

Abstract
With increasing resistance to existing antimalarials, there is an urgent need to discover new drugs at affordable prices for countries in which malaria is endemic. One approach to the development of new antimalarial drugs is to improve upon existing antimalarial agents, such as the tetracyclines. Tetracyclines exhibit potent, albeit relatively slow, action against malaria parasites, and doxycycline is used for both treatment (with other agents) and prevention of malaria. We synthesized 18 novel 7-position modified tetracycline derivatives and screened them for activity against cultured malaria parasites. Compounds with potent in vitro activity and other favorable drug properties were further tested in a rodent malaria model. Ten compounds inhibited the development of cultured Plasmodium falciparum with a 50% inhibitory concentration (IC50) after 96 h of incubation of <30 nM, demonstrating activity markedly superior to that of doxycycline (IC50 at 96 h of 320 nM). Most compounds showed little mammalian cell cytotoxicity and no evidence of in vitro phototoxicity. In a murine Plasmodium berghei model, 13 compounds demonstrated improved activity relative to that of doxycycline. In summary, 7-position modified tetracyclines offer improved activity against malaria parasites compared to doxycycline. Optimized compounds may allow lower doses for treatment and chemoprophylaxis. If safety margins are adequate, dosing in children, the group at greatest risk for malaria in countries in which it is endemic, may be feasible.
INTRODUCTION
Malaria causes hundreds of millions of illnesses and hundreds of thousands of deaths each year (1, 2). The vast majority of cases of malaria occur in developing countries, in particular many parts of Africa, Asia, Oceania, and South America. A major problem in the treatment and control of malaria, in particular that caused by Plasmodium falciparum, the most virulent human malaria parasite, is increasing resistance to available low-cost drugs (3). Resistance to older drugs, in particular chloroquine and sulfadoxine-pyrimethamine, is now widespread, such that these drugs are no longer appropriate for either the treatment or prevention of falciparum malaria. The new standard of care for the treatment of malaria is artemisinin-based combination therapy (4). A number of these regimens are highly efficacious, but their long-term utility is challenged by early signs of decreasing artemisinin efficacy in southeast Asia (5, 6). For prevention, there is renewed interest in the intermittent or regular use of drugs to protect against malaria in populations in which it is endemic (7), but the most commonly used antifolates are limited by drug resistance, optimal regimens are uncertain, and standard drugs used to prevent malaria in nonimmune travelers are probably not appropriate due to excessive cost (atovaquone-proguanil), poor tolerability (mefloquine), or unacceptability in young children (doxycycline).
Tetracyclines are active against a wide range of infectious pathogens. In limited studies, tetracycline and some tetracycline analogs were active against cultured malaria parasites (8, 9) and in murine malaria models (10). Despite limited experimental data, tetracycline and doxycycline (Fig. 1) were used for the treatment of malaria early after their introduction as antibacterial agents (11, 12), and they have become a component of some standard antimalarial regimens (13). In the United States, a standard therapy for falciparum malaria is quinine or intravenous quinidine plus doxycycline (14). Doxycycline is also recommended by the Centers for Disease Control and Prevention for chemoprophylaxis against malaria for travelers to regions where malaria is endemic, especially to areas where high levels of resistance to other agents are found (13).
Fig 1.
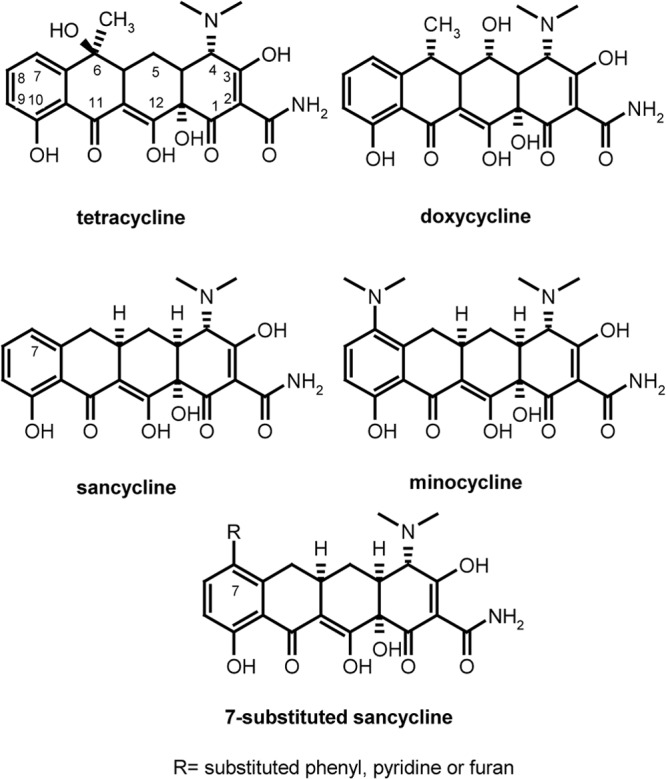
Structures of tetracyclines.
