems). Default parameters established by the manufacturer were used, and fluorescence was detected by the instrument and recorded in an amplification plot (7500 System SDS software; Applied Biosystems). The CgACT1 mRNA level was used as an internal control. The relative values obtained for the wild-type strain under control conditions were set at 1, and the remaining values are presented relative to that control. To avoid false-positive signals, the absence of nonspecific amplification with the chosen primers was confirmed by the generation of a dissociation curve for each pair of primers.
The levels of CgQDR2 expression dependent on putative transcription factor CgPdr1 binding sites were assessed in L5U1 cells harboring the pCgQDR2_lacZ fusion plasmid. This fusion plasmid was obtained as described previously (33), by using the pYEP354 expression vector and a PCR amplification fragment overlapping the CgQDR2 promoter region, the translation initiation codon, and a short portion of the coding region of CgQDR2, from bp −998 to +10. This PCR fragment was generated using the following primers: 5′-CGGAATTCGTGTCAAGAATAGTACTGTG-3′ and 5′-CGCGGATCCCCTCCATCATATGGCGCG-3′. Using site-directed mutagenesis, plasmids pCgQDR2_ΔPDRE1_lacZ and pCgQDR2_ΔPDRE2_lacZ, exhibiting point mutations in the PDRE1 and PDRE2 loci, respectively, were produced. The pairs of mutagenic oligonucleotides used for this procedure were 5′-CTTATACTCCTACGCCAAAATTACATTGTCACAGCTC-3′ and its complementary sequence and 5′-GACTTATGATTTACTCAGCCTTTAGATACCTCT-3′ and its complementary sequence, respectively. The underlined nucleotides were used to replace the core regions of the PDRE1 and PDRE2 elements, GCCATCATT and GCCGATAGA, respectively, found to occur in the CgQDR2 promoter region. The levels of CgQDR2 expression from its natural promoter were compared to those induced by point-mutated versions of the CgQDR2 promoter, based on the β-galactosidase (β-Gal) activity of CgQDR2 promoter-lacZ fusion plasmids pCgQDR2_lacZ, pCgQDR2_ΔPDRE1_lacZ, and pCgQDR2_ΔPDRE2_lacZ. L5U1 cells were transformed with these plasmids, and β-Gal activity was determined during cultivation in the presence or absence of 30 mg/liter clotrimazole. β-Gal assays were based on the method of Miller, as described previously (33), and β-Gal units were defined as (increase in A420)/(minimum OD600) × 1,000.
RESULTS
CgQdr2 is localized to the plasma membrane in C. glabrata.
C. glabrata cells harboring the pGREG576_MTII_CgQDR2 plasmid were grown to the mid-exponential phase in minimal medium and were then transferred to the same medium containing 30 μM CuSO4 to promote protein overexpression. At a standard OD600 of 0.5 ± 0.05, obtained after around 5 h of incubation, cells were inspected by fluorescence microscopy. In C. glabrata cells, the CgQdr2_GFP fusion was found to be localized predominantly to the cell periphery (Fig. 1A). Control cells harboring the pGREG576 cloning vector, on the other hand, displayed a slight and uniform distribution of fluorescence (Fig. 1A), similar to what can be observed as the host cells autofluoresce. Since CgQdr2 is predicted to be an integral membrane protein (20), these results strongly suggest a plasma membrane localization, similar to what was observed for its S. cerevisiae homologue Qdr2 (23).
Fig 1.
