

Abstract
Inflammation of central nervous system (CNS) is usually associated with trauma and infection. Neuroinflammation occurs in close relation to trauma, infection, and neurodegenerative diseases. Low-level neuroinflammation is considered to have beneficial effects whereas chronic neuroinflammation can be harmful. Innate immune system consisting of pattern-recognition receptors, macrophages, and complement system plays a key role in CNS homeostasis following injury and infection. Here, we discuss how innate immune components can also contribute to neuroinflammation and neurodegeneration.
1. Introduction
Neuroinflammation is the mechanism of CNS inflammation that occurs in response to trauma, infections, and/or neurodegenerative diseases. In neuroinflammation, cellular and molecular immune components such as specialised macrophages (microglia), cytokines, complement, and pattern-recognition receptors are the contributing players. These proinflammatory mediators are either produced locally within the CNS or recruited from the peripheral system following disruption of the blood-brain barrier. This in turn leads to the activation of the glial cells, such as microglia and astrocytes. The effect of neuroinflammation is considered neuroprotective when the inflammatory activity is for a shorter period of time whereas chronic neuroinflammation is associated with harmful consequences for the CNS.
Innate immunity is the first line of defence against the invading pathogens. Some of the components of first line of defence include epithelium (skin, gut, and lungs) that acts as a physical barrier and also produces several kinds of antimicrobial enzymes and peptides, namely, lysozyme, defensins, mucin, lectin [1]. Other components of innate immunity include the pattern-recognition receptors (PRRs) such as toll-like receptors (TLRs), nucleotide-binding, and oligomerisation domain, leucine-rich repeats containing (NOD)-like receptors (NLRs); and Scavenger receptors (SRs). Present on phagocytic and antigen-presenting cells, these receptors recognise not only exogenous pathogen-associated molecular pattern 1 (PAMP) but also endogenous modified molecules called damage-associated molecular pattern 2 (DAMP). The innate immune system launches inflammatory and regulatory responses via PRRs, phagocytes (macrophages), complement system, cytokines, and chemokines in order to counteract infection, injury, and maintenance of tissue homeostasis. Here, we discuss the role of innate immune players involved in neuroinflammation.
2. Microglia
Microglial cells are the specialised resident macrophages of the CNS. The origin of these innate immune cells is debatable but it is now widely believed that they are of myeloid lineage [2]. In mice studies, it has been found that microglia originate from primitive (yolk sac) myeloid progenitors that migrate to CNS independent of definitive progenitors and circulation (i.e., bone marrow) [3]. These cells are found in brain, spinal cord, retina, and optic nerve. Their morphology differs from “conventional” macrophages by the presence of branch-like processes (ramified appearance). This is the shape they have when in “resting” state. In this state, these cells constantly monitor and survey their area [4]. The microglial cells in resting form have been shown to be involved in other functions such as neurogenesis [5], neuroprotection [6] and synaptic pruning [7], which has been found to be complement dependent [8]. Upon environmental stimulation/challenges, the microglia become “activated” and the morphology changes to an amoeboid appearance where they retract the ramifications [9]. Activation of microglia by TLRs and NLRs is considered to be “classical” form of microglial activation where innate immune responses include production of proinflammatory cytokines like tumour necrosis factor (TNF)-α, interleukin (IL)-1 and IL-6, and chemokines. Classical activation also leads to adaptive immune response by expressing major histocompatibility class II molecules and interaction with T cells [10]. TNF-α stimulation increases phagocytic activity of microglia [11], and deficiency of TNF receptors has been found to reduce microglial activation [12]. TNF-α is associated with activation of microglial cells involved in pathogenesis of neurodegenerative diseases like Alzheimer's disease (AD) [13] and Parkinson's disease (PD) [14]. IL-1 induces expression of TNF-α and IL-6 [15] and is implicated in neuroinflammatory processes in traumatic brain injury (TBI), AD, and PD [16]. Activated microglia have also been implicated in neurotransmission [17]. In order to regulate the immune responses, anti-inflammatory cytokines IL-10 and transforming growth factor beta are produced by microglia [18–20]. Microglia also produce inhibitor of nuclear factor κβ(NF-κβ), mitogen-activated protein kinase (MAPK) phosphatases, and suppressor of cytokine signalling proteins [21], which help in immune activation regulation. Glucocorticoids have also been considered to play a regulatory role for innate immunity in CNS by regulation of microglial TNF-α [22, 23] although there are debatable views to the same [24].
There are a variety of receptors expressed on microglia related to the different functions of these cells. Some of the receptors associated with innate immunity are listed in Table 1.
Table 1.
