

Abstract
Antibiotic resistance is generally selected within a window of concentrations high enough to inhibit wild-type growth but low enough for new resistant mutants to emerge. We studied de novo evolution of resistance to ciprofloxacin in an Escherichia coli knockout library. Five null mutations had little or no effect on intrinsic antibiotic susceptibility but increased the upper antibiotic dosage to which initially sensitive populations could adapt. These mutations affect mismatch repair, translation fidelity, and iron homeostasis.
TEXT
For many antimicrobial agents—including fluoroquinolones, cephalosporins, and rifamycins—a prominent factor contributing to the evolution of resistance is the acquisition of chromosomal mutations during therapy (1–3). While a wealth of high-throughput studies have investigated the impact of individual genes on intrinsic antibiotic susceptibility (4–6) or persistence (7), no systematic large-scale study has been devoted to the identification of molecular mechanisms that promote the evolution of antibiotic resistance.
Here we systematically tested the impact of gene inactivation on the de novo evolution of antibiotic resistance. Escherichia coli is undoubtedly an ideal model prokaryote for such a study. The availability of a nearly complete single gene deletion library (the KEIO collection) enables the study of this issue in nearly all of the nonessential genes of this species (8). Our investigations concentrated on understanding the development of resistance to ciprofloxacin. It is one of the most widely deployed fluoroquinolone antibiotics in clinics, and its mechanism of action has been well studied (9–11).
We developed a simple, high-throughput protocol that allows investigation of the de novo evolution of quinolone resistance in thousands of parallel bacterial cultures. Our goal was to identify genotypes (i.e., single-gene knockout strains) that permit adaptation to high antibiotic concentrations demanding the acquisition of one or more rare mutations (12, 13). All experiments were conducted with 96-deep-well plates containing 350 μl LB medium supplemented with a toxic concentration of ciprofloxacin (200 ng/ml). The antibiotic dosage used is more than 10 times the MIC for wild-type (WT) E. coli (Table 1). About 108 cells were added to each well (two replicate populations per genotype). Following 5 days of incubation (37°C, 320 rpm), 2 μl of each culture was transferred to an agar plate containing the same concentration of the antibiotic. After 24 h, the resistant bacteria of each genotype were counted. Initial positive hits (i.e., at least one resistant population per genotype) were validated with new sets of laboratory experiments. We used the same procedure as above with 96 replicate populations per genotype. Final hits were checked for the presence of the appropriate gene deletion by PCR using site-specific primers. At such a high ciprofloxacin concentration, the toxic effect of the antibiotic is substantial and population size rapidly declines (14). Typically, WT cultures became extinct by the end of the 5-day treatment period (data not shown). Development of resistance was generally rare; it occurred only in ∼4% of parallel WT populations. The screen identified six genotypes with a massive increase in the frequency of resistant populations. In these cases, 60 to 100% of the independent populations were capable of acquiring resistance to ciprofloxacin. By using the ASKA overexpression plasmid library (15), we complemented these candidate strains with the corresponding WT alleles. In all but one case (ybgJ), we confirmed that the deletion mutation was responsible for the enhanced frequency of resistance (data not shown). The remaining five genotypes are presented in Table 1. Similar results were obtained when these five genotypes were tested against antibiotics of two other classes, chloramphenicol and streptomycin (Table 1).
Table 1.
Increased evolvability of five knockout strains under single-step antibiotic exposurea
| KEIO strain | Ciprofloxacin MIC (ng/ml) | Gene function | Frequency of resistant populations |
||
|---|---|---|---|---|---|
| Ciprofloxacin (200 ng/ml) | Chloramphenicol (12.5 μg/ml) | Streptomycin (30 μg/ml) | |||
| WT | 18.4 | 0.04 | 0.02 | 0.2 | |
| Δfur mutant | 13.9 | Fe uptake regulation | 0.6 | 0.00 | 0.73 |
| ΔmiaA mutant | 26.7 | Translational fidelity | 0.99 | 0.92 | 1 |
| ΔmutH mutant | 19.4 | Mismatch repair | 1 | 0.92 | 1 |
| ΔmutL mutant | 19.4 | Mismatch repair | 0.95 | 1 | 0.99 |
| ΔmutS mutant | 20.5 | Mismatch repair | 1 | 0.96 | 1 |
In the presence of a single antibiotic, five null mutants showed a significant increase in the frequency of resistant populations compared to the WT (BW25113, CGSC 7636). Experiments were conducted with deep-well plates using 96 parallel replicates per strain. In each well, ∼108 cells were exposed to a single antibiotic at a concentration well beyond the MIC. The MICs are 2.2 μg/ml for chloramphenicol and 2.6 μg/ml for streptomycin. After 5 days of incubation, the frequency of resistant populations was determined by transferring ∼2 μl of each culture to an agar plate supplemented with the same concentration of the antibiotic.
