

Abstract
Visceral leishmaniasis is a neglected tropical disease with significant health impact. The current treatments are poor, and there is an urgent need to develop new drugs. Primary screening assays used for drug discovery campaigns have typically used free-living forms of the Leishmania parasite to allow for high-throughput screening. Such screens do not necessarily reflect the physiological situation, as the disease-causing stage of the parasite resides inside human host cells. Assessing the drug sensitivity of intracellular parasites on scale has recently become feasible with the advent of high-content screening methods. We describe here a 384-well microscopy-based intramacrophage Leishmania donovani assay and compare it to an axenic amastigote system. A panel of eight reference compounds was tested in both systems, as well as a human counterscreen cell line, and our findings show that for most clinically used compounds both axenic and intramacrophage assays report very similar results. A set of 15,659 diverse compounds was also screened using both systems. This resulted in the identification of seven new antileishmanial compounds and revealed a high false-positive rate for the axenic assay. We conclude that the intramacrophage assay is more suited as a primary hit-discovery platform than the current form of axenic assay, and we discuss how modifications to the axenic assay may render it more suitable for hit-discovery.
INTRODUCTION
The leishmaniases are a group of parasitic diseases caused by members of the protozoan trypanosomatid genus Leishmania that have a significant health impact, primarily in the developing world (1). Current treatments for visceral leishmaniasis include antimonials, amphotericin B, miltefosine, and paromomycin. All of these drugs have substantial issues associated with them, including toxicity, emerging resistance, parenteral administration, high cost and relatively long treatment regimens. Thus, there is an urgent need for the development of new, better medicines (2). Leishmanial parasites have two major life cycle stages: the promastigote stage (inside the insect vector) and the amastigote stage (inside the mammalian host). The amastigotes reside in parasitophorous vacuoles, phagolysosome-like compartments with acidic and hydrolytic conditions (3). Leishmania amastigotes are exquisitely adapted to this environment and subvert many of the innate defense mechanisms of the host cell (4).
For the purpose of drug discovery, many different assays, using either promastigotes or amastigotes, have been developed to screen compound collections to identify new molecules that are active against Leishmania. The different assays all have advantages and drawbacks. The most straightforward assays use free-living parasites to assess the effect of compounds on cell viability. The main advantage of such assays is that they allow fast and easy screening of large compound collections, a feature that is often required for the successful identification of developable hits. Promastigotes have been used routinely for this purpose (5–10), but since they represent the insect life-stage, there is the risk of identifying compounds that do not affect the relevant disease-causing life-stage. An alternative is to use axenic amastigotes, i.e., amastigotes that have been adapted to grow outside their host cell in a growth medium that mimics the intracellular conditions (11–14). Such amastigotes can be used for straightforward high-throughput screening in a standard growth assay and have the advantage of being more similar to the disease-relevant parasite stage than promastigotes (15–17). However, several reports indicate that axenic amastigotes are different from intracellular amastigotes both in terms of protein expression and in terms of drug susceptibility (18–21). Thus, hits from screens with free-living parasites always have to be confirmed using an intramacrophage assay. Furthermore, compounds that interfere with the parasite-host interaction cannot be identified by such screens (22). With the advent of new technologies, it has now become possible to increase the throughput of the traditionally very labor-intensive intracellular amastigote assays. Two main methods are in use for the detection of intracellular parasites: plate-reader-based methods that rely on reporter constructs (23–25) and microscopy-based methods that count parasites directly (8, 26, 27). With the indirect reporter-based assays, there is a risk of artifacts; compounds may interfere with the reporter protein or with the substrate, and no information is obtained regarding the number of host cells or the distribution of amastigotes in macrophages since these are whole-well readout assays. Microscopy-based high-content technology enables direct counting and circumvent many of these problems. However, this method is complex and has only recently been described for primary screening of compound libraries (8, 26, 27). These publications show that screening with promastigotes results in a large set of hits that show no activity against the intracellular parasites (i.e., false positives) and that it is possible to identify compounds that show activity against intracellular amastigotes but not against free-living promastigotes. We present here an alternative 384-well microscopy-based assay, which we used to carry out a large-scale comparison to the axenic amastigote assay to determine the differences in hits identified by both platforms.
MATERIALS AND METHODS
Chemicals.
The following chemicals were obtained from Sigma: amphotericin B, ketoconazole, atovaquone, pentamidine, paromomycin, miltefosine, phorbol-12-myristate-13-acetate (PMA), 4′,6-diamidino-2-phenylindole (DAPI), formaldehyde, thimerosal, and resazurin. Nifurtimox was a gift from Bayer, Argentina. Nourseothricin (Lexsy NTC) was obtained from Jena Bioscience. Sodium stibogluconate was obtained from Merck. HCS Cellmask Deep Red was obtained from Invitrogen. The small-molecule library used in the present study contained 15,659 diverse compounds, dissolved at 10 mM in 100% dimethyl sulfoxide (DMSO). This library was assembled using the criteria for a diverse compound collection outlined elsewhere (28) and stored under low-oxygen and low-humidity conditions.
Cells and cell lines.
THP-1 (human monocytic leukemia) cells were obtained from the Health Protection Agency Culture Collections (catalog no. 88081201). The cells were screened for mycoplasma and maintained in RPMI plus 10% (vol/vol) heat-inactivated fetal bovine serum (FBS). MRC-5 pd30 (human fetal lung) cells were obtained from the Health Protection Agency Culture Collections (catalog no. 84101801) and tested for mycoplasma. The cells were maintained in minimal essential medium plus 10% (vol/vol) FBS. L. donovani (MHOM/SD/62/1S-CL2D, LdBOB) (11, 29) axenic amastigotes and promastigotes were maintained as described previously (29). Every 5 weeks, the parasites were cycled between developmental stages. For amastigote differentiation, 20 μl of metacyclic promastigote culture was inoculated in amastigote media. For the reverse process, 20 μl of dense amastigote culture was inoculated in promastigote media.
Cloning and expression of eGFP in LdBOB.