Innate immune receptors on microglia.
| Receptor | Functions/comments | References |
|---|---|---|
| TLR | Pattern-recognition receptors that respond to self (DAMPs) and nonself (PAMPs) activators. Microglia are known to express TLR1-9. TLRs are implicated in neuroinflammation in response to bacterial and viral infections, Alzheimer's disease, prion diseases, and amyotrophic lateral sclerosis. | [59, 69] |
|
| ||
| NLR | Cytoplasmic pattern-recognition receptors. Microglia are known to express NOD2 in response to CNS infection and NALP3 inflammasome in Alzheimer's disease. | [109, 110] |
|
| ||
| Scavenger | Another group of pattern-recognition receptors. The receptors expressed on microglia are Class A, CD36, and RAGE. | [111, 112] |
|
| ||
| RLR | RIG-I is a pattern-recognition receptor that is expressed by microglia in response to viral infections. | [110, 113] |
|
| ||
| Complement | Complement receptors expressed include CR1, CR3 and CR4. These receptors bind complement proteins and activate complement pathway which is considered to be both beneficial and detrimental depending on the level of activation. | [114] |
|
| ||
| Cytokines | Some of the cytokine receptors expressed in microglia are IL-1R, TNFR (responsible for proinflammatory actions of cytokines IL-1 and TNF-α resp.), IL-10R, TGFR (responsible for the anti-inflammatory cytokines IL-10 and TGF-β), and CCR1-5 responsible for actions of chemokines. These are expressed and produced in neuroinflammation. | [115, 116] |
TLR: toll-like receptor; DAMP: damage-associated molecular pattern; PAMP: pattern-associated molecular pattern; NLR: NOD-like receptors; NOD: nucleotide-binding and oligomerisation domain; RLR: RIG-like receptors; RIG: retinoic acid-inducible gene; CR: complement receptor; IL: interleukin; TNF: tumour necrosis factor; TGF: transforming growth factor.
TLR 1–9 receptors are known to be expressed by microglial cells (discussed in detail later). NLR form complexes called inflammasomes (for a detailed review see [25]) that have been shown to activate and recruit microglia in response to amyloid-β (Aβ) [26] and prion peptide [27]. Some of the other innate immune receptors expressed on microglia surface are CD14, CD18, CD36, CD68, mannose, and lectin (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing nonintegrin or DC-SIGN) receptors. Complement receptors found on microglia are C3a, C5a, and C1q receptors [28].
3. Astrocytes
Astrocytes are specialised glial cells and the most abundant cells of the CNS. Morphologically, astrocytes are of two types: protoplasmic (found in grey matter) and fibrous (found in white matter). The basic astrocyte morphology resembles that of a star (with multiple processes). Protoplasmic astrocytes have undistinguishable dense processes while fibrous astrocytes have clearly distinguishable processes [29]. Astrocytes have conventionally been considered to be supporting cells to the neurons. However, recently they have been shown to play an active part in the modulation of neural activity [30], potentiation of synaptic transmission [31], sleep homeostasis [32], and even long-term memory formation [33]. Any insult to the CNS is associated with changes in the structure, morphology, and hypertrophy of astrocytes, followed by cytokine and C1q secretion, leading to scar formation, collectively termed as reactive astrogliosis [34].
Like microglia, astrocytes have been shown to express innate immune PRR like TLR, NLR, scavenger, complement, and mannose receptors [35]. They have also been shown to release cytokines like TNF, IL-6, IL-1, Interferon-γ, and chemokines when stimulated with lipopolysaccharide (LPS) [36, 37]. Reactive astrogliosis is associated with a number of CNS diseases such as AD [38, 39], PD, autism, and prion diseases [40, 41].
4. Toll-Like Receptors (TLR)
4.1. Structure and Signalling Pathway
TLRsare expressed on microglia, neurons, and astrocytes similar to dendritic cells, B cells, neutrophils, epithelia, and fibroblast [42]. TLR is a type 1 membrane protein containing an extracellular leucine-rich repeat (LRR) domain and a Toll/IL-1 receptor (TIR) domain in the cytoplasmic region (Figure 1). LRR domain is involved in specific pathogen recognition [43] and TIR domain is involved in the signalling pathway. TLRs are considered to exist as dimers and bind to various ligands [44, 45]. For example, TLR2 heterodimerises with TLR1 [46] and also with TLR6 [44] and recognises bacterial lipoproteins. Upon sensing ligands, recruitment of adaptor proteins takes place which is necessary for signal transduction [47]. The adaptor proteins are (i) myeloid differentiating factor 88 (MyD88); (ii) MyD88 adaptor-like protein (Mal); (iii) TIR domain-containing adaptor inducing interferon-β (TRIF); (iv) TRIF-related adaptor molecule; and (v) sterile-α and armadillo-motif-containing protein. These adaptor proteins are recruited by TIR domain leading to activation of NF-κβ. NF-κβ then induces production of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6, and chemokines. All TLRs are activated by MyD88 except TLR3; instead MyD88 may be restricting TLR3 signalling [48]. Some of the other adaptors investigated in detail include major histocompatibility complex class II molecules [49], small heterodimer partner [50], and Dedicator of Cytokinesis 8 (DOCK8) [51].
Figure 1.
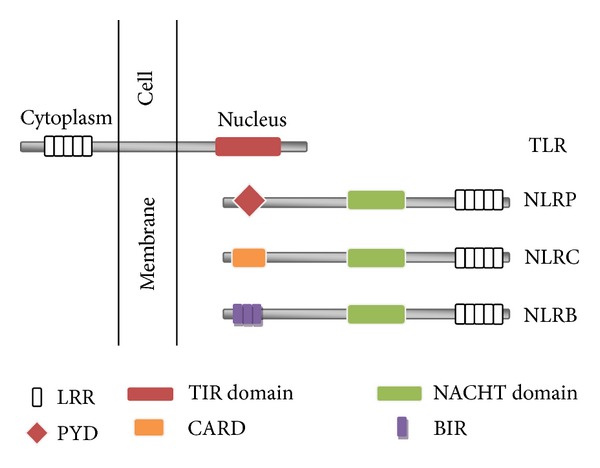
Schematic diagram showing structure of TLR and NLR family. TLR: toll-like receptor; NLRP: NOD-like receptor containing pyrin domain; NLRC: NOD-like receptor containing NLR-containing caspase activation and recruitment domain; NLRB: NOD-like receptor containing baculovirus inhibitor of apoptosis protein repeat domain; LRR: leucine-rich repeat; TIR: toll/il-1 receptor; PYD: pyrin domain; CARD: caspase activation and recruitment domain; BIR: baculovirus inhibitor of apoptosis protein repeat. The figure shows the structure of a TLR containing a TIR domain present inside nucleus which is involved in signalling pathway and an LRR domain present in the cytoplasm which is involved in pathogen recognition. NLR are intracellular receptors containing a C-terminal LRR domain, a central NACHT domain, and a variable N-terminal domain which can be a PYD, a CARD, or a BIR domain.