A number of features suggest that novel tetracyclines are well suited for inclusion in new combination regimens for the treatment of malaria. First, tetracyclines already have a proven record of safety and efficacy when used to treat bacterial infections and malaria. Second, there is currently no known clinical resistance to tetracyclines in P. falciparum or other malarial species. Third, in studies designed to select for resistance to minocycline in mice, resistance development was much slower than that to the antimalarials chloroquine, quinine, and pyrimethamine, suggesting that an improved tetracycline for malaria would have a long therapeutic lifetime (10). Finally, studies which examined the pharmacodynamic interaction of tetracyclines with artemisinin demonstrated synergism between these drugs (15). A key limitation of available tetracyclines for malaria is that they are typically contraindicated in children and pregnant women, the two groups at greatest risk of malaria in the developing world. However, recent studies have shown tetracyclines to be relatively safe in children, with dental, gastrointestinal, and dermatologic toxicity that can be limited by appropriate usage and dosing (16–19). Indeed, the American and Canadian Academies of Pediatrics now recommend doxycycline as first-line therapy for suspected or proven human granulocytic anaplasmosis, human monocytic ehrlichiosis, and Rocky Mountain spotted fever. These findings suggest that a new, more potent antimalarial tetracycline could be used safely at low doses in children. To begin to assess the antimalarial potential of new tetracyclines, we assessed the in vitro and in vivo activity of novel tetracycline derivatives. Here, we identify new antimalarial tetracyclines developed through the application of semisynthetic reactions to a sancycline core (Fig. 1) that improve upon the in vitro and, in select cases, in vivo antimalarial potency of standard tetracyclines.
MATERIALS AND METHODS
Natural and new synthetic tetracyclines.
Chemistries for the synthesis of novel tetracyclines have been previously described (20). Reagents and catalysts were used without further purification. Reactions were monitored by high-performance liquid chromatography (HPLC) with UV at 280 nm, using C18 reverse-phase 4-μm (50 by 4.5 mm) analytical columns. Compounds were purified by preparative HPLC separations. 1H nuclear magnetic resonance (NMR) spectra were recorded at 300 or 400 MHz, and the chemical shift values are expressed in δ values. Low (electron spray ionization)- and high (fast atom bombardment [FAB])-resolution spectra were recorded, while high-resolution electron impact mass spectra were via FAB analysis.
In vitro antimalarial activity assay.
In vitro activity assays were performed using the W2 (chloroquine-resistant) strain (21) of P. falciparum and a fluorescence-activated cell sorter (FACS)-based analysis of parasitemia (22). This assay, developed with modifications of published methods (23), uses FACS and nuclear staining of parasites to automate parasite counting after incubation with test compounds. Based on the known delayed activity of tetracycline against P. falciparum (24), incubations with test compounds were continued for two full parasite life cycles. Parasites cultured by standard methods were incubated with control compounds and experimental tetracyclines for 96 h, beginning at the ring stage. Parasites were then fixed with 1% formaldehyde in phosphate-buffered saline (PBS), pH 7.4, for 48 h at room temperature and then labeled with YOYO-1 (1 nM; Molecular Probes) in 0.1% Triton X-100 in PBS. Parasitemias were determined from dot plots (forward scatter versus fluorescence) acquired on a FACSort flow cytometer using CellQuest software (Becton, Dickinson). For 50% inhibitory concentration (IC50) determinations, dose-response studies were performed, values were normalized to percent control activity, and IC50s were calculated using Prism 3.0 (GraphPad Software) with data fitted by nonlinear regression. Goodness of curve fit was documented by R2 values of >0.95.
Mammalian cell cytotoxicity assay.
Nonspecific mammalian cell toxicity of the test compounds was examined using a standard nontoxic redox dye (resazurin; alamarBlue reagent) (25). At the onset of an experiment, cultures of mammalian COS-1 cells were washed, trypsinized, and harvested. Cell suspensions were prepared, seeded into 96-well black-walled microtiter plates, and incubated overnight at 37°C with 5% CO2 and approximately 95% humidity. The next day, serial dilutions of test compounds were added, and these were incubated for 24 h. Medium was removed, 50 μl of resazurin was added, and plates were incubated for 2 h in light and then 30 min in the dark at room temperature. Fluorescence was then measured (excitation, 535 nm; emission, 590 nm) and toxic effects in treated versus control cells were compared based on the degree of fluorescence in each well.
Phototoxicity assay.
Phototoxicity was measured using a modification of a previously reported protocol (26). Briefly, mouse NIH 3T3 fibroblasts were harvested and plated at 1 × 105 cells/ml. Plates were incubated overnight at 37°C with 5% CO2 and approximately 95% humidity. On the following day, medium was replaced with Hanks' balanced salt solution (HBSS). Drugs were diluted in HBSS and added to the plates. Plates were then incubated in darkness (for controls) or under UV light (1.6 to 1.8 mW/cm2) for 50 min. Cells then were washed with HBSS, fresh medium was added, and plates were incubated overnight as described above. On the following day, neutral red was added as an indicator of cell viability, plates were incubated for an additional 3 h, and then cells were washed with HBSS, blotted on absorbent paper, and incubated with 50% ethanol, 10% glacial acetic acid for 20 min. Absorbance was read at 535 nm using a Victor 5 spectrophotometer.
In vitro antibacterial susceptibility testing.