Using a standard microdilution method (16) with 1.4-fold dilution steps, we found that the MIC for these genotypes is comparable to that for the WT (Table 1). Further support comes from a systematic chemo-genomic study that exposed a transposon library of E. coli to 17 different antibiotics at sublethal concentrations (17). None of our candidate genes influences growth rates in the presence of any of these 17 antibiotics. The only exception is a null mutation of miaA that appears to enhance the growth rate on nalidixic acid, another quinolone. We next asked whether the survival rate upon toxic antibiotic exposure is especially high in the knockout populations. These experiments were conducted with 96-deep-well plates (350 μl LB medium). Large cell numbers (∼108) were transferred to fresh medium containing 200 ng/ml ciprofloxacin. Persistence was measured by determining survival rates upon antibiotic exposure. Viable cell numbers were determined every hour during the 5 h of ciprofloxacin treatment by plating dilutions onto 24-well LB agar macroplates and counting growing colonies. We confirmed that during the first 5 h of antibiotic exposure, the number of resistant cells remained below the detection level (below 1 per 1.4 × 107 cells). As demonstrated previously (14, 18), the surviving fraction of a WT E. coli culture treated with ciprofloxacin produces a typical biphasic pattern, reflecting rapid killing of most of the cells (99%) except for a small persister subpopulation. None of the five genotypes showed a statistically significant increase in survival compared with that of the WT (Fig. 1). We conclude that the capacity of these genotypes to evolve resistance is not due to changes in intrinsic antibiotic susceptibility or elevated persister formation (19). What else might be the cause? Genotypes with increased constitutive mutation rates (mutators) are generally considered to have an important role during microbial evolution (20, 21). They are frequently found in evolving natural and experimental populations and facilitate rapid adaptation during periods of stress, such as antibiotic exposure (22–25). To measure mutation rates, we used a classic lac reversion screen (26). We investigated the rates of six major types of nucleotide substitutions (Table 2) by using six indicator E. coli strains with different inactivating mutations at the same coding position in the lacZ gene. Each strain is Lac− and reverts to Lac+ only when the appropriate codon is restored. The appropriate gene deletion mutations were introduced into each of the six indicator strains by P1 transduction and checked by PCR with site-specific primer pairs. We followed established protocols (27) to measure the frequency of Lac+ revertants for each type of point mutation. Mutation rates were calculated by using the MSS maximum-likelihood method (FALCOR package) (28, 29). Remarkably, all of the knockout strains have an elevated mutation rate and their mutations partially influence different aspects of mutagenesis (Table 2).
Fig 1.
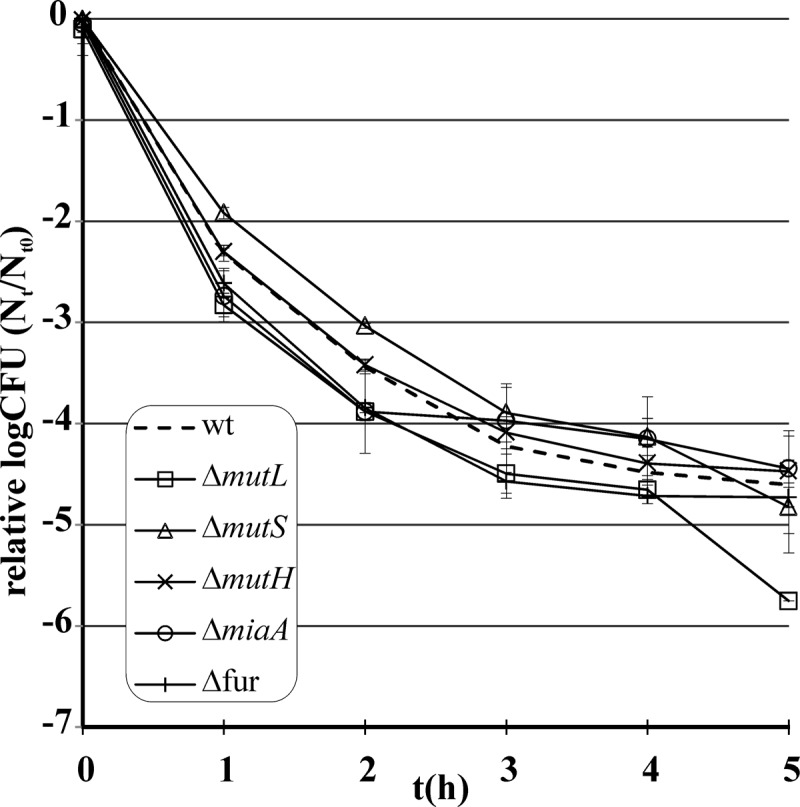
Survival of WT (BW25113, CGSC 7636) and mutant strains under ciprofloxacin (200 ng/ml) treatment. We followed standard protocols. Strains were exposed to ciprofloxacin in the late exponential phase, and viable cell numbers were determined by counting colonies on agar plates (see references 14 and 18). None of the single-knockout mutants showed enhanced survival under ciprofloxacin stress (P > 0.05, Wilcoxon test). Error bars indicate 95% confidence intervals.