It has recently been shown that oligomerisation of TLR4 with myeloid differentiation protein-2 by morphine causes neuroinflammation [52]. Necrotic neurons have been shown to activate microglia via MyD88 pathway leading to increased neuroinflammation [53]. In mouse models, both MyD88 and TRIF pathways have been implicated in regulation of IL-6 and IL-10 after cerebral ischaemia [54] as well as regulation of IL6, TNF-α, and IL-1β following intracerebral haemorrhage [55]. MyD88 pathway also plays an important role in CNS infection and consequent astrocyte activation [56]. MyD88 pathway may also be involved in PD [57] and optic nerve injury [58].
4.2. Ligands
Some of the exogenous and endogenous ligands of TLR are listed in Table 2 [59–62].
Table 2.
Exogenous and endogenous ligands of toll-like receptors.
| Ligand | TLR | Implications/comments | References |
|---|---|---|---|
| Lipopolysaccharide | TLR4 | Recognition of Gram (−) bacteria | [117] |
| Triacylated lipopeptides | TLR1 and TLR2 | Recognition of Gram (−) bacteria and mycobacteria | [118] |
| Diacylated lipopeptides | TLR2 and TLR6 | Recognition of Gram (+) bacteria and mycoplasma | [119, 120] |
| Lipoteichoic acid | TLR2 | Recognition of Gram (+) bacteria | [121] |
| Zymosan | TLR2 | Recognition of fungi | [122] |
| Double-stranded RNA | TLR3 | Recognition of virus | [123] |
| Single-stranded RNA | TLR7 and TLR8 | Recognition of virus | [124, 125] |
| Flagellin | TLR5 | Recognition of Gram (−) bacteria | [126] |
| Unmethylated CpG DNA | TLR9 | Recognition of bacteria and virus | [127, 128] |
| β-amyloid | TLR2; TLR4; TLR4 and TLR6 |
Neuroinflammation in Alzheimer's disease | [95, 96, 129, 130] |
| Mitochondrial DNA | TLR9 | Pathogenesis of myocarditis and heart failure | [128] |
| Lung surfactant protein-A and -D | TLR4 TLR2 |
Innate immune component of lung. Act as opsonin and macrophage activator. Physiological implications of excessive activation by TLR is not known | [131–133] |
| Tenascin-C | TLR4 | Maintenance and pathogenesis of inflammation in rheumatoid arthritis | [134, 135] |
| Fibrinogen | TLR4 | Present normally in serum and activation has been implicated in rheumatoid arthritis and atherosclerosis | [136, 137] |
| Oxidised low-density lipoprotein | TLR4 | Pathogenesis of atherosclerosis | [95] |
| MicroRNA let-7 | TLR7 | Pathogenesis of neurodegeneration | [138] |
4.3. Response in CNS to Ligands of TLR
In vivo studies have shown that the administration of LPS (peripheral/intraperitoneal) leads to expression of genes coding for proinflammatory cytokines in the microglial cells [63, 64]. CD14 has been found to be required for LPS-induced endocytosis of TLR4 [65]. Injection of LPS directly into brain has been shown to produce an increased expression of genes of proinflammatory cytokines, chemokines, and complement proteins and receptors such as CD14 [66, 67]. Production of TNF by microglial cells upon LPS stimulation has been found to cause death of dopaminergic neurons [68]. TLR2 ligands stimulation of microglial and astrocytic cells leads to an increase in production of IL-6, chemokines, and IFN-β [69]. In mice studies, TLR9 ligand CpG has been found to be neuroprotective in cerebral ischaemia [70] while similar findings have been reported in TLR4 knockout mice [71]. TLR2 activation has been shown to be involved in neurogenesis [72] while TLR8 induces apoptosis of neurons [73]. TLR3 impairs plasticity and working memory [74] while TLR7 and TLR9 have been found to be associated with the development of mouse brain [75]. Interestingly, increased peripheral responses of TLR2, TLR4, TLR8, and TLR9 have been detected in psychosis [76] while TLR9 is associated with posttraumatic anxiety [77].