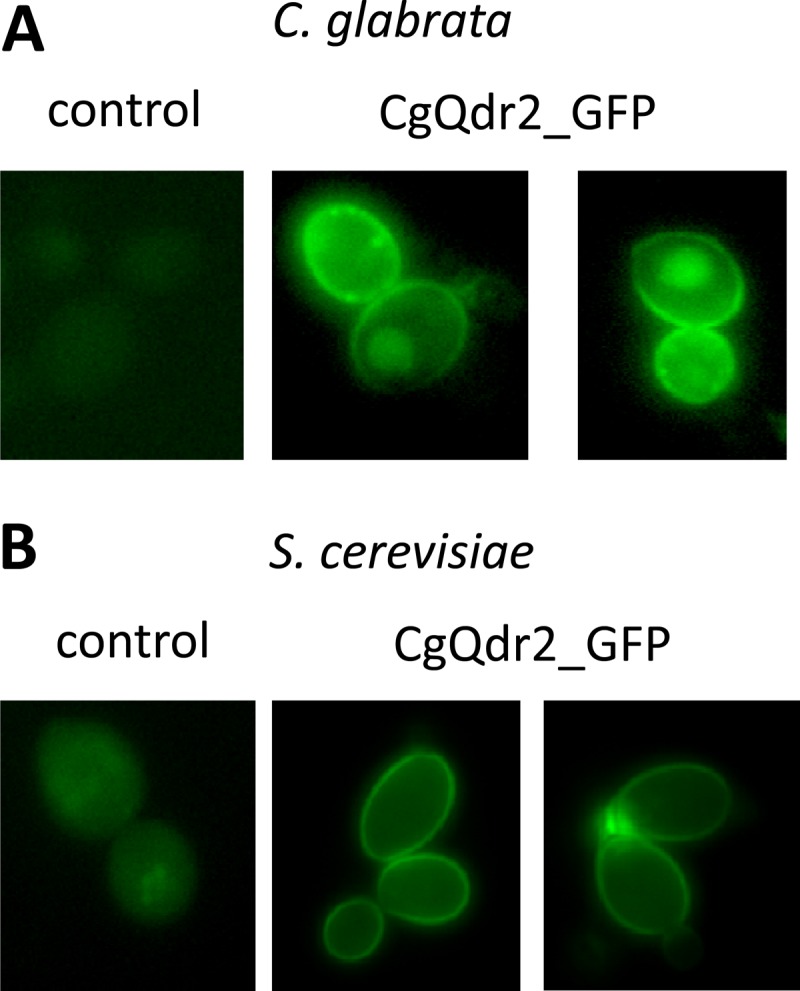
Fluorescence of exponential-phase C. glabrata L5U1 (A) and S. cerevisiae BY4741 (B) cells harboring either the cloning vector pGREG576 (control) or the pGREG576_MTII_CgQDR2 or pGREG576_CgQDR2 plasmid (CgQdr2_GFP) after 5 h of copper- or galactose-induced recombinant protein production, respectively. The results indicate that the CgQdr2-GFP fusion protein localizes to the plasma membranes of both S. cerevisiae and C. glabrata cells.
CgQDR2 expression confers resistance to azoles and quinidine, similarly to its S. cerevisiae counterpart.
Based on spot assays, the deletion of the CgQDR2 gene in Candida glabrata KUE100 was found to increase the susceptibility of this pathogen not only to the imidazole antifungal drugs miconazole, clotrimazole, and ketoconazole but also to quinidine (Fig. 2A). Given that the subsequent studies were focused on clotrimazole, the corresponding MIC50 was determined, according to the standard NCCLS method, for strains KUE100 (0.25 mg/liter) and KUE100_Δcgqdr2 (0.125 mg/liter), reinforcing the identification of CgQdr2 as a clotrimazole resistance determinant. No effect of CgQDR2 deletion on fluconazole or itraconazole resistance could be detected (Fig. 2A). In a different C. glabrata strain, L5U1, bearing auxotrophy toward uracil, the introduction of a recombinant plasmid expressing CgQDR2 increased the natural resistance toward, at least, miconazole, clotrimazole, ketoconazole, and quinidine, over that of the same strain harboring the corresponding cloning vector (Fig. 2B), reinforcing the finding that CgQDR2 is a determinant of azole drug resistance in C. glabrata.
Fig 2.

(A) Comparison by spot assays of the susceptibilities of C. glabrata strains KUE100 and KUE100_Δcgqdr2 to miconazole, clotrimazole, ketoconazole, fluconazole, itraconazole, and quinidine, at the concentrations indicated, in BM agar plates. (B) Comparison by spot assays of the susceptibilities of C. glabrata strain L5U1, harboring the pGREG576 cloning vector (v) or the pGREG576_CgQDR2 plasmid, to miconazole, clotrimazole, ketoconazole, and quinidine, at the indicated concentrations, in BM agar plates. The inocula were prepared as described in Materials and Methods. The cell suspensions used to prepare the spots were 1:5 (b) and 1:25 (c) dilutions of the cell suspension used for column a. The images displayed are representative of at least three independent experiments.