The enhanced green fluorescent protein (eGFP) gene from Aequorea victoria (Swiss-Prot accession no. AAB02576) was submitted to GenScript (Piscataway, NJ) for OptimumGene codon optimization to facilitate expression in Leishmania spp. The resulting synthetic gene (see Data File S1 in the supplemental material), cloned into pUC57, was flanked by SmaI sites at the 5′ and 3′ ends of the gene. The pUC57-eGFP construct was then digested with SmaI, and the fragment was cloned into the pIR1SAT expression vector (30), resulting in a pIR1SAT-eGFP construct. Mid-log-phase L. donovani promastigotes (WT, LdBOB) were transfected with pIR1SAT-eGFP using the human T cell Nucleofector kit and Nucleofector (Amaxa, program V-033). After transfection, the cells were allowed to grow 16 to 24 h in modified M199 medium (29) with 10% FBS prior to drug selection with nourseothricin (100 μg ml−1). Cloned cell lines were generated by limiting dilution and maintained in selective medium. Clones expressing high levels of eGFP were then selected using fluorescence microscopy and converted to axenic amastigotes for use in the screening assays.
Assays. (i) Axenic L. donovani assay.
LdBOB axenic amastigotes were incubated for 72 h with compounds, followed by a resazurin-based readout. A more detailed description of the assay can be found in the supplemental material.
(ii) Intramacrophage L. donovani assay.
An intracellular Leishmania assay using eGFP expressing LdBOB amastigotes was performed. PMA differentiated THP-1 cells were infected overnight with axenic amastigotes at a multiplicity of infection of 5. After infection, compounds were added to the plates, followed by a 3-day incubation and microscopy-based readout. Further details can be found in the supplemental material.
(iii) MRC-5 assay.
MRC-5 cells were incubated for 72 h with compounds, followed by a resazurin-based readout. A more detailed description of the assay can be found in the supplemental material.
Data analysis.
All data were processed using IDBS ActivityBase. Raw data were converted into percent inhibitions through linear regression by setting the high-inhibition control as 100% and the no-inhibition control as 0%. Quality control criteria for passing plates were as follows: z′ > 0.5, signal-to-background ratio (S:B) > 3, and percent coefficient of variation (%CV) of the no-inhibition control < 15. The formula used to calculate z′ is as follows: z′ = 1 − {[3 × (SDhigh + SDlow)]/[ABS(Meanhigh − Meanlow)]} (31), where SDhigh is the standard deviation of the maximum inhibition controls, SDlow is the standard deviation of the vehicle-treated controls, ABS stands for absolute value, Meanhigh is the mean of the maximum inhibition controls, and Meanlow is the mean of the vehicle-treated controls. Curve fitting was carried out using the following four-parameter equation: y = A + {[B − A]/[1 + (C/x)D]}, where A is the percent inhibition at the bottom, B is the percent inhibition at the top, C is the 50% effective concentration (EC50), D is the slope, x is the inhibitor concentration, and y is the percent inhibition. If curve definition was poor, B was fixed to 100. For the determination of the reference compound panel potency, all experiments were carried out with a minimum of three independent repeats.
RESULTS
High-content assay design.
Figure 1A shows an overview of the assay, a more detailed description of which can be found in Materials and Methods. Plates were imaged on an automated microscope, and the images were analyzed with an image analysis algorithm that we designed using GE Incell Investigator software (Fig. 1B and see also the supplemental material). Examples of curves obtained for the standard compounds amphotericin B and miltefosine are given in Fig. 1C. Note that the increase in THP-1 counts after treatment with 0.1 to 1 μM amphotericin B is a reproducible, compound-specific effect. The DMSO tolerance of the assay was determined, and there was no detectable effect of DMSO on the assay at 0.5% DMSO, the concentration routinely used in our screens (see Fig. S1 in the supplemental material).
Fig 1.

Intracellular Leishmania assay. (A) Graphical representation of assay protocol. The x axis represents the time in hours. Details regarding each step can be found in Materials and Methods. (B) Image analysis. Pseudocolored raw images are shown and marked with their respective dyes/fluorophores (CMdr, cytoplasm marker [HCS Cellmask Deep Red]). The result of image analysis is shown in the bottom right panel (cytoplasm, green outline; amastigotes, yellow outline). (C) Representative dose-response curves for miltefosine and amphotericin B. Ten-point potency curves for the indicated compounds were created and tested in the intracellular Leishmania assay. Potency values for the compounds are given in Table 2. The data are from at least four replicates, shown as purple diamonds, and the curve is fitted to the average values (blue circles). The data points for Leishmania inhibition that are omitted from the curve fit for miltefosine because of toxicity to THP-1 cells are marked as boxed crosses.
Assay characterization.
To further characterize the assay, the level of Leishmania infection was assessed by looking at the number of amastigotes per cell and the number of infected cells. Figure 2A shows a typical distribution for the number of amastigotes per macrophage at 72 h postinfection (mean = 7.2 and mode = 2 amastigotes/infected macrophage). The assay also allows determination of amastigote replication inside the host cell during the time frame of the assay. Replication was assessed over 7 days by counting the average number of amastigotes per macrophage at multiple time points. Since there is effectively no division of the differentiated THP-1 cells, this gives a good indication of intracellular parasite replication. Over the 7 days we only observed limited Leishmania growth, as shown in Fig. 2B (doubling time ∼ 12 days). In contrast, cell division is rapid and exponential in the axenic system (doubling time, 5.8 h, Fig. 2C).
Fig 2.

Assay characterization. (A) Frequency distribution of intracellular amastigote count. At the end of the intracellular Leishmania assay, the number of amastigotes in 19,738 infected cells was counted using the automated image analysis algorithm, and a frequency distribution was plotted. (B) Growth curve for intracellular amastigotes. Cells were plated and infected in 384-well plates, and one plate was fixed at each time point. The values represent the number of amastigotes per infected cell (i.e., the averages and standard deviations from the entire plate). A linear regression was calculated (R2 = 0.93). (C) Growth curve for axenic amastigotes in 384-well plates. Axenic amastigotes were counted at the indicated time points. The y axis shows the number of amastigotes per ml (×1,000). An exponential curve was fitted to time points 0 to 55 h (R2 = 0.999). The results from triplicate experiments are shown. (D) Replicate potencies. The potency of 85 compounds was tested on two separate occasions in the intracellular Leishmania assay, and the potencies of the replicates were plotted against each other. A linear regression was calculated (R2 = 0.88).