4.4. TLR Response to Pathogens
Pneumococcal infection leads to innate immune response in brain and this depends on TLR2 and TLR4 [78]. Deficiency of TLR2 causes an increased TNF gene expression in the brain [79]. TLRs have been found to be involved in pneumococcal infection in HIV-associated neurocognitive disorders [80]. TLR signalling is also associated with virulence of intracellular pathogens [81]. TLR2 and TLR9 initiate immune response against herpes simplex virus (HSV) [82] and also control HSV infection in the brain [83]. TLR3 is protective for the CNS in HSV1 infection [84]. In mice models, TLR3 in astrocytes may be protective in HSV2 infection [85] and has been reported to mediate entry of West Nile virus (WNV) into the CNS, causing encephalitis [86]. TLRs have also been implicated in CNS parasitic infections like toxoplasmosis, 3 sleeping sickness, 4 cerebral malaria, 5 and neurocysticercosis 6 [87]. TLR2 is associated with protection from cerebral malaria [88] and therapeutic targeting of TLRs has been shown to prevent experimental cerebral malaria [89, 90].
4.5. Neurodegenerative Diseases
In mouse model of AD, MyD88 has been found to prevent memory [91] and cognitive deficits [92] while another study found MyD88 deficiency to improve AD-related pathology [93]. TLR2 clears Aβ and delays cognitive decline, again in mouse model of disease [94]. TLR4 causes Aβ-induced microglial activation [95] and Aβ-induced neuronal apoptosis [96]. A loss-of-function mutation of TLR4 has been found to reduce microglial activation and increase Aβ deposits with increased cognitive deficits [97]. Intracranial injection of LPS (a TLR4 ligand) reduces Aβ levels in brain [98]. TLR9 may have a protective role in AD by improving cognitive functions [99], reducing Aβ-toxicity [100], and clearing Aβ [101]. In amyotrophic lateral sclerosis 7 (ALS), MyD88 has been shown to activate microglia due to mutant SOD1 [102] and in vitro studies show enhanced microglial activation and neurotoxicity when stimulated with TLR2 and TLR4 ligands [103, 104]. MyD88 pathway may also be involved in PD [57] where α-synuclein directly activates microglia and alters expression of TLRs [105]. TLR signalling has been found to interfere with prion disease pathogenesis. Studies involving mice possessing mutant gene which prevents TLR4 signalling was found to have a shorter time for scrapie pathogenesis [106] while administration of TLR9 agonist in prion-infected mice leads to delayed onset of the disease [107]. However, MyD88 knockout mice (lacking TLR signalling) were found to develop prion disease similar to wild-type mice both in terms of time and severity [108].
5. NOD-Like Receptors
5.1. Structure
Like TLRs, NOD-like receptors (NLRs) also detect PAMPs and DAMPs. NLRs are intracellular receptors thereby monitoring intracellular environment. They consist of a central nucleotide-binding and oligomerisation (NACHT) domain and a C-terminal LRRs. Their N-terminal component may be variable based on which NLRs are further subdivided. It can be caspase activation and recruitment domain (CARD); a pyrin domain (PYD), or baculovirus inhibitor of apoptosis protein repeat (BIR) termed, respectively, as NLRC, NLRP, and NLRB [139]. Upon binding to agonists, NLR can lead to the activation of NF-κβ or MAPK signalling pathways and production of cytokines and chemokines. NLR binding to agonist also causes the activation of procaspase-1 leading to inflammasome formation; pyroptosis; autophagy; and IFN-1 signalling [140–145] (Figure 1) [141–145].
5.2. Inflammasomes
Inflammasomes are multiprotein complexes that activate caspase-1, which in turn leads to processing and secretion of proinflammatory cytokines such as IL-1β and IL-18. The members of NLR family that are capable of forming inflammasomes are PYD-containing NLRP1, NLRP3, NLRP6, and CARD-containing NLRC4 [146]. Inflammasome complex formation occurs when a ligand binds to NLR and thereby induces a conformational change, leading to ATP binding at NACHT domain which causes receptor oligomerisation and recruitment of other complex members [141]. Inflammasomes have been implicated in various diseases such as gout, pseudogout, contact dermatitis, allergic dermatitis, vitiligo, hydatidiform mole 8 [147], Muckle-Wells syndrome 9 [148], atherosclerosis, type 2 diabetes mellitus, obesity [149], metabolic syndrome 10 [150], acute myocardial infarction [151], coeliac disease, inflammatory bowel disease [152], asthma, pulmonary fibrosis [153], and viral [154] and bacterial infections [155].
5.3. Role in Neuroinflammation
NLRP3 inflammasome is involved in the innate immune response to Aβ [156] leading to AD pathology. In multiple sclerosis (MS), NLRP3 knockout mice model of disease shows reduced demyelination [157], while another study shows NLRP3 involvement in migration of T-helper cells into CNS [158]. IFN-β therapy is effective in treating inflammasome-dependent disease in mouse models of MS [159]. NLRP1 has been found to be involved in TBI and neutralising its effect or formation was found to have beneficial effects [160]. Inflammasome complex inhibition has also been found to reduce inflammation and improve pathology in mouse models of stroke [161]. NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis [162] and is associated with inflammation in Japanese encephalitis [163]. Both NLRP1 and NLRP3 are increased in postmortem alcoholic human brains and inhibition of these inflammasomes was found to be beneficial in reversing ethanol-mediated neuroinflammation [164].