By use of S. cerevisiae as a heterologous expression system, the effect of CgQDR2 expression on the resistance of yeasts to antifungal drugs and other xenobiotics was further tested, in order to determine whether or not CgQDR2 is able to functionally complement its S. cerevisiae homologue. S. cerevisiae cells harboring the pGREG576_CgQDR2 plasmid were tested for the subcellular localization of CgQdr2 in order to verify that in these cells, the C. glabrata transporter was still localized to the plasma membrane. Cells were grown to the mid-exponential phase in minimal medium containing 0.5% glucose and 0.1% galactose and were then transferred to the same medium containing 0.1% glucose and 1% galactose to promote protein overexpression. At a standard OD600 of 0.5 ± 0.05, obtained after ca. 5 h of incubation, cells were inspected by fluorescence microscopy, and plasma membrane localization was verified (Fig. 1B). Based on spot assays, CgQDR2 was found to confer resistance to imidazole antifungals, particularly miconazole and tioconazole, as well as to quinidine, on S. cerevisiae (Fig. 3A). No role in this yeast's resistance to triazoles, such as fluconazole, to polyenes, including amphotericin B, or to the fluoropyrimidine drug flucytosine could be detected either for the S. cerevisiae QDR2 gene or for CgQDR2. Significantly, CgQdr2 expression appears to complement, at least partially, the susceptibility phenotype exhibited upon the deletion of its S. cerevisiae homologue Qdr2 (Fig. 3). Based on analysis of the growth curves of the same populations in liquid medium in the presence of the selected antifungals, it is possible to see that deletion of the S. cerevisiae QDR2 gene dramatically increases the susceptibility of this yeast to miconazole, tioconazole, and, as observed previously (23), quinidine, increasing the length of the lag phase induced upon drug exposure. The expression of CgQdr2 partially complements the susceptibilities of the Δqdr2 mutant strain to the three drugs tested and increases the resistance of the wild-type strain to an otherwise toxic concentration of (at least) tioconazole (Fig. 3B).
Fig 3.

(A) Comparison by spot assays of the susceptibilities of S. cerevisiae strains BY4741 and BY4741_Δqdr2, harboring the cloning vector pGREG576 (v) or the CgQDR2 expression plasmid pGREG576_CgQDR2, to miconazole, tioconazole, and quinidine, at the indicated concentrations, in MM4 agar plates. The cell suspensions used to prepare the spots were 1:5 (b) and 1:25 (c) dilutions of the cell suspension used for column a. (B) Comparison of the susceptibilities of S. cerevisiae strains BY4741 (□, ◆) and BY4741_Δqdr2 (○, ▲), harboring the cloning vector pGREG576 (◆, ▲) or the CgQDR2 expression plasmid pGREG576_CgQDR2 (□, ○), under control conditions (A) and under stress induced by 7 mg/liter miconazole (B), 5 mg/liter tioconazole (C), or 4.5 mg/liter quinidine (D) in MM4 liquid medium. The inocula were prepared as described in Materials and Methods. Growth curves and spot assay images are representative of at least three independent experiments.
CgQDR2 plays a role in [3H]clotrimazole efflux in C. glabrata.
Since the C. glabrata gene CgQDR2, encoding a plasma membrane MDR transporter, was identified as a determinant of resistance to imidazole drugs, its possible involvement in reducing their accumulation in challenged yeast cells was examined by focusing on the antifungal drug clotrimazole. The accumulation of 3H-labeled clotrimazole in nonadapted C. glabrata cells suddenly exposed to the presence of 30 mg/liter cold clotrimazole, which induces mild growth inhibition in both the parental strain and Δcgqdr2 cells, was tested and found to be 2-fold higher in cells devoid of CgQDR2 than in parental KUE100 cells (Fig. 4A). Furthermore, the accumulation of 3H-labeled clotrimazole in strain L5U1 overexpressing the CgQDR2 gene upon 30 min of incubation was found to be 2-fold lower than that for the same strain harboring the cloning vector (results not shown). This result strongly suggests that CgQdr2 activity increases the resistance of yeast to clotrimazole by reducing its accumulation within yeast cells, presumably by catalyzing the direct extrusion of this antifungal drug. To assess this possibility, the same cells were deenergized and, in the absence of glucose, were exposed to 3H-labeled clotrimazole. The passive accumulation of this radiolabeled antifungal drug reached similar levels in KUE100 and KUE100_Δcgqdr2 cells. Upon a glucose pulse, to drive energy-dependent transport mechanisms, [3H]clotrimazole was found to be extruded from these preloaded C. glabrata cells (Fig. 4B). However, drug efflux was found to be much more efficient in wild-type KUE100 cells, expressing CgQDR2, than in the Δcgqdr2 deletion mutant (Fig. 4B). To assess whether the efflux of [3H]clotrimazole was dependent on the proton gradient across the plasma membrane, efflux assays were further carried out in the presence of the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP). [3H]clotrimazole efflux was found to be severely decreased in the presence of CCCP (Fig. 4B), a result consistent with the predicted role of CgQdr2 as a drug:H+ antiporter. Taken together, these results suggest that CgQDR2 plays a direct role in catalyzing the active export of clotrimazole from C. glabrata cells in a proton gradient-dependent manner.