Table 2.
Mutational spectra of gene knockout strains with enhanced evolutionary potentiala
| Mutation | Mutation rate/generation (10−8) |
|||||
|---|---|---|---|---|---|---|
| CC101 (A·T → C·G) | CC102 (G·C → A·T) | CC103 (G·C → C·G) | CC104 (G·C → T·A) | CC105 (A·T → T·A) | CC106 (A·T → G·C) | |
| None (WT) | —b | 1.6 | — | 2.3 | 0.4 | — |
| Δfur | — | 4 | — | 7.3 | — | 0.9 |
| ΔmiaA | — | — | 4.5 | 95.3 | 68.2 | — |
| ΔmutH | — | 88.3 | 1.5 | 6.7 | 19.5 | 82.1 |
| ΔmutL | — | 203.5 | 2.3 | 2.9 | 12 | 80.6 |
| ΔmutS | — | 18.2 | 2.6 | 28.6 | 0.5 | 312.7 |
Deletions from KEIO mutants were transferred into the WT (P90C, CGSC 8083) containing different types of lacZ mutations (CC101 to CC106, CGSC 8095 to CGSC 8100) by P1 transduction. Mutational frequencies were determined by lacZ reversion method. Mutation rates were calculated by the MSS maximum-likelihood method (see reference 29).
—, below detection limit (0.4/generation/108 cells).
The corresponding mutator genes have diverse molecular functions. First, our list includes central components of methyl-directed mismatch repair (mutS, mutH, and mutL) (30, 31). Defects in and downregulation of these genes are frequently associated with pathogenic populations, indicating a central role for this pathway during bacterial adaptation in nature (22, 24, 32). Second, defects in tRNA modification due to miaA deletion cause reduced fidelity and efficiency of translation (33). This gene encodes a tRNA dimethylallyltransferase and is involved in hypermodification of the A37 base of certain tRNAs (34). Removal of miaA results in an ∼50-fold increase in GC → TA transversions (31) (Table 2). While the cascade of events that promotes mutations in ΔmiaA mutant populations is far from clear, several clues indicate that it is associated with translational stress-induced mutagenesis (33). Our screen also identified a central regulator of iron homeostasis (fur) (35) whose removal yields a mutator phenotype. Inactivation of fur leads to an increase in the intracellular concentration of ferrous iron (Fe2+) (36), which accelerates the Fenton reaction and potentiates oxidative damage-induced mutagenesis (37). This possibility will be explored in a future work.
There are many further known null mutations that confer a mild mutator phenotype, none of which appeared as positive hits in our screen (31). We briefly investigated two well-described genes (mutD and mutT). Despite the moderate mutator phenotypes the corresponding null mutations confer (31), these genes had no or only a minor effect on either resistance evolution (Fig. 2) or intrinsic susceptibility to ciprofloxacin (data not shown).
Fig 2.