6. Scavenger Receptors
6.1. Types
Scavenger receptors (SRs) are members of PRRs and are transmembrane glycoprotein PRRs [165]. SRs are expressed on macrophages, dendritic cells, microglia, and endothelial cells [111, 112]. Recently, SR expression on astrocytes has been reported [166]. The family of SRs include class A (macrophage receptors, MARCO), class B (CD36, SR-BI), CD68 and endothelial or LOX-1, CD163, and receptor for advanced glycation end products (RAGE) [167, 168]. Some of the ligands that SRs bind to are pathogen-specific: LPS, lipoteichoic acid, Streptococcus pneumoniae, Staphylococcus aureus, Mycoplasma pneumoniae, Neisseriia meningitides, Escherichia coli [169], apoptotic cells [170], and erythrocytes infected with Plasmodium [171–173]. SRs have been implicated in atherosclerosis [174], lung inflammation [175], cystic fibrosis [176], SLE [170], and AD [112].
6.2. Role in Neuroinflammation
Microglia express SR and thus bind to Aβ fibrils [177] which is associated with AD plaques [178]. Class A SR (SR-A) has also been shown to play an important role in cerebral injury due to ischemia. Mice deficient in SR-A showed reduced expression levels of TNF-α and IL-1β as well as decreased infarct size [179]. In experimental model of MS, SR-A knockout mice showed significantly reduced demyelination as well as reduced proinflammatory cytokines production [180]. However, deficiency of SR-A in AD mouse models was not found to impact amyloid plaque deposition or clearance [181]. In vitro studies have shown that astrocytes express SR-A and thus play a role in neuroinflammation [166]. Class B SR Type I (SR-BI) has been shown to be produced in vivo in AD brains [182] with increased expression being observed in cerebellum and cortex [183]. In mice studies, SR-BI has also been shown to impair perivascular macrophages leading to AD pathology such as increased amyloid deposition, cerebral amyloid angiopathy (deposition of Aβ in cerebral arteries), and memory deficits [184]. CD36 appears to be involved in neurovascular dysfunction due to Aβ [185] and promotes cerebral amyloid angiopathy leading to cognitive deficits [186]. RAGE is a receptor for Aβ and expressed on neurons, microglia, astrocytes, and endothelial cells [187]. RAGE signalling in microglia due to p38 MAPK signalling pathway leads to neuroinflammation and cognitive disturbances in AD [188] as well as synaptic [189] and neuronal [190] dysfunction.
7. Complement
7.1. Three Activation Pathways of the Complement System
The complement system comprises of more than 30 proteins in the serum as well as membrane-bound receptors and regulators. The complement system consists of 3 different initiating or activation pathways culminating into a final common lytic pathway, leading to the formation of membrane attack complex (MAC) (Figure 2). MAC are pores that penetrate cell membrane (lipid bilayers) of pathogens or abnormal cells, thereby causing their lysis. The three initiating pathways are called (i) classical pathway which is mostly antibody mediated (C1q being the first subcomponent) and is activated by C1 complex (C1q-C1r-C1s); (ii) alternative pathway (AP) which is activated spontaneously involving low-level hydrolysis of C3 to C3 (H20); and (iii) lectin pathway where activation occurs through binding of a carbohydrate pattern present on microorganisms called mannan, with mannan-binding lectin (MBL) and Ficolins (ficolin-1, -2 and -3). They circulate in the serum in combination with zymogen serine proteases called MBL-associated serine proteases (MASPs) [191–196]. All the 3 pathways ultimately converge to lead to formation of C3 convertase. C3 convertases then cleaves C3 into C3a and C3b. This C3b binds to C3 convertase and leads to the formation of C5 convertase. This C5 convertase cleaves C5 into C5a and C5b. C3a and C5a are called anaphylatoxins and are chemoattractants. The C5b formed associates with C6, C7, C8, and C9 to form MAC [197]. The functions of the complement system include opsonisation of pathogens, direct lysis of foreign cells, chemotaxis and activation of leukocytes, and clearance of apoptotic cells. The complement system also interacts with TLRs [198] and plays a role in the regulation of humoral immunity [199]. The complement system is kept in check by regulators in order to prevent overactivation leading to damage to tissues and autoimmune diseases. The regulators can be grouped into fluid-phase: factor H (fH) and properdin for alternative pathway, C1 inhibitor and C4b-binding protein (C4BP) for classical and MBL pathway; host cell membrane-bound: CR1, CR2, CD55, CD46, CD59; cell surface-attached complement regulators: fH, factor H-like protein 1 (FHL-1), C4BP and clusterin [200, 201]. For certain ligands, factor H can also regulate C1q-mediated classical pathway [202–205].
Figure 2.

The complement system. Complement regulators are indicated in red. MBL: mannan-binding lectin; MASP: MBL-associated serine protease; C4BP: C4b-binding protein; CR1: complement receptor 1. The complement system consists of 3 initiating pathways: classical pathway, lectin pathway, and alternative pathway. The classical pathway is usually activated by antigen-antibody complexes, the lectin pathway is activated by microbes with MBL-MASP complex, and the alternative pathway is activated spontaneously by hydrolysis of C3 to C3(H2O). All 3 pathways lead to formation of C3 convertase, followed by C5 convertase, ultimately leading to formation of membrane attack complex. In this process, anaphylatoxins C3a and C5a are also released. The complement system is kept in check by a number of regulators.
7.2. Role in CNS Physiology
Complement is produced mainly in the liver and, over the years, it was thought that the brain was an immune-privileged organ due to the presence of blood-brain barrier. Now, it is well known that components of innate immunity like complement are present and even produced within the CNS. Neuronal cells [206–209], astrocytes [210, 211], and microglia [212–214] have been shown to produce complement and also express complement receptors. Role of complement in CNS is considered to be dual-neurotoxic and/or neuroprotective, depending on the level of its activation.