MICs of tetracyclines were determined according to Clinical and Laboratory Standards Institute (CLSI) methodology. Serial dilutions of compounds were prepared in microdilution plates using a Tecan robotic workstation. Mueller-Hinton broth cultures of Gram-negative (Escherichia coli strain ML308-225) and Gram-positive (Staphylococcus aureus strain RN450) bacterial strains were adjusted to match the turbidity of a 0.5 McFarland standard. Dilutions (1:200) were made in appropriate broth. Plates were incubated at 35°C in ambient air for 18 to 24 h, read spectrophotometrically, and checked manually for evidence of bacterial growth. The lowest dilution of compound that inhibited growth was recorded as the MIC.
Evaluation of antimalarial efficacy in an early-stage P. berghei model of murine malaria.
Female Swiss Webster mice (average of 20 g body weight) were infected by intraperitoneal injection with 1 × 106 Plasmodium berghei (NK65)-infected erythrocytes collected from a previously infected animal. For screening experiments, animals were treated with once-daily intraperitoneal injection at a single concentration of test compounds beginning 12 h after initial infection for a total of 4 days. All compounds were dissolved in PBS and administered in a total volume of 100 μl. Giemsa-stained blood smears were made from tail vein bleeds, and parasitemias were counted using a microscope. All counts within a single experiment were performed by the same investigator. When parasitemias exceeded 50%, animals were euthanized. No mice developed signs or symptoms suggestive of drug toxicity.
In animal survival experiments, mice were treated twice daily by oral gavage with various concentrations of test compound beginning 12 h after initial infection, for a total of 4 days. All compounds were dissolved in PBS and administered in a total volume of 100 μl. Animal survival and morbidity was monitored for 15 days postinfection. On days animals were found moribund, they were sacrificed and recorded as dead. Survival was evaluated by calculating the 50% protective dose (PD50) on two early endpoints, days 10 and 15 postinfection. The PD50 is defined as the daily dose required to achieve 50% survival and is estimated by the formula y = 1/{1 + 10[log(k) − log(x)] × 4.2}, where k = 0.5, by nonlinear regression analysis with Prism 3.0 software. All animal protocols were critically reviewed and approved by the University of California, San Francisco, Institutional Animal Care and Use Committee.
Pharmacokinetic analysis.
MAL5166 and doxycycline were formulated in sterile water for intravenous (i.v.) and oral (p.o.) dosing. Mice were given a single i.v. or p.o. dose of 10 mg/kg MAL5166 or doxycycline, and plasma was drawn at 0.0833, 0.166, 0.25, 0.5, 1, 2, 4, 8, and 16 h after dosing. Cynomolgus monkeys were given a single i.v. or p.o. dose of 5 mg/kg of MAL5166 or doxycycline, and plasma was drawn at 0, 0.083, 0.25, 0.5, 1, 3, 5, 7, 16, 24, and 48 h after dosing. Compounds were extracted from the plasma matrix with acetonitrile–0.1% formic acid. The extract was taken to dryness and reconstituted in methanol-water (1:1). Samples were analyzed on an API 3000 liquid chromatography-tandem mass spectrometer (LC-MS/MS) (Applied Biosystems) in positive ion mode using minocycline as the internal standard. All quantitative values were calculated using Analyst 1.2 software (Applied Biosystems). Statistical analyses were employed to generate means, standard deviations, coefficients of variation, and regression parameters where applicable. All pharmacokinetic parameters were calculated using WinNonlin 4.0.1 utilizing a noncompartmental approach.
RESULTS
In vitro antimalarial activity of new synthetic tetracyclines.
We evaluated a set of novel 7-substituted sancycline derivatives and three clinically used tetracyclines for in vitro activity against cultured P. falciparum parasites. Activities were compared after a 96-h incubation due to the known slow antimalarial activity of tetracyclines (24). Minocycline was the most active established tetracycline against malaria parasites. Tetracycline and doxycycline also demonstrated nanomolar antimalarial activities. Numerous experimental tetracyclines were much more active than minocycline, doxycycline, and tetracycline against cultured parasites (Table 1). Cytotoxicity against COS-1 cells was not seen up to the medium- to high-micromolar-concentration limits of solubility for the test compounds (Table 1), indicating an excellent therapeutic index for the compounds with nanomolar antiparasitic activity.
Table 1.
Antimalarial activity and cytotoxicity of selected new tetracyclinesa
| Compound (n) | IC50 for: |
|
|---|---|---|
| Malaria (nM) | Cytotoxicity (μM) | |
| Tetracycline (2) | 330 | >100 |
| Doxycycline (29) | 321 (78) | >50 |
| Minocycline (34) | 102 (49) | >100 |
| MAL0919 (1) | 22 | >100 |
| MAL1570 (4) | 53 (8) | >10 |
| MAL5001 (1) | 28 | >50 |
| MAL5002 (4) | 18 (4) | >50 |
| MAL5003 (2) | 34 | >10 |
| MAL5004 (1) | 46 | >100 |
| MAL5008 (1) | 25 | >100 |
| MAL5011 (1) | 44 | >100 |
| MAL5012 (1) | 37 | >100 |
| MAL5021 (3) | 19 (7) | >50 |
| MAL5022 (1) | 21 | >100 |
| MAL5028 (1) | 27 | >100 |
| MAL5061 (1) | 47 | >10 |
| MAL5064 (2) | 43 | >10 |
| MAL5086 (1) | 26 | >10 |
| MAL5113 (3) | 37 (26) | >10 |
| MAL5155 (4) | 52 (12) | >100 |
| MAL5166 (11) | 21 (7) | >50 |
Compound names beginning with MAL are novel compounds. Results, selected from among all active compounds, are means from comparisons of control and treated P. falciparum cultures, assessed by FACS analysis after a 96-h incubation. Values were normalized to percent control activity, and IC50s were calculated by nonlinear regression. n is the number of times antimalarial activity was assayed. Where n is greater than 1, the values shown are means. Where n is greater than 3, the values shown are means (standard deviations). For cytotoxicity, the IC50 indicates the concentration, up to limits of solubility, that killed 50% of COS-1 cells.