To assess the suitability of the assay for high-throughput screening, we determined standard screening statistics across a large number of plates (Table 1). The data show that the assay is robust and has low variability and a high S:B. We also monitored the potency of amphotericin B, which was always included as reference on potency plates, and found it to be highly reproducible. Using 384-well plates, we achieved a throughput of 7,680 wells per batch while maintaining high-quality performance statistics. The reproducibility of the assay was further assessed by carrying out a set of potency determinations on two separate occasions, and a high level of correlation was observed, as shown in Fig. 2D (R2 = 0.88, slope = 0.9).
Table 1.
Performance statistics of the intramacrophage assay
| Performance statistic | Avg ± SD (n)a |
|---|---|
| Z-factor | 0.68 ± 0.09 (518) |
| %CV | 9.8 ± 2.8 (518) |
| S:B | 90 ± 51 (518) |
| Amphotericin B potency (pEC50) | 6.74 ± 0.18 (41) |
| % Infected cells | 81 (19,738) |
| Throughput | 20 × 384 wells per batch |
Values are averages ± the standard deviation except as noted otherwise in column 1. n is the number of plates for the Z-factor, the percent coefficient of variation (%CV), and the signal-to-background ratio (S:B), the number of curves for amphotericin B potency, and the number of cells for the percent infected cells.
Reference compound panel.
A panel of reference compounds (amphotericin B, atovaquone, miltefosine, paromomycin, sodium stibogluconate, ketoconazole, pentamidine, and nifurtimox) was tested against intracellular amastigotes, as well as axenic amastigotes, and MRC-5 cells, a human cell-line used for initial toxicity determinations. The results are shown in Table 2 and are broadly in line with previously reported values (15, 20, 32, 33, 34). The clinically used compounds amphotericin B, miltefosine, and paromomycin show very similar activities against axenic and intracellular amastigotes, whereas most other compounds show a significant dropoff from the axenic to the intramacrophage assay (Fig. 3).
Table 2.
Potencies for reference compound panel
| Compound | pEC50 {−log(EC50[M])}a |
|||
|---|---|---|---|---|
| Axenic | INMAC | THP1 | MRC5 | |
| Amphotericin B | 7.06 (0.17) | 7.13 (0.05) | 5.68 (0.1) | 5.08 (0.12) |
| Miltefosine | 5.61 (0.2) | 6.03 (0.17) | 4.33 (0.08) | 4.3 (0.09) |
| Atovaquone | 5.37 (0.11) | 5.36 (0.12) | <3.8 | 4.56 (0.13) |
| Paromomycin | 5.29 (0.37) | 5.18 (0.14) | <2.5 | <2.5 |
| Ketoconazole | 7.57 (0.22) | 4.89 (0.1) | 4.33 (0.07) | 4.29 (0.07) |
| Pentamidine | 6.3 (0.25) | 4.88 (0.21) | 4.09 (0.07) | 4.8 (0.1) |
| Nifurtimox | 5.39 (0.34) | 4.62 (0.09) | <3.8 | 3.98 (0.06) |
| Sodium stibogluconate | <2.9 | 3.82 (0.17) | 2.98 (0.05) | 3.09 (0.13) |
Results are expressed as the pEC50 with standard deviations given in parentheses, except for sodium stibogluconate, where the result is given as the −log(EC50[μg of Sb/ml]).
Fig 3.

Comparison of reference compound activity between the axenic and intramacrophage assays. The fold differences between the potencies, as reported in Table 2, are displayed.
Axenic versus intramacrophage assay.
In order to compare the axenic L. donovani assay with the intramacrophage assay, we screened a diverse compound set (∼16,000 compounds) in both assays. For the axenic screen, a top concentration of 3 μM drug was used. Preliminary experiments with the intramacrophage assay showed that the hit rate at this concentration was very low, and instead we screened the set at 50 μM in this assay. Figure 4A shows the hit frequency distributions for both assays. Hits were defined as showing >70% inhibition of amastigote growth, with the additional criterion of <50% inhibition of THP-1 cell counts for the intramacrophage assay. In the axenic screen 381 hits were identified (a hit rate of 2.4% at 3 μM). In the intracellular assay, a total of 213 compounds showed >70% inhibition of the amastigote count (1.4% of the compounds at 50 μM). Of these, 128 showed >50% inhibition of THP-1 cell counts, thus leaving 85 hits and a hit rate of 0.5% (Fig. 4B). Only 17 compounds were hits in both the axenic and the intramacrophage screens, whereas 85 compounds showed activity against the THP-1 cells and the axenic amastigotes, indicating that these may be generally toxic compounds (Fig. 4C).
Fig 4.

Screening cascades. (A) Frequency distributions for 15,659 compound diverse library screen against axenic (left panel) and intracellular (center and right panels) Leishmania. The abscissa shows the percent inhibition (PI). Three populations are shown on each frequency diagram: negative controls (red), positive controls (green), and test compounds (blue). Positive controls on the left panel cannot be seen since they are fully covered by the blue line for the test compounds at 100%. (B) Venn diagram for intramacrophage primary screen hits. INMAC, compounds that showed >70% inhibition of intracellular Leishmania; THP-1, compounds that showed >50% inhibition of THP-1 cell count. (C) Comparison of hits from intramacrophage and axenic primary screen. THP-1, compounds that showed >50% inhibition of THP-1 cell count; axenic, compounds that showed >70% inhibition in the axenic primary screen; INMAC*, compounds that showed >70% inhibition of intracellular Leishmania and <50% inhibition of the THP-1 cell count. (D and E) Hit selection in the axenic (D) and intracellular (E) screening cascades. Numbers in green are compounds that progressed to the next step; numbers in red are compounds that were removed from the cascade (no activity, low activity, or toxicity). SP, single point primary screen; Pot, ten-point dose-response curve in duplicate; EC50, concentration at which a 50% effect is seen; tox, <10-fold window between antileishmanial EC50 and THP-1 EC50.