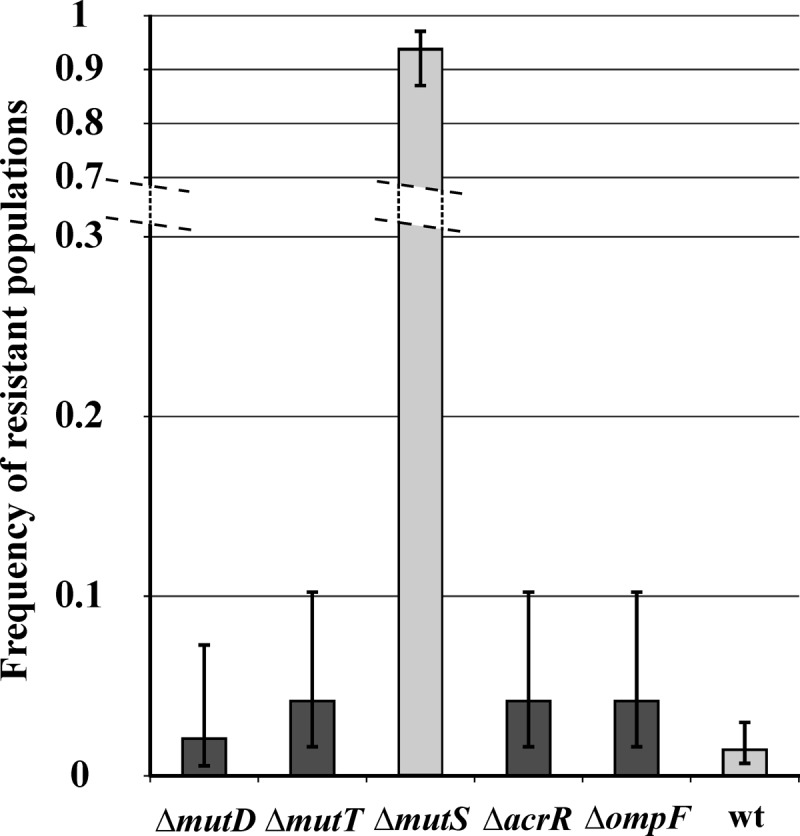
Evolvability of mild mutators and null mutants with decreased antibiotic susceptibility exposed to a high ciprofloxacin concentration (200 ng/ml, 96 parallel populations per strain). Error bars correspond to 95% confidence intervals of a proportion.
Why should this be so? As noted previously, adaptation to a high ciprofloxacin dosage demands one or more specific mutations (12). To investigate this issue, we isolated 10 ciprofloxacin-resistant clones revealed by our screen (7 and 3 in the ΔmutS mutant and WT genetic backgrounds, respectively). We sequenced the quinolone resistance-determining regions (QRDRs) of the gyrA and parC genes and the marR gene. These genes are known to bear mutations in ciprofloxacin-resistant isolates (38, 39). The major target protein (GyrA) of ciprofloxacin was regularly mutated, and the same amino acid substitution occurred in all 10 isolates (S83L substitution, Table 3). The very same mutation is regularly observed in ciprofloxacin-resistant laboratory (40) and clinical (40) E. coli strains. While multiple mutations were observed in one clone only (Table 3), there are two reasons why other unknown loci were also mutated. First, the measured MICs for these clones are above the value the single S83L mutation confers (41). This was achieved by engineering a single point mutation in the WT background (by single-stranded oligonucleotide-mediated recombineering [42]) and then measuring the MIC for this strain (Table 3).
Table 3.
Characterization of 10 ciprofloxacin-resistant clones isolated after 5 days of ciprofloxacin treatmenta
| Strain | Ciprofloxacin MIC (μg/ml) | Mutation |
Membrane permeabilityb | ||
|---|---|---|---|---|---|
| gyrA | parC | marR | |||
| ΔmutS clone 1 | 1 | S83L | —c | — | 1.08d |
| ΔmutS clone 2 | 2 | S83L | A140T | — | 1.01 |
| ΔmutS clone 3 | 0.75 | S83L | — | — | 0.90d |
| ΔmutS clone 4 | 1 | S83L | — | — | 0.94d |
| ΔmutS clone 5 | 2 | S83L | — | — | 0.74d |
| ΔmutS clone 6 | 2 | S83L | — | — | 1.30d |
| ΔmutS clone 7 | 2 | S83L | — | — | 0.91d |
| WT clone 1 | 1.5 | S83L | — | — | 0.91d |
| WT clone 2 | 1 | S83L | — | — | 0.91d |
| WT clone 3 | 0.25 | S83L | — | — | 0.98 |
| WT S83L | 0.25 | S83L | — | — | 1.01 |
| WT | 0.008 | — | — | — | 1.00 |
Mutants were selected in the presence of 200 ng/ml ciprofloxacin. MICs were determined by using E tests (BioMérieux). The QRDRs of gyrA, parC, and the marR gene were sequenced following PCR amplification. Membrane permeability was measured by a previously established Hoechst 33342 fluorescent dye accumulation assay (see reference 43).