Phototoxicity of new synthetic tetracyclines.
An additional factor to monitor with the development of new tetracyclines is phototoxicity, a known concern for certain members of the tetracycline class. Studies of a select group of compounds demonstrated that they were similar in toxicity and phototoxicity to minocycline, which is generally considered nonphototoxic (Table 2).
Table 2.
Phototoxic potential of new antimalarial tetracyclinesa
| Compound | IC50 (μM) |
|
|---|---|---|
| Dark cytotoxicity | UV cytotoxicity | |
| Doxycycline | >100 | 16 |
| Minocycline | >100 | >100 |
| COL-3 | >100 | 0.8 |
| MAL0919 | >100 | >100 |
| MAL1570 | >100 | >100 |
| MAL5001 | >100 | >100 |
| MAL5002 | >100 | 32 |
| MAL5003 | >100 | >100 |
| MAL5004 | >100 | >100 |
| MAL5008 | >100 | >100 |
| MAL5011 | >100 | >100 |
| MAL5012 | >100 | >100 |
| MAL5021 | >100 | >100 |
| MAL5022 | >100 | >100 |
| MAL5028 | >100 | >100 |
| MAL5061 | >100 | >100 |
| MAL5064 | >100 | 30.45 |
| MAL5086 | >100 | 5.6 |
| MAL5113 | >100 | 6.47 |
| MAL5155 | >100 | >100 |
| MAL5166 | >100 | 25 |
Phototoxicity (UV cytotoxicity) was assessed by exposing NIH 3T3 cells in the presence of the test compound to a UV light source at ∼1.7 mW/cm2 for 50 min and assessing cell survival 24 h later relative to a control plate that was kept in the dark (dark cytotoxicity). The highly phototoxic tetracycline derivative COL-3 as well as doxycycline and minocycline were tested as controls.
Antibacterial activity of new synthetic tetracyclines.
We explored the possibility of creating new tetracyclines with antimalarial, but not antibacterial, activity. Most 7-substituted sancycline compounds maintained both Gram-positive and Gram-negative antibacterial activity (Table 3). In one instance, with compound MAL5155, antibacterial activity was decreased relative to the other compounds, despite potent antimalarial activity.
Table 3.
Separation of antimalarial and antibacterial activitya
| Compound | MIC (μM [μg/ml]) |
|
|---|---|---|
| Gram positive | Gram negative | |
| Doxycycline | <0.1 (0.06) | 0.5 (0.25) |
| Minocycline | <0.1 (0.06) | 1.0 (0.5) |
| MAL0919 | <0.1 (0.06) | 0.8 (0.5) |
| MAL1570 | <0.1 (0.06) | 0.5 (0.25) |
| MAL5001 | <0.1 (0.06) | 0.4 (0.25) |
| MAL5002 | <0.1 (0.06) | 0.8 (0.5) |
| MAL5003 | <0.1 (0.06) | 3.3 (2) |
| MAL5004 | <0.1 (0.06) | 1.6 (1) |
| MAL5008 | <0.1 (0.06) | 0.8 (0.5) |
| MAL5011 | <0.1 (0.06) | 0.8 (0.5) |
| MAL5012 | <0.1 (0.06) | 0.4 (0.25) |
| MAL5021 | <0.1 (0.06) | 0.4 (0.25) |
| MAL5022 | <0.1 (0.06) | 0.2 (0.13) |
| MAL5028 | <0.1 (0.06) | 0.2 (0.13) |
| MAL5061 | <0.1 (0.06) | 1.6 (1) |
| MAL5064 | <0.1 (0.06) | 1.7 (1) |
| MAL5086 | <0.1 (0.06) | 0.8 (0.5) |
| MAL5113 | <0.1 (0.06) | 1.6 (1) |
| MAL5155 | 0.5 (0.25) | 7.4 (4) |
| MAL5166 | <0.1 (0.06) | <0.1 (0.06) |
Antibacterial MICs were determined using CLSI methodology for antibacterial susceptibility testing. Gram-positive activity is reported for S. aureus strain RN450. Gram-negative activity is reported for E. coli strain ML308-225.
In vivo antimalarial activity of new synthetic tetracyclines.