The results of screening cascades performed after the primary screens are shown in Fig. 4D and E. Hits from the axenic screen were first tested for potency in both the axenic model and the MRC-5 counterscreen. Compounds showing selectivity over the MRC-5 cells (319 compounds in total) were then progressed to potency testing in the macrophage assay. To quickly remove inactive compounds at this stage, we triaged compounds by assessing their potency using a single-replicate, 5-point, one-in-three dilution curve. Compounds showing activity were then further progressed to full 10-point duplicate potency testing. The majority of the axenic hits dropped out at this stage, showing no activity in the macrophage assay (278 compounds, 87% of the axenic hits). After application of selectivity rules comparing intramacrophage potency with THP-1 and MRC-5 cell activity, six hits remained.
The 85 hits from the primary intramacrophage screen were progressed into 10-point potency testing and MRC-5 counterscreening, after which 7 hits were retained. Of these, 6 were the same as the ones identified using the axenic cascade. The additional hit (compound 2) was not very potent against the intracellular amastigotes, with a pEC50 of 5 (EC50 × 10 μM). Since axenic potency testing was initially carried out with a top concentration of 16.6 μM, we may not have picked up such a low level of activity in the axenic primary screen. As a result, this compound was retested in the axenic assay with a top concentration of 50 μM and returned a potency value in the same range as seen in the intracellular assay (pEC50 = 4.7). The structures for all 7 hits from both screening campaigns with their potencies in the various assays are given in Table 3.
Table 3.
Hits identified in screening campaign
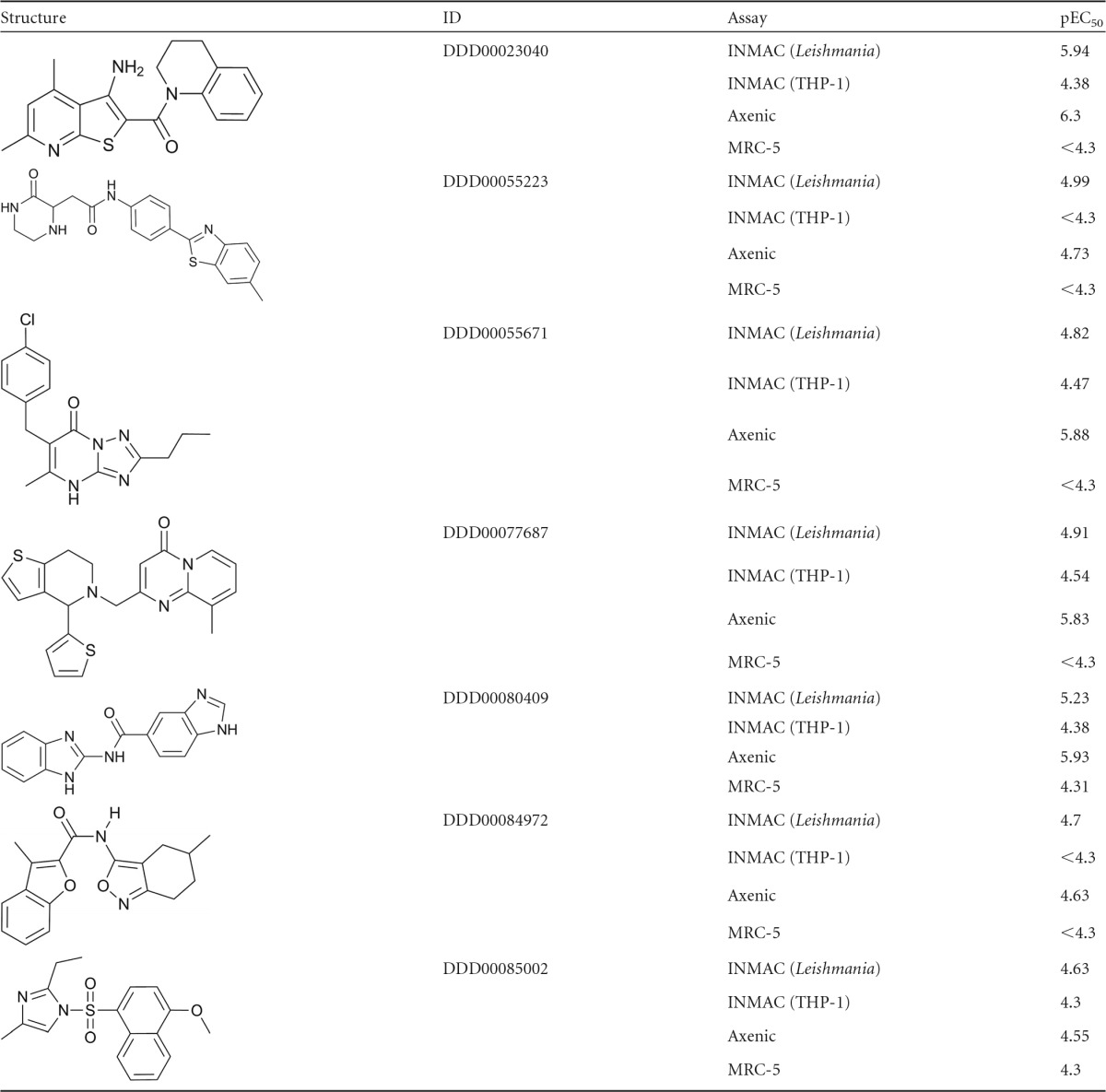
For a total of 110 compounds from this screen, we have carried out full 10-point potency determinations in both the axenic and the intramacrophage systems. Comparison of these shows that for the majority of compounds there was a significant reduction in potency from the axenic to the intramacrophage assay, with 80 compounds showing a >10-fold drop in activity from the axenic to the intramacrophage assay (includes compounds with no activity intramacrophage assay) (Fig. 5A). No compound was markedly more potent in the intramacrophage assay.
Fig 5.