Fig 4.

(A) Time course accumulation ratio of [3H]clotrimazole in nonadapted cells of KUE100 (filled diamonds) or KUE100_Δcgqdr2 (filled squares) strains during cultivation in liquid BM in the presence of 30 mg/liter unlabeled clotrimazole. (B) Time course efflux ratio of [3H]clotrimazole in preloaded KUE100 (diamonds) or KUE100_Δcgqdr2 (squares) cells, upon a glucose pulse, in the presence (open symbols) or absence (filled symbols) of the protonophore CCCP, given after 30 min of passive accumulation of the radiolabeled drug. The accumulation ratio values are averages for at least three independent experiments. Error bars represent standard deviations.
CgQDR2 transcript levels are upregulated under chemical stress, under the control of the pleiotropic drug resistance transcription factor CgPdr1.
The effect on CgQDR2 transcription of exposure of C. glabrata cells to the chemical stressors to which CgQdr2 confers resistance was evaluated. The transcript levels of the CgQDR2 gene were seen to increase upon 1 h of exposure of an unadapted Candida glabrata population to inhibitory concentrations of clotrimazole (3-fold) or quinidine (4-fold) (Fig. 5B). Given its implication in antifungal drug resistance, the participation of the transcription factor CgPdr1 in the control of the observed upregulation was also tested. Indeed, deletion of the CgPDR1 gene, although it has no apparent role in CgQDR2 basal expression, completely abrogates the induction of CgQDR2 under clotrimazole or quinidine stress (Fig. 5B). CgPDR1 deletion was also found to dramatically increase the susceptibility of C. glabrata to the drugs tested over that of the corresponding parental strain (Fig. 5A).
Fig 5.

(A) Comparison by spot assays of the susceptibilities of C. glabrata strains 66032 and 66032_Δcgpdr1 to clotrimazole and quinidine, at the indicated concentrations, in BM agar plates. The inocula were prepared as described in Materials and Methods. The cell suspensions used to prepare the spots were 1:5 (b) and 1:25 (c) dilutions of the cell suspension used in column a. The images displayed are representative of at least three independent experiments. (B) Comparison of the differences in CgQDR2 transcript levels in C. glabrata 66032 wild-type (filled bars) and Δcgpdr1 (shaded bars) cells before (control) and after 1 h of exposure to stress induced by quinidine or clotrimazole at the indicated concentrations. Transcript levels were determined by quantitative RT-PCR, as described in Materials and Methods, and are expressed as CgQDR2/CgACT1 mRNA ratios. The value reported for wild-type cells under control conditions was considered equal to 1. The values shown are averages for at least three independent experiments. Error bars represent standard deviations.
To assess whether or not the action of CgPdr1 on CgQDR2 is direct, a plasmid containing a CgQDR2 promoter-lacZ fusion was constructed, and site-directed mutagenesis was used to disrupt the two putative Pdr1 binding sites occurring within the 600 bp before the CgQDR2 start codon, GCCATCATT and GCCGATAGA, identified by use of the YEASTRACT database tools as exhibiting a single nucleotide change from the CgPdr1 binding site described elsewhere (34). CgQDR2 expression levels, measured in terms of β-galactosidase activity, were confirmed to be upregulated upon exposure to 30 mg/liter of clotrimazole (Fig. 6A). The disruption of either PDRE1 or PDRE2 (Fig. 6B) was found to lead to a reduction in the basal expression level of CgQDR2 and to completely abrogate the upregulation of CgQDR2 under clotrimazole stress (Fig. 6A).
Fig 6.

(A) CgQDR2 promoter analysis based on a CgQDR2 promoter-lacZ fusion. The changes in β-galactosidase activity, expressed from wild-type, ΔPDRE1, and ΔPDRE2 CgQDR2 promoter-lacZ fusions, in wild-type L5U1 cells under control conditions (filled bars) or upon exposure for 2 h (shaded bars) or 4 h (open bars) to 30 mg/liter clotrimazole were compared. The values obtained are averages for at least three independent experiments. Error bars represent standard deviations. (B) Schematic representation of the occurrence of predicted CgPdr1 binding sites (GCCATCATT and GCCGATAGA [34]) in the CgQDR2 promoter region, as analyzed with the YEASTRACT database tools (44).