New tetracyclines were tested in a mouse model of P. berghei malaria. Doxycycline offered potent antimalarial activity when dosed intraperitoneally at 50 mg/kg once daily, but activity was suboptimal at 10 mg/kg (Table 4). More than 100 compounds were screened for in vivo activity, and several showed improved activity relative to doxycycline (Table 4). These data suggest that new compounds with better potency could be used at doses substantially lower than those used for the currently available tetracyclines.
Table 4.
In vivo antimalarial activity of selected new tetracyclines after intraperitoneal administrationa
| Compound | IC50c (nM) | Dose (mg/kg) | Avg parasitemia score on dayb: |
||
|---|---|---|---|---|---|
| 7 | 8 | 10 | |||
| PBS (control) | 3 | 4 | 4* | ||
| Doxycycline | 321 | 10 | 1.4 | 1.6 | 4 |
| 50 | 0 | 0 (day 9) | 0.6 (day 11) | ||
| Minocycline | 102 | 20 | 0.2 | 1 | 1.6 (day 9) |
| MAL0919 | 22 | 10 | 0 | 0.2 | 1.2 |
| MAL1570 | 53 | 10 | 0.4 | 0.8 | 4 |
| MAL5001 | 28 | 10 | 0 | 0 | 0 |
| MAL5002 | 18 | 10 | 0 | 0 | 0.2 |
| MAL5003 | 34 | 10 | 0.6 | 1.4 | 4 |
| MAL5004 | 46 | 10 | 0.2 | 0.2 | 1.6 |
| MAL5008 | 25 | 10 | 0 | 0 | 0 |
| MAL5011 | 44 | 10 | 0 | 0 | 0.4 |
| MAL5012 | 37 | 10 | 0.2 | 1.6 | 4 |
| MAL5021 | 19 | 10 | 0 | 0 | 0 |
| MAL5022 | 21 | 10 | 0 | 0 | 0 |
| MAL5028 | 27 | 10 | 0 | 0 | 0.2 |
| MAL5061 | 47 | 10 | 0.4 | 0.6 | 4 |
| MAL5064 | 43 | 10 | 0.6 | 0.8 | 4 |
| MAL5086 | 26 | 10 | 0 | 0 | 0.6 |
| MAL5113 | 22 | 10 | 0 | 0 | 0.2 |
| MAL5155 | 52 | 50 | 0 | 0 | 0 (day 9) |
| MAL5166 | 21 | 20 | 0 | 0 | 0 (day 9) |
Selected tetracyclines were tested in a P. berghei-based model of rodent malaria. Compounds were given by intraperitoneal administration once a day for 4 days at 10 mg/kg.
Percent parasitemia was scored on days 7, 8, and 10. Score 0, no parasites detected; 1, less than 1% parasitemia; 2, between 1 and 10% parasitemia; 3, greater than 10%; and 4, >50% parasitemia, at which point the animal was euthanized. Data are presented as the average score on a particular day for 5 animals. In some instances, parasitemia was evaluated on day 9 or 11 instead of day 10. The asterisk indicates that all control animals were euthanized on day 8.
IC50 data are in vitro activity against P. falciparum as shown in Table 1.
Selected compounds were also studied for activity when administered orally to mice. Despite the improved efficacy that many compounds displayed with intraperitoneal administration, most compounds from the series were not substantially better than doxycycline when given orally. Only MAL5166 (Fig. 2) demonstrated improved activity compared to that of doxycycline with oral dosing (Fig. 3A). At 15 days postinfection, the PD50 of MAL5166 was 46 mg/kg/day and the PD50 for doxycycline was >100 mg/kg/day (Fig. 3B). Pharmacokinetic studies of MAL5166 in mice revealed very modest oral uptake (Table 5), explaining the discrepancy between intraperitoneal and oral efficacy of this compound. However, studies in cynomolgus monkeys revealed more substantial oral bioavailability, but it was still less than that of doxycycline (Table 6).
Fig 2.
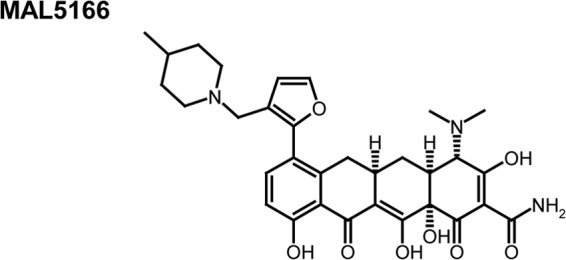
Structure of antimalarial tetracycline active with oral dosing.
Fig 3.