(A) Comparison of compound potency in axenic and intracellular assays. Compounds on the blue line show equal activity in both assays, and compounds on the right-hand side of the line are more potent in the axenic assay. Red diamonds, no activity in intramacrophage assay (pEC50<4.78, 82 compounds); green squares, activity in both assays (28 compounds). (B) Comparison of compound potency against THP-1 cells and MRC-5 cells. Red diamonds, active against MRC-5 and no activity against THP-1 (pEC50<4.78) (47 compounds); green triangles, activity against both cell lines (10 compounds); blue squares, active against THP-1 and no activity against MRC-5 (pEC50 < 4.78) (4 compounds).
A total of 206 compounds were tested for potency in the intramacrophage Leishmania assay and the human counterscreen line MRC-5. Of these, 145 showed no activity against both THP-1 and MRC-5 cell lines. Comparison of the remaining 61 compounds revealed that, in general, the MRC-5 cells are more sensitive than the THP-1 cells, with 47 compounds showing no activity against THP-1 cells while being active against MRC-5 cells, and with 28 compounds being >3-fold more active against MRC-5 cells than THP-1 cells (Fig. 5B).
DISCUSSION
A 384-well intramacrophage Leishmania donovani assay.
There is an urgent need for new medicines for leishmaniasis. In terms of hit discovery, this parasitic disease poses a significant challenge, since the disease-causing stage resides in an acidic vacuole (the parasitophorous vacuole) inside the host's macrophages. High-content screening using automated microscopy provides a suitable platform for assessing such intracellular parasites, and recently both a 96-well and a 384-well high-content assay have been reported (26, 27). We have also developed a 384-well microscopy-based intracellular Leishmania assay with sufficient capacity to allow primary screening and potency follow-up of medium-sized small-molecule libraries. To achieve sufficient throughput, robustness, and reproducibility, a number of alterations were made to the traditional direct counting assay (reviewed in reference 35). Typically, ex vivo amastigotes are used to infect primary macrophages. However, this is difficult to achieve for a large-scale screening assay. Instead, we opted to use axenic amastigotes. The advantage of axenic amastigotes over metacyclic promastigotes is that they do not elicit an oxidative burst when entering the macrophage (4). This oxidative burst may result in the killing or sensitizing of the phagocytosed promastigotes, which may potentiate drug action. In the host, most spreading of the disease occurs through amastigotes and, as such, they may provide a better disease model (36). We used L. donovani strain 1S-CL2D, which was shown to retain in vivo infectivity and exhibits characteristics of ex vivo amastigotes (11). In terms of the mammalian host cell, it would be preferable to use primary macrophages, ideally of human origin. However, it is difficult to obtain a reliable and homogeneous supply at a scale suitable for routine screening. Instead, PMA-differentiated THP-1 cells were chosen, which are considered a suitable model for human macrophages (37–39). The use of nonprimary cells for our assay necessitates follow-up experiments using ex vivo amastigotes and primary macrophages during the hit-to-lead stage of a compound series since cell-line-specific effects have been described both for Leishmania strains and host cell lines (32, 40).
A key challenge in developing the intramacrophage assay proved to be the optimization of the washing step after infection. Since the axenic amastigotes were able to proliferate outside the macrophages, it was crucial to remove the majority of amastigotes that had not been phagocytosed without damaging the THP-1 cells. They would otherwise provide a reservoir for continuous infection, which would make interpretation of the results difficult. In addition, their presence would complicate the imaging of the intracellular parasites. As detailed above, there are two significant differences with the previously published 384-well high-content Leishmania assay (27): the use of axenic amastigotes for infection instead of metacyclic promastigotes and the removal of the infection inoculum by a washing step. Whether either of these changes has a major impact on the relevance of the assay is difficult to say without careful side-by-side comparison, but it is worth pointing out that the clinically used compounds paromomycin and sodium stibogluconate show activity in the assay presented here, while they do not in the assay described by Siqueira-Neto et al. (27). This may indicate a higher physiological relevance for the assay presented here.
Advantages of a high-content approach.
A microscopy-based assay, as presented here, has many advantages over plate-reader-based assays. A microscopy-based assay mimics the traditional direct-counting assay and provides cell-by-cell analysis. This allows the determination of parameters such as the number of infected cells and the number of amastigotes per individual cell. This information provides a more direct understanding of the assay results and reduces the occurrence of artifacts. The assay also reports the number of THP-1 cells that may give a first indication of toxicity of the compounds tested. However, since differentiated THP-1 cells are less sensitive to compound toxicity than actively growing human cells, a secondary mammalian counterscreen is still advisable (see Fig. 5B). For correct interpretation of the Leishmania inhibition data, it is essential to take into account the THP-1 cell count, since the Leishmania percent inhibition value becomes unreliable at drug concentrations that kill the majority of the macrophages. This is demonstrated at the top concentration of the miltefosine dose-response curve shown in Fig. 1C. This figure also reveals that amphotericin B has a beneficial effect on the number of THP-1 cells in the 0.1 to 1 μM range. This could potentially be explained by the known immunomodulatory effects of amphotericin B on THP-1 cells (41) or its ability to activate the survival- and growth-promoting kinase Akt/PKB (42, 43).
Although the high-content assays discussed here provide a large amount of information, they do not take into account the host microenvironment, which could influence parasite behavior and drug action. Recently, a study demonstrated that, by using splenic explants and a reporter gene, it is possible to further improve the physiological relevance of the intracellular assay, albeit at a lower throughput (44).
Comparison of intracellular assay with axenic amastigote assay.
Other groups have shown that assaying free-living promastigotes results in high false-positive rates (i.e., the promastigote activity does not translate into activity against intracellular amastigotes) and may not identify intracellular stage-specific compounds (8, 26, 27, 44). Here, we compared the intracellular assay to the axenic assay, which has been proposed as an alternative model to promastigotes for primary screening (15, 17, 45). The rationale behind this comparison was to see whether the axenic assay has a place as a primary screening assay to allow for a higher throughput, followed by confirmation in the macrophage assay. In the first instance, we tested a panel of reference compounds in both assays. We found that all clinically used compounds show similar activity in the axenic and intracellular assays, except for sodium stibogluconate, which shows no activity in our axenic assay (Table 2). The literature contains a lot of discussion, and conflicting results, regarding the effect of pentavalent antimony (SbV) on axenic amastigotes, with some groups showing activity against all stages (46) and others showing activity against axenic and intracellular amastigotes but not promastigotes (15, 47, 48), while others showed activity only against intracellular amastigotes (20, 36, 49). The evidence indicates that the reduction of SbV to trivalent antimony (SbIII) is critical for toxicity and that this only happens in fully differentiated amastigotes, so it is possible that our axenic amastigotes retain some promastigote traits, including an inability to reduce SbV.