DISCUSSION
In this study, the functional characterization of the Candida glabrata drug:H+ antiporter CgQdr2 was carried out. CgQdr2 was found to be the first of its family of 10 members to be associated with antifungal drug resistance in C. glabrata, playing a role in imidazole drug resistance.
The imidazole antifungal drugs to which CgQdr2 confers resistance include miconazole, tioconazole, ketoconazole, and clotrimazole. These antifungals are widely used in the treatment of fungal skin infections, including vaginal or oral candidemia. Acquired resistance to azole drugs has been shown to rely on point mutations or overexpression of their specific target gene, ERG11, and also on the action of multidrug transporters. In C. glabrata clinical isolates, resistance to fluconazole has been shown to depend on the action of the ABC drug efflux pumps encoded by CgCDR1 and CgCDR2. The action of these transporters in the resistance to imidazole antifungals, particularly ketoconazole, has been demonstrated as well (12, 35). Furthermore, the molecular basis for the intrinsically low azole susceptibility of C. glabrata has been at least partially linked to the expression of the putative sterol transporter CgAus1, which has been shown to protect cells against azoles in the presence of serum (36). The participation of the drug:H+ antiporter family members in this phenomenon in C. glabrata had been investigated previously only for CgFlr1, but no role in antifungal drug resistance was reported for this DHA (18). However, in C. albicans, two of the DHA, CaMdr1 and, much less significantly, CaFlu1, were related to antifungal drug activity. CaMDR1 expression has been detected both in azole-resistant clinical isolates (37, 38) and in in vitro-derived fluconazole-resistant strains (39), while no evidence for a role of CaFlu1 in clinical azole resistance has been obtained so far (40). Interestingly, a strong association between the expression of DHA Mdr1 from Candida dubliniensis and fluconazole resistance has also been reported (19). This study adds CgQdr2 to the number of characterized Candida DHA involved in azole drug resistance, making it the first DHA characterized with such a role in C. glabrata. Significantly, CgQdr2 has close homologues in other pathogenic Candida species—C. albicans (orf19.8138, orf19.6992, and orf19.508), C. dubliniensis (CD36_85540), C. tropicalis (CTRG_00385), C. guilliermondii (PGUG_00335), C. parapsilosis (CPAG_05683), and C. lusitaniae (CLUG_05479)—which may also play a role in azole drug resistance in these species.
Although many of the characterized drug efflux pumps have been shown to confer resistance to a wide variety of drugs and chemicals (9, 10, 41), the molecular mechanisms behind their apparent promiscuity remains elusive and a topic of debate (5–10). Based on its high degree of homology and functional similarity to the S. cerevisiae QDR2 gene, CgQDR2 was examined for a possible physiological role linked to yeast survival under conditions of potassium limitation (22). However, CgQdr2 expression did not seem to improve the fitness of C. glabrata or S. cerevisiae under conditions of low potassium availability or to complement the S. cerevisiae Δqdr2 susceptibility phenotype observed under these conditions. Since CgQdr2 was found to play no role in K+ homeostasis, no link between the K+ concentration and CgQdr2-mediated drug resistance was looked for. The possibility that CgQdr2 may play a physiological role that contributes indirectly to its action as a multidrug resistance determinant cannot be excluded, given that this seems to be the case for the majority of the ABC and MFS MDR transporters characterized in S. cerevisiae (5, 9, 42). Nonetheless, the results of this study point out to a direct role of CgQdr2 in clotrimazole efflux. CgQdr2 was found to be localized to the plasma membrane both in S. cerevisiae and in C. glabrata. The observation that a CgQdr2-GFP fusion protein exhibits some degree of vacuolar accumulation in C. glabrata suggests that CgQdr2 may be internalized more rapidly in its native environment than in the heterologous host. Nonetheless, the higher level of clotrimazole resistance observed in cells expressing the CgQDR2 gene than in Δcgqdr2 deletion mutant cells appears to correlate with the observation that the level of accumulation of this antifungal drug is 2-fold higher in the deletion mutant than in the corresponding parental strain. Furthermore, the efflux of radiolabeled clotrimazole from preloaded cells was demonstrated to be impaired in the Δcgqdr2 deletion mutant relative to that in the parental strain and to depend on the maintenance of the proton gradient across the plasma membrane.