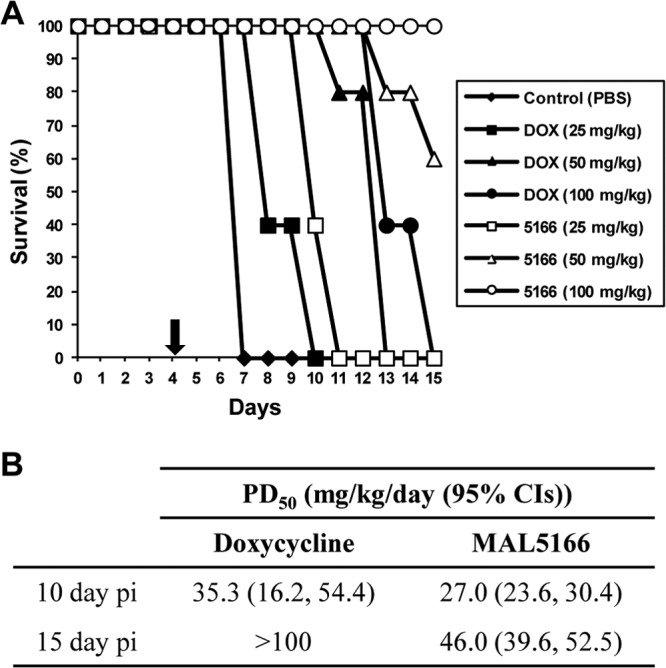
Oral activity of MAL5166 in vivo. Doxycycline (DOX) and MAL5166 (5166) were tested in an early-stage P. berghei model of murine malaria. Compounds were given at the indicated dose by oral administration for 4 days after infection (n = 5/group). Animals were sacrificed at 15 days after infection; thus, data on long-term survival are not available. (A) Survival curves are presented as percent survival/group. The arrow denotes the end of treatment. (B) The PD50 (50% protective dose) on days 10 and 15 postinfection (pi) were calculated as an early endpoint evaluation of efficacy. CI, confidence interval.
Table 5.
Pharmacokinetics of doxycycline and MAL5166 in micea
| Compound | i.v. dose (mg/kg) | Cmax (μg/ml) | Tmax (h) | AUC(0-∞) (h · μg/ml) | t1/2 (h) | Vss (ml/kg) | CL (ml/min/kg) | %Fb |
|---|---|---|---|---|---|---|---|---|
| Doxycycline | 10 | 4.51 (0.021) | 0.0833 (0.00) | 14.4 (0.146) | 4.41 (0.063) | 2892 (44.2) | 11.6 (7.34) | 25.3 |
| MAL5166 | 10 | 1.65 (0.151) | 0.194 (0.049) | 6.82 (0.514) | 6.19 (2.71) | 13843 (1542) | 24.5 (1.91) | 1.64 |
Pharmacokinetic studies were performed as described in Materials and Methods. All pharmacokinetic values, except %F (oral bioavailability), were calculated using data from the i.v. dose. Data shown are means (standard deviations). Cmax, maximum concentration of drug in serum; Tmax, time to maximum concentration of drug in serum; AUC(0-∞), area under the concentration-time curve from 0 h to infinity; t1/2, half-life; Vss, volume of distribution at steady state; CL, clearance.
%F was calculated based on ratio between the i.v. and oral AUC exposure values.
Table 6.
Pharmacokinetics of doxycycline and MAL5166 in monkeysa
| Compound | i.v. dose (mg/kg) | Cmax (μg/ml) | Tmax (h) | AUC(0-∞) (h · μg/ml) | t1/2 (h) | Vss (ml/kg) | CL (ml/min/kg) | %Fb |
|---|---|---|---|---|---|---|---|---|
| Doxycyclinec | 5 | 11.1 | 0.25 | 19.9 | 2.86 | 780 | 4.2 | 101 |
| MAL5166 | 5 | 42.5 (5.00) | 0.0 (0.00) | 51.5 (5.72) | 11.8 (0.785) | 1,309 (190) | 1.6 (0.173) | 35.5 |
Pharmacokinetic studies were performed as described in Materials and Methods. All pharmacokinetic values, except %F, were calculated using data from the i.v. dose. The i.v. dose was administered as a 15-min infusion. Data are means (standard deviations).
%F (oral bioavailability) was calculated based on ratio between the i.v. and oral AUC exposure values.
Insufficient animal numbers did not allow for meaningful statistical analysis of the doxycycline data.
DISCUSSION
The efficacy of tetracyclines for treatment and prophylaxis of malaria is proven, and in practice doxycycline is an important component of our antimalarial armamentarium. However, use of doxycycline and other tetracyclines for malaria is limited by concerns regarding toxicity in children. Our current results, demonstrating marked improvement in the antimalarial potency of 7-substituted sancyclines compared to doxycycline, suggest that improved antimalarials can be obtained from this new class of tetracyclines. Ideal new antimalarial drugs will be safe, orally bioavailable, efficacious with once-daily dosing, inexpensive, and readily partnered with other drugs. Our data suggest that a new, more potent 7-substituted sancycline will allow much lower dosing than that for prior antimalarial tetracyclines, and as a result it may be safe in children.
A number of antibiotics, including clindamycin, macrolides, chloramphenicol, and tetracyclines, are slow-acting antimalarial agents. The antimalarial effects of antibiotics are unusual in that activity is much greater when assessed in the parasite life cycle after that initially exposed to the drug. This “delayed death” phenomenon appears to be the result of specific effects on protein synthesis in the apicoplast, a prokaryote-like organelle of malaria parasites (24). New tetracyclines appear to share mechanisms of action with doxycycline, as they exhibit much more potent activity after 96- than 48-h incubations with cultured parasites (data not shown). Considering these results and the apparent need for two parasite life cycles to exert the full effects of apicoplast inhibitors, it is likely that all new tetracyclines will exhibit slow antimalarial activity. This property will limit the use of tetracyclines for severe malaria or to treat uncomplicated malaria as a monotherapy. However, this feature should not be seen as a limitation for development of new tetracyclines as combination chemotherapy, which is now the rule for the treatment of malaria. As a class, the tetracyclines are good candidates for partnering with other compounds, since they have a low potential for drug-drug interactions and are inexpensive to manufacture. Ideally, new, more potent antimalarial tetracyclines with appropriate pharmacokinetics and documented safety at therapeutic doses will be studied in combination with more rapidly acting agents, such as artemisinin derivatives, in the hope of identifying effective combination regimens.