This small panel of reference compounds does not allow us to draw any conclusions regarding the general relationship between the axenic and intracellular assays. To do this, we carried out a parallel primary screen of a library of nearly 16,000 diverse compounds in both systems. We observed high hit rates in the axenic assay compared to the intracellular assay, even though we screened at much lower compound concentrations. As expected, many of the axenic hits did not show activity in the intracellular assay, and the compounds that did show activity across both platforms showed a significant dropoff of activity from the axenic assay to the intracellular assay, without any clear correlation. After progressing the compounds through the two screening cascades (Fig. 4D and E), we obtained the same hit compounds. Our observations allowed us to draw several important conclusions. (i) The axenic assay is much more sensitive than the intracellular assay, and the lack of correlation between the two assays limits the use of the axenic assay as a predictor of intracellular activity. (ii) The use of the axenic assay as a primary platform, followed by intracellular follow-up, is highly inefficient. The cascade takes a lot longer (7 weeks versus 4 weeks), and the majority of the compounds that progressed to the intracellular assay showed no activity, so that valuable throughput is wasted. (iii) When stringent potency criteria are applied to axenic hits before progression to the intracellular assay, there is a significant risk of losing interesting compounds, since there is no correlation between axenic and intracellular potency. Thus, it seems clear that running the intracellular assay as a primary platform is the preferred route for finding new hits.
Assay type versus compound mode of action.
It is worth considering why so few axenic hits translate into intracellular activity. There are inherent differences between the transcriptomes and proteomes of axenic and intracellular amastigotes (19, 50), and these could affect drug action. Two other obvious differences are rate of replication and free-living versus intracellular localization. In the assays described here, we see fast exponential growth of the axenic amastigotes and almost no growth of the intracellular amastigotes (Fig. 2C and D). This is likely to have a dramatic effect on their respective drug sensitivities, with the replicating cells being much more sensitive to inhibitors of cell division, energy metabolism, etc. In addition, a starting density significantly below the detection limit is used in the axenic assay because of the high growth rate. This results in the assay not only identifying cytocidal compounds but also growth-slowing compounds (51), which will also contribute to the high false-positive rate, since only cytocidal compounds were identified in our intramacrophage assay. We anticipate that to achieve the current target product profile for visceral leishmaniasis compounds need to be cytocidal rather than static or growth slowing (52). Comparing in vitro growth to the situation in vivo is difficult, since many other factors play a role (in particular, the clearance of amastigotes by the immune system and differences between rodent and human physiology). Bradley and Kirkley (53) reported an ∼20-fold increase in tissue parasite counts over the first 8 days of infection in mice, which means that a minimum of four rounds of cell division take place during this window. Hence, the rate of amastigote division in vivo is likely to be somewhere in between what we see in the intramacrophage and axenic assays. When considering false-positive rates for the axenic assay, it is important to keep in mind that these rates should be based on the gold standard comparator. Here, we used the intracellular Leishmania assay as the comparator, but it would be better to compare the findings to those obtained in an in vivo animal model (or to human clinical data). However, we did not have sufficient compounds with appropriate efficacy and absorption, distribution, metabolism, and excretion properties to carry out such a comparison.
The localization of the parasites inside the parasitophorous vacuole presents a serious hurdle for small molecules; to exert their action, they need to cross three membranes and do this at both neutral and acidic pH. This means that even if a compound could kill intracellular amastigotes, it may not be able to reach them. Although such compounds will not be identified using the intracellular assay as a primary platform, they may nevertheless be of interest since further chemical optimization may allow these molecules to reach the vacuole and exert their effect, or alternative delivery strategies could be used. It is likely that for these reasons the intracellular assay may not detect a subset of potentially interesting compounds and, in view of the dearth of new leads for leishmaniasis, we may have to consider alternative assays that will yield more hits. To understand the effect of the fast axenic growth on the hit rate in this assay, it would be very informative to devise an axenic amastigote assay under much less stimulating growth conditions. Such an assay may have lower false-positive rates and could be of use for high-throughput screening. It would also allow the use of a higher starting density so that it only reports cidal compounds, again potentially reducing the false-positive rate.
Conclusion.
Combining high throughput with physiological relevance is a challenge for organisms with a complicated life cycle such as Leishmania. Ultimately, the only relevant readout is success in the clinic. The intramacrophage assay presented here reports activity for all clinically used compounds and, as such, appears to be a good in vitro model for predicting clinical activity. The acquisition of clinical data for new drugs that derive from the current leishmaniasis drug discovery drive will allow further assessment of the suitability of this assay as a primary drug discovery platform.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniel James for data management, Michael Thomas for help with preparing Table 3, and Mascha Brinkkötter for assistance in the generation of the L. donovani fluorescent reporter strain. We thank Jean-Robert Ioset and Eric Chatelain at DNDi for their advice and for critically reading the manuscript.
This study was funded by the Drugs for Neglected Diseases Initiative, through grants from several donors (Department for International Development, United Kingdom; Swiss Agency for Development and Cooperation, Switzerland; Médecins Sans Frontières/Doctors Without Borders; International and Gesellschaft für Technische Zusammenarbeit, Germany). This study was also supported by Wellcome Trust strategic award 083481. A.H.F., S.W., and S.C. are supported by the Wellcome Trust (079838 and 083481).
Footnotes
Published ahead of print 9 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02398-12.