CgQDR2 transcript levels were seen to be upregulated in C. glabrata cells upon exposure to chemical stress induced by quinidine or clotrimazole. Since the multidrug resistance phenotype can be seen as a long-term genetic stabilization of the normal transient drug response, the upregulation of CgQDR2 under clotrimazole-induced stress further suggests that the action of CgQdr2 may be relevant to the clinical acquisition of azole drug resistance. This idea is reinforced by the fact that the upregulation observed was found to depend on the transcription factor CgPdr1, the main regulator of multidrug resistance in C. glabrata. Indeed, gain-of-function mutations of the CgPDR1 gene have been found in azole-resistant clinical isolates, and very recently, the CgQDR2 gene was found to be upregulated due to the L946S amino acid substitution in CgPDR1 in a resistant clinical isolate exhibiting the same genotype as a susceptible isolate (43). Other CgPDR1 gain-of function mutations identified so far were found to have no effect on CgQDR2 expression (43). The results obtained are also consistent with the occurrence in the CgQDR2 promoter region of two predicted Pdr1 binding sites (GCCATCATT and GCCGATAGA [34]) (Fig. 6B) that were found to be required for the clotrimazole-induced upregulation of CgQDR2 expression. These results strongly suggest that CgQDR2 is a direct target of the multidrug resistance transcription factor CgPdr1. The observed effect of CgPDR1 deletion on the susceptibilities of C. glabrata to clotrimazole and quinidine is certainly due to the effect of CgPDR1 on a broad range of targets, including ABC multidrug efflux pumps. However, it appears reasonable to say that this effect may also be due to the CgPdr1-controlled upregulation of CgQDR2 occurring under these stresses. It is also interesting that although CgQDR2 and ScQDR2 confer resistance to the same drugs, their transcriptional control appears to be quite diverse. Indeed, while ScQDR2 expression is not induced upon exposure to the drugs to which this gene confers resistance (23), CgQDR2 transcript levels appear to be highly responsive to drugs. This is consistent with a role for CgPdr1 in CgQDR2 gene regulation, while ScQDR2 appears to be controlled mostly by ScGcn4 (22), a transcription factor involved in the response to amino acid starvation.
Although in general a much stronger association between azole resistance and the expression of ABC multidrug transporters has been revealed so far (40), the results described here reinforce the notion that multidrug transporters of the MFS contribute to the overall resistance phenotype. Indeed, this study, characterizing the first C. glabrata DHA involved in azole drug resistance, highlights the importance of studying the remaining members of this family in C. glabrata in this context. Such investigations may be expected to have an impact on the treatment of the increasing number of azole-resistant fungal infections.
ACKNOWLEDGMENTS
This work was supported by FEDER and “Fundação para a Ciência e a Tecnologia” (FCT) (contracts PTDC/BIA-MIC/72577/2006 and PTDC/EBB-BIO/119356/2010) and by Ph.D. and postdoctoral grants to C.C. and T.R.C., respectively).
We acknowledge Thomas Edlind (Department of Microbiology and Immunology, Drexel University, College of Medicine, Philadelphia, PA) and John Bennett (Clinical Mycology Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD) for kindly providing the 66032 and L5U1 strains, respectively.