A second important potential use for a new tetracycline antimalarial is in the prevention of malaria. Drugs are typically used to prevent malaria in nonimmune travelers, and first-line agents for this purpose are limited to mefloquine, which is not as well tolerated as other agents and entails neurological toxicity, atovaquone-proguanil, which is expensive and may fail due to drug resistance, and doxycycline, which is somewhat limited by gastrointestinal and dermatological toxicity and is not indicated in children. A more potent tetracycline might offer lower dosing and a more acceptable toxicity profile. In addition, our results demonstrate that many of the new 7-sancycline derivatives have a low potential for phototoxicity, which would eliminate dermatological safety issues. Strategies to enterically coat tetracyclines have proven effective at addressing gastrointestinal side effects (27). In the developing world, antimalarial drugs are increasingly considered components of malaria control. In particular, the use of intermittent preventive therapy and seasonal malaria chemoprevention in high-risk groups has offered benefits to children and pregnant women (7, 28, 29), and regular use of trimethoprim-sulfamethoxazole in patients with HIV infection has led to dramatic reductions in malaria incidence (30). However, to date these approaches are highly dependent on antifolates, which are losing efficacy because of parasite resistance. If safe, an effective new tetracycline would be an important addition to preventive regimens, with either regular or intermittent dosing in high-risk groups.
To date, the in vivo efficacy of all studied tetracyclines, including both standard agents and new compounds, has been somewhat less than that expected based on in vitro results. As antibacterial agents, the primary pharmacodynamic driver for tetracycline efficacy is the ratio of the area under the curve (exposure) to the MIC (31). In studies of P. berghei-infected mice, we found it necessary to use substantially higher doses of tetracyclines than those needed to achieve efficacy in bacterial infection models. The activity of tetracyclines against P. falciparum is parasite stage specific (24). As such, with the relatively short half lives of the drugs in rodents, it may be necessary to use larger doses in order to achieve adequate drug levels for 24 to 48 h. Our animal model may have required higher-than-expected dosing of tetracyclines, because the rodent parasite P. berghei may differ from P. falciparum in drug sensitivity. In any event, multiple new 7-sancyclines offered markedly improved in vivo efficacy over that of doxycycline when dosed parenterally. One compound was clearly superior to doxycycline with oral dosing. Improved in vivo activity was likely due principally to about 10-fold improved potency of optimal 7-sancyclines. These studies offer promise that new tetracyclines of the 7-sancycline class or other classes can be developed as new antimalarial drugs. Future investigative efforts will need to focus on identifying compounds which maintain improved antimalarial potency and increased oral bioavailability.
ACKNOWLEDGMENTS
This work was supported by funding from the Medicines for Malaria Venture and by Public Health Service Grant AI051800-02A1 from the National Institute of Allergy and Infectious Diseases.
P.J.R. is a Distinguished Clinical Scientist of the Doris Duke Charitable Foundation.
Footnotes
Published ahead of print 29 April 2013
REFERENCES
- 1. Breman JG. 2001. The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am. J. Trop. Med. Hyg. 64:1–11 [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization 2012. World malaria report 2012. World Health Organization, Geneva, Switzerland [Google Scholar]
- 3. Olliaro P, Cattani J, Wirth D. 1996. Malaria, the submerged disease. JAMA 275:230–233 [PubMed] [Google Scholar]
- 4. Nosten F, White NJ. 2007. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 77:181–192 [PubMed] [Google Scholar]
- 5. Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noedl H, Socheat D, Satimai W. 2009. Artemisinin-resistant malaria in Asia. N. Engl. J. Med. 361:540–541 [DOI] [PubMed] [Google Scholar]
- 7. Bardají A, Bassat Q, Alonso PL, Menéndez C. 2012. Intermittent preventive treatment of malaria in pregnant women and infants: making best use of the available evidence. Expert Opin. Pharmacother. 13:1719–1736 [DOI] [PubMed] [Google Scholar]
- 8. Divo AA, Geary TG, Jensen JB. 1985. Oxygen- and time-dependent effects of antibiotics and selected mitochondrial inhibitors on Plasmodium falciparum in culture. Antimicrob. Agents Chemother. 27:21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geary TG, Jensen JB. 1983. Effects of antibiotics on Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 32:221–225 [DOI] [PubMed] [Google Scholar]