REFERENCES
- 1. World Health Organization 2010. Control of the leishmaniases. World Health Organ. Tech. Rep. Ser. 2010:xii–xiii; 1–186; back cover [PubMed] [Google Scholar]
- 2. Singh N, Kumar M, Singh RK. 2012. Leishmaniasis: current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 5:485–497 [DOI] [PubMed] [Google Scholar]
- 3. Burchmore RJ, Barrett MP. 2001. Life in vacuoles–nutrient acquisition by Leishmania amastigotes. Int. J. Parasitol. 31:1311–1320 [DOI] [PubMed] [Google Scholar]
- 4. Kima PE. 2007. The amastigote forms of Leishmania are experts at exploiting host cell processes to establish infection and persist. Int. J. Parasitol. 37:1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bodley AL, McGarry MW, Shapiro TA. 1995. Drug cytotoxicity assay for African trypanosomes and Leishmania species. J. Infect. Dis. 172:1157–1159 [DOI] [PubMed] [Google Scholar]
- 6. Mikus J, Steverding D. 2000. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar blue. Parasitol. Int. 48:265–269 [DOI] [PubMed] [Google Scholar]
- 7. Nolan LL, Bouchard B. 1991. A rapid in vitro system for screening the effect of experimental compounds on nonadhering cell lines. Curr. Microbiol. 23:277–279 [Google Scholar]
- 8. Siqueira-Neto JL, Song OR, Oh H, Sohn JH, Yang G, Nam J, Jang J, Cechetto J, Lee CB, Moon S, Genovesio A, Chatelain E, Christophe T, Freitas-Junior LH. 2010. Antileishmanial high-throughput drug screening reveals drug candidates with new scaffolds. PLoS Negl. Trop. Dis. 4:e675. 10.1371/journal.pntd.0000675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. St George S, Bishop JV, Titus RG, Selitrennikoff CP. 2006. Novel compounds active against Leishmania major. Antimicrob. Agents Chemother. 50:474–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharlow ER, Close D, Shun T, Leimgruber S, Reed R, Mustata G, Wipf P, Johnson J, O'Neil M, Grogl M, Magill AJ, Lazo JS. 2009. Identification of potent chemotypes targeting Leishmania major using a high-throughput, low-stringency, computationally enhanced, small molecule screen. PLoS Negl. Trop. Dis. 3:e540. 10.1371/journal.pntd.0000540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Debrabant A, Joshi MB, Pimenta PF, Dwyer DM. 2004. Generation of Leishmania donovani axenic amastigotes: their growth and biological characteristics. Int. J. Parasitol. 34:205–217 [DOI] [PubMed] [Google Scholar]
- 12. Bates PA. 1993. Axenic culture of Leishmania amastigotes. Parasitol. Today 9:143–146 [DOI] [PubMed] [Google Scholar]
- 13. Gupta N, Goyal N, Rastogi AK. 2001. In vitro cultivation and characterization of axenic amastigotes of Leishmania. Trends Parasitol. 17:150–153 [DOI] [PubMed] [Google Scholar]
- 14. Teixeira MC, de Jesus Santos R, Sampaio RB, Pontes-de-Carvalho L, dos Santos WL. 2002. A simple and reproducible method to obtain large numbers of axenic amastigotes of different Leishmania species. Parasitol. Res. 88:963–968 [DOI] [PubMed] [Google Scholar]
- 15. Callahan HL, Portal AC, Devereaux R, Grogl M. 1997. An axenic amastigote system for drug screening. Antimicrob. Agents Chemother. 41:818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monte-Alegre A, Ouaissi A, Sereno D. 2006. Leishmania amastigotes as targets for drug screening. Kinetoplastid Biol. Dis. 5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimony O, Jaffe CL. 2008. Rapid fluorescent assay for screening drugs on Leishmania amastigotes. J. Microbiol. Methods 75:196–200 [DOI] [PubMed] [Google Scholar]
- 18. Holzer TR, McMaster WR, Forney JD. 2006. Expression profiling by whole-genome interspecies microarray hybridization reveals differential gene expression in procyclic promastigotes, lesion-derived amastigotes, and axenic amastigotes in Leishmania mexicana. Mol. Biochem. Parasitol. 146:198–218 [DOI] [PubMed] [Google Scholar]
- 19. Pescher P, Blisnick T, Bastin P, Spath GF. 2011. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cell Microbiol. 13:978–991 [DOI] [PubMed] [Google Scholar]
- 20. Vermeersch M, da Luz RI, Tote K, Timmermans JP, Cos P, Maes L. 2009. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 53:3855–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Q., Zhao Y, Ni B, Yao C, Zhou Y, Xu W, Wang Z, Qiao Z. 2008. Comparison of the expression profiles of promastigotes and axenic amastigotes in Leishmania donovani using serial analysis of gene expression. Parasitol. Res. 103:821–828 [DOI] [PubMed] [Google Scholar]
- 22. Sereno D, Cordeiro da Silva A, Mathieu-Daude F, Ouaissi A. 2007. Advances and perspectives in Leishmania cell based drug-screening procedures. Parasitol. Int. 56:3–7 [DOI] [PubMed] [Google Scholar]
- 23. Buckner FS, Wilson AJ. 2005. Colorimetric assay for screening compounds against Leishmania amastigotes grown in macrophages. Am. J. Trop. Med. Hyg. 72:600–605 [PubMed] [Google Scholar]
- 24. Lang T, Goyard S, Lebastard M, Milon G. 2005. Bioluminescent Leishmania expressing luciferase for rapid and high throughput screening of drugs acting on amastigote-harboring macrophages and for quantitative real-time monitoring of parasitism features in living mice. Cell. Microbiol. 7:383–392 [DOI] [PubMed] [Google Scholar]
- 25. Mandal S, Maharjan M, Ganguly S, Chatterjee M, Singh S, Buckner FS, Madhubala R. 2009. High-throughput screening of amastigotes of Leishmania donovani clinical isolates against drugs using a colorimetric beta-lactamase assay. Indian J. Exp. Biol. 47:475–479 [PMC free article] [PubMed] [Google Scholar]