Complement has been shown to play a role in adult neurogenesis. Complement receptors C3aR and C5aR are expressed on neural stem cells and reduced neurogenesis is observed in the absence of C3aR signalling [215]. Another complement receptor CR2 has been found to be expressed in neural progenitor cells and also negatively regulates hippocampal neurogenesis [216]. An emerging area for complement involvement in CNS is in relation to synapse (reviewed in [217]). C1q, initiating component of classical pathway and widely expressed by postnatal neurons and immature astrocytes [218], mediates the elimination of synapse [219, 220]. C1q knockout mice show increased synaptic connectivity and spontaneous epilepsy [221]. Synapse remodelling by microglia involves CR3 [8]. In vitro studies show that C1q also promotes neuronal viability and survival [222]. In vitro and in vivo studies implicate a role for C3aR and C5aR in the development of cerebellum [223]. Many other in vitro and in vivo studies show neuroprotective functions for C3a and C5a that include protection against NMDA-induced apoptosis [224] and protection against glutamate-induced apoptosis [225] via MAPK-dependent inhibition of caspase 3 [226] as well as regulation of glutamate receptor subunit 2 [227].
7.3. Role in CNS Pathology
CNS can be infected by bacteria, virus, fungus, or protozoa. Deficiency of C3 is associated with increased susceptibility to meningococcal and pneumococcal infections [228]. Meningococcus binds to Factor H (fH), a negative regulator of alternative pathway, and evades host innate immune system [229, 230]. Neisseria meningitidis recruits host fH using protein mimicry [231]. Individuals with deficiency of properdin (positive regulator of alternative pathway) are susceptible to meningitis and individuals with combined properdin and MBL deficiency are at increased risk of infection with Neisseria meningitidis [232]. Streptococcus pneumonia infection of CNS is kept in check by complement system (mainly C1q and C3) [233]. C1q and C3 genetically deficient mice each showed considerably high bacterial titres in CNS as compared to wild-type mice. Escherichia coli, a cause for neonatal meningitis, crosses the blood-brain barrier by surviving in the serum where it binds to C4BP [234].
Viruses have also evolved mechanisms to evade complement system [235]. Gamma-herpesvirus encode for proteins that regulate and inhibit host C3-mediated resistance [236]. Complement controls antibody response in WNV infection [237] with lectin pathway activation being found to be protective in WNV infection [238]. C3 has been found to participate in seizure induction during viral encephalitis [239]. Increased MBL is seen in postmortem HIV encephalitis brains [240].
Fungal infection like cerebral aspergillosis leads to increased complement production seen in astrocytes, neurons, and oligodendrocytes, especially C1q production by infiltrating macrophages [241]. Some of the defence mechanisms developed by Aspergillus fumigatus to avoid complement include secreting fungal proteases [242] as well as production and recruitment of complement inhibitors [243]. In cerebral malaria, C1q and C5 levels have been found to be increased in mice studies [244] while another murine study also points to the requirement of MAC in the pathogenesis of cerebral malaria [245]. Infectious particles called prions cause CNS disorders like Creutzfeldt-Jakob disease and Bovine Spongiform Encephalopathy. These prion particles which activate classical complement pathway [246] are thought to bind to C1q and subsequently transported to the CNS [247]. C1q, C3b have been detected in postmortem brains of individuals with prion diseases [248], and MAC deposition was found to co-relate with prion disease severity [249].
Complement activation occurs in TBI and act as mediators of secondary brain injury [250, 251]. Following injury, levels of MAC corelate with blood-brain barrier (BBB) disruption [252]. In mice studies, absence of CD59 (a regulator of MAC formation) leads to increased neuropathology [253]. Postmortem studies on TBI brains show upregulation of C1q, C3b, and MAC [251]. Studies involving mice overexpressing complement inhibitor CR-related protein y (Crry) show reduced neurological impairment following TBI [254]. Hence, targeting complement activity in TBI may have therapeutic implications [255].
Cerebral ischemia can lead to the activation of the complement cascade leading to inflammation [256]. Systemic complement activity is also found to be enhanced in ischaemic stroke [257]. Complement system is implicated in ischemia reperfusion injury [258]. Ischaemic neurons have been found to produce C5a which causes apoptosis of neurons [259]. Better outcome is seen in individuals with low levels of MBL activity and mice lacking MBL [260]. Immunohistochemistry on brains of stroke patients shows C1q deposition while complement regulator CD59 was found to be absent [261]. Studies involving C5- [262] and C3-deficient mice [263] as well as C1 inhibition [264] have been successful in having beneficial effects in stroke therapy by targeting complement [256, 265].