- 10. Jacobs RL, Koontz LC. 1976. Plasmodium berghei: development of resistance to clindamycin and minocycline in mice. Exp. Parasitol. 40:116–123 [DOI] [PubMed] [Google Scholar]
- 11. Colwell EJ, Hickman RL, Kosakal S. 1973. Quinine-tetracycline and quinine-bactrim treatment of acute falciparum malaria in Thailand. Ann. Trop. Med. Parasitol. 67:125–132 [DOI] [PubMed] [Google Scholar]
- 12. Colwell EJ, Hickman RL, Kosakal S. 1972. Tetracycline treatment of chloroquine-resistant falciparum malaria in Thailand. JAMA 220:684–686 [PubMed] [Google Scholar]
- 13. Tan KR, Magill AJ, Parise ME, Arguin PM. 2011. Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am. J. Trop. Med. Hyg. 84:517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenthal PJ. 2009. Antiprotozoal drugs. In Basic and clinical pharmacology. Lange Medical Books/McGraw Hill, New York, NY [Google Scholar]
- 15. Sponer U, Prajakwong S, Wiedermann G, Kollaritsch H, Wernsdorfer G, Wernsdorfer WH. 2002. Pharmacodynamic interaction of doxycycline and artemisinin in Plasmodium falciparum. Antimicrob. Agents Chemother. 46:262–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grossman ER, Walchek A, Freedman H. 1971. Tetracyclines and permanent teeth: the relation between dose and tooth color. Pediatrics 47:567–570 [PubMed] [Google Scholar]
- 17. Lochary ME, Lockhart PB, Williams WT., Jr 1998. Doxycycline and staining of permanent teeth. Pediatr. Infect. Dis. J. 17:429–431 [DOI] [PubMed] [Google Scholar]
- 18. Purvis JJ, Edwards MS. 2000. Doxycycline use for rickettsial disease in pediatric patients. Pediatr. Infect. Dis. J. 19:871–874 [DOI] [PubMed] [Google Scholar]
- 19. Volovitz B, Shkap R, Amir J, Calderon S, Varsano I, Nussinovitch M. 2007. Absence of tooth staining with doxycycline treatment in young children. Clin. Pediatr. (Philadelphia) 46:121–126 [DOI] [PubMed] [Google Scholar]
- 20. Nelson ML, Ismail MY, McIntyre L, Bhatia B, Viski P, Hawkins P, Rennie G, Andorsky D, Messersmith D, Stapleton K, Dumornay J, Sheahan P, Verma AK, Warchol T, Levy SB. 2003. Versatile and facile synthesis of diverse semisynthetic tetracycline derivatives via Pd-catalyzed reactions. J. Org. Chem. 68:5838–5851 [DOI] [PubMed] [Google Scholar]
- 21. Singh A, Rosenthal PJ. 2001. Comparison of efficacies of cysteine protease inhibitors against five strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 45:949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sijwali PS, Koo J, Singh N, Rosenthal PJ. 2006. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Mol. Biochem. Parasitol. 150:96–106 [DOI] [PubMed] [Google Scholar]
- 23. Barkan D, Ginsburg H, Golenser J. 2000. Optimization of flow cytometric measurement of parasitaemia in plasmodium-infected mice. Int. J. Parasitol. 30:649–653 [DOI] [PubMed] [Google Scholar]
- 24. Dahl EL, Shock JL, Shenai BR, Gut J, DeRisi JL, Rosenthal PJ. 2006. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 50:3124–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahmed SA, Gogal RM, Jr, Walsh JE. 1994. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J. Immunol. Methods 170:211–224 [DOI] [PubMed] [Google Scholar]
- 26. Zerler B, Roemer E, Raabe H, Reeves A, Sizemore A, Harbell JW. 2000. Evaluation of the phototoxic potential of chemical modified tetracyclines with the 3T3 neutral red uptake phototoxicity test, p 545–554 In Balls M, van Zeller A-M, Halder M. (ed), Progress in the reduction, refinement and replacement of animal experimentation. Elsevier Sciences, Amsterdam, the Netherlands [Google Scholar]
- 27. Berger RS. 1988. A double-blind, multiple-dose, placebo-controlled, cross-over study to compare the incidence of gastrointestinal complaints in healthy subjects given Doryx and Vibramycin. J. Clin. Pharmacol. 28:367–370 [DOI] [PubMed] [Google Scholar]
- 28. Cairns M, Roca-Feltrer A, Garske T, Wilson AL, Diallo D, Milligan PJ, Ghani AC, Greenwood BM. 2012. Estimating the potential public health impact of seasonal malaria chemoprevention in African children. Nat. Commun. 3:881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. ter Kuile FO, van Eijk AM, Filler SJ. 2007. Effect of sulfadoxine-pyrimethamine resistance on the efficacy of intermittent preventive therapy for malaria control during pregnancy: a systematic review. JAMA 297:2603–2616 [DOI] [PubMed] [Google Scholar]
- 30. Sandison TG, Homsy J, Arinaitwe E, Wanzira H, Kakuru A, Bigira V, Kalamya J, Vora N, Kublin J, Kamya MR, Dorsey G, Tappero JW. 2011. Protective efficacy of co-trimoxazole prophylaxis against malaria in HIV exposed children in rural Uganda: a randomised clinical trial. BMJ 342:d1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Ogtrop ML, Andes D, Stamstad TJ, Conklin B, Weiss WJ, Craig WA, Vesga O. 2000. In vivo pharmacodynamic activities of two glycylcyclines (GAR-936 and WAY 152,288) against various gram-positive and gram-negative bacteria. Antimicrob. Agents Chemother. 44:943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]