Relative Hoechst dye accumulation.
—, no mutation.
P < 0.05 (Mann-Whitney U test).
In addition, six strains showed slight but significant decreases in intracellular levels of the fluorescent probe Hoechst 33342 (43), suggesting either decreased porin or increased efflux pump activity (Table 3).
Contrary to our initial expectations, enhanced evolutionary capacity is not due to changes in intrinsic antibiotic susceptibility. This is somewhat surprising, as numerous E. coli single knockouts have elevated growth rates under low quinolone stress (17), including those lacking members of the general bacterial porin family (e.g., OmpF) and a regulator of the AcrAB efflux pump (AcrR). Although null mutations in the genes for these two proteins enhance viability under mild quinolone stress (44, 45), they had no major effect on the frequency of resistant populations (Fig. 2). Additionally, we failed to identify gene deletions that overlap genes previously recognized as modulators of intrinsic antibiotic tolerance (7). This result suggests that a minor variation in antibiotic tolerance has a relatively small impact on the evolution of clinically significant resistance. As the main objective of this work was to identify genes that mold the upper antibiotic dosage to which populations can adapt, we expect that further genes with mild positive or negative effects on the rate of resistance evolution remain to be identified. Single gene deletions may also fail to uncover phenotypes if the underlying mutational pathways are redundant. Regardless of these limitations, our work clearly demonstrates that at high antibiotic concentrations, an enhanced mutation supply can dramatically alter the outcome of selection.
ACKNOWLEDGMENTS
This work was supported by grants from the European Research Council (202591), the Welcome Trust, and the Lendület Program of the Hungarian Academy of Sciences.
Footnotes
Published ahead of print 13 May 2013
REFERENCES
- 1. Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050 [DOI] [PubMed] [Google Scholar]
- 2. Martinez JL, Baquero F. 2000. Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 44:1771–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spratt B. 1994. Resistance to antibiotics mediated by target alterations. Science 264:388–393 [DOI] [PubMed] [Google Scholar]
- 4. Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. 2010. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob. Agents Chemother. 54:1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breidenstein EBM, Khaira BK, Wiegand I, Overhage J, Hancock REW. 2008. Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob. Agents Chemother. 52:4486–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansen S, Lewis K, Vulic M. 2008. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52:2718–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.2008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolfson JS, Hooper DC. 1985. The fluoroquinolones: structures, mechanisms of action and resistance, and spectra of activity in vitro. Antimicrob. Agents Chemother. 28:581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hooper DC, Wolfson JS, Ng EY, Swartz MN. 1987. Mechanisms of action of and resistance to ciprofloxacin. Am. J. Med. 82:12–20 [PubMed] [Google Scholar]
- 11. Drlica K. 1999. Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 2:504–508 [DOI] [PubMed] [Google Scholar]
- 12. Hooper DC. 2001. Emerging mechanisms of fluoroquinolone resistance. Emerg. Infect. Dis. 7:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olofsson SK, Marcusson LL, Komp Lindgren P, Hughes D, Cars O. 2006. Selection of ciprofloxacin resistance in Escherichia coli in an in vitro kinetic model: relation between drug exposure and mutant prevention concentration. J. Antimicrob. Chemother. 57:1116–1121 [DOI] [PubMed] [Google Scholar]
- 14. Eliopoulos GM, Gardella A, Moellering RC., Jr 1984. In vitro activity of ciprofloxacin, a new carboxyquinoline antimicrobial agent. Antimicrob. Agents Chemother. 25:331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299 [DOI] [PubMed] [Google Scholar]
- 16. Wiegand I, Hilpert K, Hancock REW. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175 [DOI] [PubMed] [Google Scholar]
- 17. Girgis HS, Hottes AK, Tavazoie S. 2009. Genetic architecture of intrinsic antibiotic susceptibility. PLoS One 4:e5629. 10.1371/journal.pone.0005629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levin BR, Rozen DE. 2006. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 4:556–562 [DOI] [PubMed] [Google Scholar]
- 19. Dörr T, Lewis K, Vulić M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760. 10.1371/journal.pgen.1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. 1997. Role of mutator alleles in adaptive evolution. Nature 387:700–702 [DOI] [PubMed] [Google Scholar]
- 21. Sniegowski PD, Gerrish PJ, Lenski RE. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387:703–705 [DOI] [PubMed] [Google Scholar]