Footnotes
Published ahead of print 29 April 2013
REFERENCES
- 1. Jarvis WR. 1995. Epidemiology of nosocomial fungal infections, with emphasis on Candida species. Clin. Infect. Dis. 20:1526–1530 [DOI] [PubMed] [Google Scholar]
- 2. Fidel PL, Jr, Vazquez JA, Sobel JD. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12:80–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mishra NN, Prasad T, Sharma N, Payasi A, Prasad R, Gupta DK, Singh R. 2007. Pathogenicity and drug resistance in Candida albicans and other yeast species. A review. Acta Microbiol. Immunol. Hung. 54:201–235 [DOI] [PubMed] [Google Scholar]
- 4. Hayes JD, Wolf CR. (ed). 1997. Modern genetics, vol 3 Molecular genetics of drug resistance. Harwood Academic, Amsterdam, Netherlands [Google Scholar]
- 5. Jungwirth H, Kuchler K. 2006. Yeast ABC transporters—a tale of sex, stress, drugs and aging. FEBS Lett. 580:1131–1138 [DOI] [PubMed] [Google Scholar]
- 6. Paulsen IT. 2003. Multidrug efflux pumps and resistance: regulation and evolution. Curr. Opin. Microbiol. 6:446–451 [DOI] [PubMed] [Google Scholar]
- 7. Prasad R, Panwar SL, Smriti 2002. Drug resistance in yeasts-an emerging scenario. Adv. Microb. Physiol. 46:155–201 [DOI] [PubMed] [Google Scholar]
- 8. Roepe PD, Wei LY, Hoffman MM, Fritz F. 1996. Altered drug translocation mediated by the MDR protein: direct, indirect, or both? J. Bioenerg. Biomembr. 28:541–555 [DOI] [PubMed] [Google Scholar]
- 9. Sá-Correia I, Santos SC, Teixeira MC, Cabrito TR, Mira NM. 2009. Drug:H+ antiporters in chemical stress response in yeast. Trends Microbiol. 17:22–31 [DOI] [PubMed] [Google Scholar]
- 10. Sá-Correia I, Tenreiro S. 2002. The multidrug resistance transporters of the major facilitator superfamily, 6 years after disclosure of Saccharomyces cerevisiae genome sequence. J. Biotechnol. 98:215–226 [DOI] [PubMed] [Google Scholar]
- 11. Prasad R, De Wergifosse P, Goffeau A, Balzi E. 1995. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr. Genet. 27:320–329 [DOI] [PubMed] [Google Scholar]
- 12. Sanglard D, Ischer F, Monod M, Bille J. 1997. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 143(Part 2):405–416 [DOI] [PubMed] [Google Scholar]
- 13. Torelli R, Posteraro B, Ferrari S, La Sorda M, Fadda G, Sanglard D, Sanguinetti M. 2008. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 68:186–201 [DOI] [PubMed] [Google Scholar]
- 14. Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. 1996. Life with 6000 genes. Science 274:546, 563–567 [DOI] [PubMed] [Google Scholar]
- 16. Goldway M, Teff D, Schmidt R, Oppenheim AB, Koltin Y. 1995. Multidrug resistance in Candida albicans: disruption of the BENr gene. Antimicrob. Agents Chemother. 39:422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Calabrese D, Bille J, Sanglard D. 2000. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology 146(Part 11):2743–2754 [DOI] [PubMed] [Google Scholar]
- 18. Chen KH, Miyazaki T, Tsai HF, Bennett JE. 2007. The bZip transcription factor Cgap1p is involved in multidrug resistance and required for activation of multidrug transporter gene CgFLR1 in Candida glabrata. Gene 386:63–72 [DOI] [PubMed] [Google Scholar]
- 19. Sullivan DJ, Moran GP, Pinjon E, Al-Mosaid A, Stokes C, Vaughan C, Coleman DC. 2004. Comparison of the epidemiology, drug resistance mechanisms, and virulence of Candida dubliniensis and Candida albicans. FEMS Yeast Res. 4:369–376 [DOI] [PubMed] [Google Scholar]
- 20. Gbelska Y, Krijger JJ, Breunig KD. 2006. Evolution of gene families: the multidrug resistance transporter genes in five related yeast species. FEMS Yeast Res. 6:345–355 [DOI] [PubMed] [Google Scholar]
- 21. Tenreiro S, Vargas RC, Teixeira MC, Magnani C, Sá-Correia I. 2005. The yeast multidrug transporter Qdr3 (Ybr043c): localization and role as a determinant of resistance to quinidine, barban, cisplatin, and bleomycin. Biochem. Biophys. Res. Commun. 327:952–959 [DOI] [PubMed] [Google Scholar]
- 22. Vargas RC, Garcia-Salcedo R, Tenreiro S, Teixeira MC, Fernandes AR, Ramos J, Sá-Correia I. 2007. Saccharomyces cerevisiae multidrug resistance transporter Qdr2 is implicated in potassium uptake, providing a physiological advantage to quinidine-stressed cells. Eukaryot. Cell 6:134–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vargas RC, Tenreiro S, Teixeira MC, Fernandes AR, Sá-Correia I. 2004. Saccharomyces cerevisiae multidrug transporter Qdr2p (Yil121wp): localization and function as a quinidine resistance determinant. Antimicrob. Agents Chemother. 48:2531–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teixeira MC, Cabrito TR, Hanif ZM, Vargas RC, Tenreiro S, Sá-Correia I. 2011. Yeast response and tolerance to polyamine toxicity involving the drug:H+ antiporter Qdr3 and the transcription factors Yap1 and Gcn4. Microbiology 157:945–956 [DOI] [PubMed] [Google Scholar]