- 26. De Muylder G, Ang KK, Chen S, Arkin MR, Engel JC, McKerrow JH. 2011. A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 5:e1253. 10.1371/journal.pntd.0001253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siqueira-Neto JL, Moon S, Jang J, Yang G, Lee C, Moon HK, Chatelain E, Genovesio A, Cechetto J, Freitas-Junior LH. 2012. An image-based high-content screening assay for compounds targeting intracellular Leishmania donovani amastigotes in human macrophages. PLoS Negl. Trop. Dis. 6:e1671. 10.1371/journal.pntd.0001671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. 2008. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3:435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goyard S, Segawa H, Gordon J, Showalter M, Duncan R, Turco SJ, Beverley SM. 2003. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 130:31–42 [DOI] [PubMed] [Google Scholar]
- 30. Gaur U, Showalter M, Hickerson S, Dalvi R, Turco SJ, Wilson ME, Beverley SM. 2009. Leishmania donovani lacking the Golgi GDP-Man transporter LPG2 exhibit attenuated virulence in mammalian hosts. Exp. Parasitol. 122:182–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
- 32. Seifert K, Escobar P, Croft SL. 2010. In vitro activity of anti-leishmanial drugs against Leishmania donovani is host cell dependent. J. Antimicrob. Chemother. 65:508–511 [DOI] [PubMed] [Google Scholar]
- 33. Wang MZ, Zhu X, Srivastava A, Liu Q, Sweat JM, Pandharkar T, Stephens CE, Riccio E, Parman T, Munde M, Mandal S, Madhubala R, Tidwell RR, Wilson WD, Boykin DW, Hall JE, Kyle DE, Werbovetz KA. 2010. Novel arylimidamides for treatment of visceral leishmaniasis. Antimicrob. Agents Chemother. 54:2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yardley V, Croft SL. 2000. A comparison of the activities of three amphotericin B lipid formulations against experimental visceral and cutaneous leishmaniasis. Int. J. Antimicrob. Agents 13:243–248 [DOI] [PubMed] [Google Scholar]
- 35. Gupta S. 2011. Visceral leishmaniasis: experimental models for drug discovery. Indian J. Med. Res. 133:27–39 [PMC free article] [PubMed] [Google Scholar]
- 36. Sereno D, Cavaleyra M, Zemzoumi K, Maquaire S, Ouaissi A, Lemesre JL. 1998. Axenically grown amastigotes of Leishmania infantum used as an in vitro model to investigate the pentavalent antimony mode of action. Antimicrob. Agents Chemother. 42:3097–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Auwerx J. 1991. The human leukemia cell line, THP-1: a multifaceted model for the study of monocyte-macrophage differentiation. Experientia 47:22–31 [DOI] [PubMed] [Google Scholar]
- 38. Gebre-Hiwot A, Tadesse G, Croft SL, Frommel D. 1992. An in vitro model for screening antileishmanial drugs: the human leukaemia monocyte cell line, THP-1. Acta Trop. 51:237–245 [DOI] [PubMed] [Google Scholar]
- 39. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. 1980. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. cancer 26:171–176 [DOI] [PubMed] [Google Scholar]
- 40. Fernandez O, Diaz-Toro Y, Valderrama L, Ovalle C, Valderrama M, Castillo H, Perez M, Saravia NG. 2012. Novel approach to in vitro drug susceptibility assessment of clinical strains of Leishmania spp. J. Clin. Microbiol. 50:2207–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rogers PD, Stiles JK, Chapman SW, Cleary JD. 2000. Amphotericin B induces expression of genes encoding chemokines and cell adhesion molecules in the human monocytic cell line THP-1. J. Infect. Dis. 182:1280–1283 [DOI] [PubMed] [Google Scholar]
- 42. Gao Y, Deng K, Cao Z, Graziani EI, Gilbert AM, Koehn FE, Wood A, Doherty P, Walsh FS. 2010. Amphotericin B, identified from a natural product screen, antagonizes CNS inhibitors to promote axon growth via activation of an Akt pathway in neurons. J. Neurochem. 113:1331–1342 [DOI] [PubMed] [Google Scholar]
- 43. Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Osorio Y, Travi BL, Renslo AR, Peniche AG, Melby PC. 2011. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 5:e962. 10.1371/journal.pntd.0000962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sereno D, Lemesre JL. 1997. Axenically cultured amastigote forms as an in vitro model for investigation of antileishmanial agents. Antimicrob. Agents Chemother. 41:972–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carrio J, de Colmenares M, Riera C, Gallego M, Arboix M, Portus M. 2000. Leishmania infantum: stage-specific activity of pentavalent antimony related with the assay conditions. Exp. Parasitol. 95:209–214 [DOI] [PubMed] [Google Scholar]
- 47. Ephros M, Bitnun A, Shaked P, Waldman E, Zilberstein D. 1999. Stage-specific activity of pentavalent antimony against Leishmania donovani axenic amastigotes. Antimicrob. Agents Chemother. 43:278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shaked-Mishan P, Ulrich N, Ephros M, Zilberstein D. 2001. Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 276:3971–3976 [DOI] [PubMed] [Google Scholar]
- 49. Mookerjee Basu J, Mookerjee A, Sen P, Bhaumik S, Banerjee S, Naskar K, Choudhuri SK, Saha B, Raha S, Roy S. 2006. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob. Agents Chemother. 50:1788–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rochette A, Raymond F, Corbeil J, Ouellette M, Papadopoulou B. 2009. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Mol. Biochem. Parasitol. 165:32–47 [DOI] [PubMed] [Google Scholar]
- 51. De Rycker M, O'Neill S, Joshi D, Campbell L, Gray DW, Fairlamb AH. 2012. A static-cidal assay for Trypanosoma brucei to aid hit prioritization for progression into drug discovery programmes. PLoS Negl. Trop. Dis. 6:e1932. 10.1371/journal.pntd.0001932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Freitas-Junior LH, Chatelain E, Kim HA, Siqueira-Neto JL. 2012. Visceral leishmaniasis treatment: what do we have, what do we need and how to deliver it? Int. J. Parasitol. Drugs Drug Resist. 2:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bradley DJ, Kirkley J. 1977. Regulation of Leishmania populations within the host. I. the variable course of Leishmania donovani infections in mice. Clin. Exp. Immunol. 30:119–129 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.