- 22. Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J, LeClerc JE, Cebula TA. 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277:1833–1834 [DOI] [PubMed] [Google Scholar]
- 23. Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, Matic I. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J. Bacteriol. 184:605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiegand I, Marr AK, Breidenstein EBM, Schurek KN, Taylor P, Hancock REW. 2008. Mutator genes giving rise to decreased antibiotic susceptibility in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52:3810–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersson DI, Hughes D. 2011. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 35:901–911 [DOI] [PubMed] [Google Scholar]
- 26. Cupples CG, Miller JH. 1989. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl. Acad. Sci. U. S. A. 86:5345–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seier T, Padgett DR, Zilberberg G, Sutera VA, Toha N, Lovett ST. 2011. Insights into mutagenesis using Escherichia coli chromosomal lacZ strains that enable detection of a wide spectrum of mutational events. Genetics 188:247–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbrück fluctuation analysis. Bioinformatics 25:1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sarkar S, Ma WT, Sandri GH. 1992. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85:173–179 [DOI] [PubMed] [Google Scholar]
- 30. Junop MS, Yang W, Funchain P, Clendenin W, Miller JH. 2003. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair 2:387–405 [DOI] [PubMed] [Google Scholar]
- 31. Horst J-P, Wu T, Marinus MG. 1999. Escherichia coli mutator genes. Trends Microbiol. 7:29–36 [DOI] [PubMed] [Google Scholar]
- 32. LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211 [DOI] [PubMed] [Google Scholar]
- 33. Zhao J, Leung HE, Winkler ME. 2001. The miaA mutator phenotype of Escherichia coli K-12 requires recombination functions. J. Bacteriol. 183:1796–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jenner LB, Demeshkina N, Yusupova G, Yusupov M. 2010. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat. Struct. Mol. Biol. 17:555–560 [DOI] [PubMed] [Google Scholar]
- 35. Hantke K. 1981. Regulation of ferric iron transport in Escherichia coli K12: isolation of a constitutive mutant. Mol. Gen. Genet. 182:288–292 [DOI] [PubMed] [Google Scholar]
- 36. Touati D, Jacques M, Tardat B, Bouchard L, Despied S. 1995. Lethal oxidative damage and mutagenesis are generated by iron in delta fur mutants of Escherichia coli: protective role of superoxide dismutase. J. Bacteriol. 177:2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nunoshiba T, Obata F, Boss AC, Oikawa S, Mori T, Kawanishi S, Yamamoto K. 1999. Role of iron and superoxide for generation of hydroxyl radical, oxidative DNA lesions, and mutagenesis in Escherichia coli. J. Biol. Chem. 274:34832–34837 [DOI] [PubMed] [Google Scholar]
- 38. Bagel S, Hullen V, Wiedemann B, Heisig P. 1999. Impact of gyrA and parC mutations on quinolone resistance, doubling time, and supercoiling degree of Escherichia coli. Antimicrob. Agents Chemother. 43:868–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Komp Lindgren P, Marcusson LL, Sandvang D, Frimodt-Moller N, Hughes D. 2005. Biological cost of single and multiple norfloxacin resistance mutations in Escherichia coli implicated in urinary tract infections. Antimicrob. Agents Chemother. 49:2343–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cirz RT, Chin JK, Andes DR, De Crécy-Lagard V, Craig WA, Romesberg FE. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 3:e176. 10.1371/journal.pbio.0030176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marcusson LL, Frimodt-Møller N, Hughes D. 2009. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 5:e1000541. 10.1371/journal.ppat.1000541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ellis HM, Yu D, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 98:6742–6746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coldham NG, Webber M, Woodward MJ, Piddock LJV. 2010. A 96-well plate fluorescence assay for assessment of cellular permeability and active efflux in Salmonella enterica serovar Typhimurium and Escherichia coli. J. Antimicrob. Chemother. 65:1655–1663 [DOI] [PubMed] [Google Scholar]
- 44. Kern WV, Oethinger M, Jellen-Ritter AS, Levy SB. 2000. Non-target gene mutations in the development of fluoroquinolone resistance in Escherichia coli. Antimicrob. Agents Chemother. 44:814–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cohen SP, McMurry LM, Hooper DC, Wolfson JS, Levy SB. 1989. Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (Mar) Escherichia coli selected by tetracycline or chloramphenicol: decreased drug accumulation associated with membrane changes in addition to OmpF reduction. Antimicrob. Agents Chemother. 33:1318–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]