- 25. Ueno K, Uno J, Nakayama H, Sasamoto K, Mikami Y, Chibana H. 2007. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata. Eukaryot. Cell 6:1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 48:3773–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen G, Wu C, Schade B, Thomas DY, Whiteway M. 2005. Drag&Drop cloning in yeast. Gene 344:43–51 [DOI] [PubMed] [Google Scholar]
- 28. Cabrito TR, Teixeira MC, Duarte AA, Duque P, Sá-Correia I. 2009. Heterologous expression of a Tpo1 homolog from Arabidopsis thaliana confers resistance to the herbicide 2,4-D and other chemical stresses in yeast. Appl. Microbiol. Biotechnol. 84:927–936 [DOI] [PubMed] [Google Scholar]
- 29. Ueno K, Matsumoto Y, Uno J, Sasamoto K, Sekimizu K, Kinjo Y, Chibana H. 2011. Intestinal resident yeast Candida glabrata requires Cyb2p-mediated lactate assimilation to adapt in mouse intestine. PLoS One 6:e24759. 10.1371/journal.pone.0024759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. NCCLS 2002. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 2nd ed M27-A2 NCCLS, Wayne, PA [Google Scholar]
- 31. Rodríguez-Tudela JL, Barchiesi F, Bille J, Chryssanthou E, Cuenca-Estrella M, Denning D, Donnelly JP, Dupont B, Fegeler W, Moore C, Richardson M, Verweij PE. 2003. Method for the determination of minimum inhibitory concentration (MIC) by broth dilution of fermentative yeasts. Clin. Microbiol. Infect. 9(8):i–viii. 10.1046/j.1469-0691.2003.00789.x [DOI] [Google Scholar]
- 32. Rosa MF, Sá-Correia I. 1996. Intracellular acidification does not account for inhibition of Saccharomyces cerevisiae growth in the presence of ethanol. FEMS Microbiol. Lett. 135:271–274 [DOI] [PubMed] [Google Scholar]
- 33. Teixeira MC, Dias PJ, Monteiro PT, Sala A, Oliveira AL, Freitas AT, Sá-Correia I. 2010. Refining current knowledge on the yeast FLR1 regulatory network by combined experimental and computational approaches. Mol. Biosyst. 6:2471–2481 [DOI] [PubMed] [Google Scholar]
- 34. Paul S, Schmidt JA, Moye-Rowley WS. 2011. Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot. Cell 10:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakayama H, Tanabe K, Bard M, Hodgson W, Wu S, Takemori D, Aoyama T, Kumaraswami NS, Metzler L, Takano Y, Chibana H, Niimi M. 2007. The Candida glabrata putative sterol transporter gene CgAUS1 protects cells against azoles in the presence of serum. J. Antimicrob. Chemother. 60:1264–1272 [DOI] [PubMed] [Google Scholar]
- 37. Perea S, Lopez-Ribot JL, Kirkpatrick WR, McAtee RK, Santillan RA, Martinez M, Calabrese D, Sanglard D, Patterson TF. 2001. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 45:2676–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White TC. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 41:1482–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Albertson GD, Niimi M, Cannon RD, Jenkinson HF. 1996. Multiple efflux mechanisms are involved in Candida albicans fluconazole resistance. Antimicrob. Agents Chemother. 40:2835–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cannon RD, Lamping E, Holmes AR, Niimi K, Baret PV, Keniya MV, Tanabe K, Niimi M, Goffeau A, Monk BC. 2009. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 22:291–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paulsen IT, Sliwinski MK, Nelissen B, Goffeau A, Saier MH., Jr 1998. Unified inventory of established and putative transporters encoded within the complete genome of Saccharomyces cerevisiae. FEBS Lett. 430:116–125 [DOI] [PubMed] [Google Scholar]
- 42. Cabrito TR, Teixeira MC, Singh A, Prasad R, Sá-Correia I. 2011. The yeast ABC transporter Pdr18 (ORF YNR070w) controls plasma membrane sterol composition, playing a role in multidrug resistance. Biochem. J. 440:195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. 2011. Genome-wide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell 10:373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teixeira MC, Monteiro P, Jain P, Tenreiro S, Fernandes AR, Mira NP, Alenquer M, Freitas AT, Oliveira AL, Sá-Correia I. 2006. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 34:D446–D451 [DOI] [PMC free article] [PubMed] [Google Scholar]