A major role for complement is also seen in neurodegenerative diseases like AD. The neuropathology in AD includes loss of neurons, extracellular amyloid plaques, and intracellular neurofibrillary tangles consisting of abnormally phosphorylated tau protein [266]. Aβ activates complement [267], most notably via the classical pathway. Activated complement components C1q, C3d, and C4d have been detected in amyloid plaques [268, 269] by immunohistochemistry. C1q binds to Aβ [270, 271] and modulates phagocytosis of Aβ by microglia [272]. Upon exposure to Aβ, C1q is expressed in neurons (hippocampus) [273], and it has been found that inhibiting the binding of C1q to Aβ leads to protection of hippocampal cells [274]. In mouse models of AD, absence of C1q shows less neuropathology [275]. Complement regulators factor H, FHL-1, and C4BP have also been localised in amyloid plaques and fH and C4BP have been shown to bind Aβin vitro [276–278]. These regulators could be involved in regulation of excessive complement activation. Another interesting feature is the presence of microglia expressing complement receptors found in close proximity to plaques. Microglia are found in and around plaques of AD brains [279] and are found to express C1q [280] and complement receptors C1qR, CR3, CR4, and C5aR, which help in the phagocytosis of Aβ [281, 282]. Complement activation is therefore also considered to be neuroprotective [266]. C3 deficiency in mouse model shows accelerated amyloid plaque deposition [283]. Furthermore, inhibition of complement was found to be associated with an increased formation of plaque and neurodegeneration [284]. Amyloid precursor protein transgenic mouse models of AD that lack the ability to activate classical pathway (APPQ−/−) (i.e., C1q−/− phenotype) show less neuropathology as compared to APPQ+/+ mice. However, APPQ−/− mice also show increased C3 levels, providing evidence for alternative pathway activation in AD [285]. In mice models, deficiency of sCrry increases tau pathology [286]. Genetic association of AD and complement involves complement genes CR1 and CLU [287]. Micro-RNAs 11 (miRNAs) −9, −125b, −146a, and −155 are found to be upregulated in AD and these miRNAs target gene encoding fH [288].
An emerging role for complement in MS has become evident recently [289]. C3d is localized along with microglia in MS tissues [290]. Priming of microglia in MS has been found to be C3-dependent and, in the same study, it was found that in animal model of MS, Crry-deficient mice show exacerbated and accelerated disease progression [291]. Serum factor H has been found to be a useful biomarker for MS [292]. Pathological studies of MS lesions have found presence of complement components C3d, C4b, C1q, and MAC on myelin sheath, surrounding vessel walls, microglia, and astrocytes [293–296].
There is evidence for neuroinflammation in PD as well [297] with the presence of reactive microglia and activated components of complement. Elevated mRNA levels of activated complement and markers of reactive microglia are also seen in PD [298]. Pathological studies show the presence of MAC components intracellularly on the characteristic Lewy Bodies [299, 300]. The cerebrospinal fluid levels of C3 and factor H have been observed to correlate with severity of PD [301]. An interesting study found a role for C1q in PD. Neuromelanin (NM) is a pigment that accumulates in dopaminergic neurons in normal aging process. In PD, these dopaminergic neurons are susceptible to degeneration [302] which is thought to be caused by activation of microglia by NM [303]. Furthermore, this NM pigment is found to be opsonised by C1q and phagocytosed by C1q-positive microglia [304].
Huntington's disease (HD) is another neurodegenerative disorder and a genetic cause of dementia. It is inherited as an autosomal-dominant trait characterised by abnormal (at least 36) CAG repeats on the coding sequence of huntingtin gene [305]. Neuropathological studies in HD brains show presence of complement components C1q, C4, and C3 along with upregulation of complement regulators C1 inhibitor, clusterin, CD59, and CD55. In this study, microglial expression of higher levels of C3 and C9 was also observed [306].
There has been increasing evidence for involvement of complement in schizophrenia. Schizophrenia is a psychiatric illness characterised by thought insertion, thought withdrawal, hallucinations, delusions, and negative symptoms such as apathy, speech problems, and slow cognition. There is an increase in serum levels of classical pathway complement proteins such as C1q, C1, C3, and C4; increased total complement activity (CH50), CR1 levels; and decreased C4BP levels [307–309]. The alternative pathway is also involved with increased factor B levels and increased activity in serum [310]. MBL pathway shows increased activity as well (increased MBL and MASP-2 levels) [311, 312]. Genetic studies have shown C1QB gene polymorphism, CSMD1 and CSMD2 (code for complement regulatory proteins), C3, MBL2, and MASP2 gene association [313–316].
8. Conclusion
A role for innate immunity in inflammation of CNS is being increasingly evidenced. Cells of the CNS such as neurons, astrocytes, and microglia along with pattern recognition receptors, cytokines, chemokines, complement, peripheral immune cells, and signal pathways form the basis for neuroinflammation. Local synthesis of a number of innate immune humoral factors within CNS offers an opportunity for therapeutic intervention. Furthermore, excessive activation of immune system is thought to be destructive to tissues whereas, simultaneously, it opens up possibilities to harness this activation in a controlled manner to obtain desired therapeutic or preventive strategies in CNS diseases. A detailed understanding of the processes and mechanisms involved in the etiopathogenesis of CNS diseases as well as normal functioning of CNS immunity is essential and can pave the way for reducing excessive neuroinflammation and its effects. Modulation of cellular processes, phenotypes, and functions looks increasingly likely to be a way forward in combating CNS disorders.
Abbreviations
- Aβ:
Amyloid-β
- AD:
Alzheimer's disease
- BIR:
Baculovirus inhibitor of apoptosis protein repeat
- C4BP:
C4b-binding protein
- CARD:
Caspase activation and recruitment domain
- CNS:
Central nervous system
- Crry:
Complement receptor 1-related protein-y
- DAMP:
Damage-associated molecular pattern
- DOCK8:
Dedicator of cytokinesis 8
- HSV:
Herpes simplex virus
- HD:
Huntington's disease
- IL:
Interleukin
- LPS:
Lipopolysaccharide
- MAPK:
Mitogen-activated protein kinase
- MBL:
Mannan-binding lectin
- MASP:
MBL-associated serine protease
- MyD88:
Myeloid differentiating factor 88
- NF-κβ:
Nuclear factor-κβ
- NOD:
Nucleotide-binding and oligomerisation domain
- NLR:
NOD-like receptors
- NM:
Neuromelanin
- PAMP:
Pathogen-associated molecular pattern
- PD:
Parkinson's disease
- PRR:
Pattern-recognition receptor
- PYD:
Pyrin domain
- RAGE:
Receptor for advanced glycation endproducts
- SR:
Scavenger receptor
- SR-BI:
Class B SR type I
- TBI:
Traumatic brain injury
- TLRs:
Toll-like receptors
- TNF:
Tumour necrosis factor
- WNV:
West Nile virus.
Endnotes
PAMPs are conserved sequences or structural fragments on pathogens (nonself) that are recognised by PRRs. Examples of PAMP include bacterial, viral, fungal, and parasitic-derived lipids (lipopolysaccharide, lipoteichoic acid), proteins (flagellin), carbohydrates (mannan, zymosan), and nucleic acids (dsRNA, CpG).
DAMPs are endogenous molecules released from damaged cells (altered self). Examples of DAMP include heat shock proteins, ATP, and uric acid.
Toxoplasmosis is caused by Toxoplasma gondii. Cats are the definitive hosts and humans being intermediate hosts of T. gondii. Infection to humans spreads with contamination of food and water by cat faeces as well as eating undercooked meat infected with the parasitic cyst. Clinically, swelling of lymph nodes may occur but, interestingly, toxoplasmosis is associated with psychiatric disorders like schizophrenia, bipolar disorder, anxiety, and personality disorders.
Sleeping sickness is also known as Human African trypanosomiasis. It is caused by Trypanosoma brucei and is transmitted by tsetse fly. Prevalence is mainly in West, Central, and East Africa. It is characterised by intermittent fever and CNS manifestations in late stages including tremors, encephalopathy, and sleep disturbances which is mainly daytime somnolence.
Cerebral malaria is encephalopathy caused by sequelae of Plasmodium falciparum infection. Neurological features include coma, seizures, and upper-motor neuron lesion features (muscle spasticity and rigidity).
Neurocysticercosis is an infection of the CNS caused by the tapeworm Taenia solium. Pig is the intermediate host while humans are the definitive hosts of T. solium. Most common clinical presentation is seizures (an important and leading cause for acquired epilepsy) and focal neurological signs depending on the site and localisation of the cysts.
ALS is also known as motor neurone disease and Lou Gehrig's disease. Majority of the cases are idiopathic with however a small percentage (5–10%) being familial. Mutations in genes SOD1 (codes for Superoxide dismutase 1, an antioxidant); TARDBP (codes for Transactive response DNA-binding protein 43, a nuclear protein); and FUS (codes for Fused in Sarcoma, another cellular protein) are involved in familial ALS. It is a fatal, progressive neurodegenerative disease characterised by muscle spasticity, wasting and fasciculations as well as dysphagia and dysarthria. Interestingly, ALS is associated with frontotemporal dementia and this lead to discovery of mutation in C9ORF72 gene (abnormal nucleotide repeats) in familial and sporadic forms of ALS [317–319].
Hydatidiform mole is a gestational trophoblastic disease. Trophoblasts are precursors to placental cells. The products of conception will completely or partially comprise of grape-like vesicles or sacs (villous trophoblast). Most pregnancies are not viable with presenting symptom being vaginal bleeding. Early diagnosis can be established by ultrasonography (“snowstorm” appearance).
Muckle-Wells syndrome is an autosomal dominant disease characterised by the presence of intermittent fevers, rashes, sensorineural hearing loss, and amyloidosis. Mutation occurs in gene CIAS1.
Metabolic syndrome refers to a combination of hyperglycemia, obesity, dyslipidaemia, and hypertension.
MicroRNAs are 22 nucleotide RNAs that are noncoding and repress expression of mRNAs.
References
- 1.Murphy K. Innate Immunity: The First Lines of Defense. Janeway's Immunobiology. 8th edition. Abingdon, UK: Garland Science, Taylor & Francis Group; 2012. [Google Scholar]
- 2.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 3.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 5.Sierra A, Encinas JM, Deudero JJP, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinet J, Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. Journal of Neuroinflammation. 2012;9:p. 27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 8.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 10.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. The Journal of Immunology. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 11.Von Zahn J, Möller T, Kettenmann H, Nolte C. Microglial phagocytosis is modulated by pro-and anti-inflammatory cytokines. NeuroReport. 1997;8(18):3851–3856. doi: 10.1097/00001756-199712220-00003. [DOI] [PubMed] [Google Scholar]
- 12.Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-α . The FASEB Journal. 2006;20(6):670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- 13.Combs CK, Colleen Karlo J, Kao SC, Landreth GE. β-amyloid stimulation of microglia anti monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. The Journal of Neuroscience. 2001;21(4):1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barcia C, Ros CM, Annese V, et al. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death and Disease. 2011;2(4, article e142) doi: 10.1038/cddis.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basu A, Krady JK, Levison SW. Interleukin-1: a master regulator of neuroinflammation. Journal of Neuroscience Research. 2004;78(2):151–156. doi: 10.1002/jnr.20266. [DOI] [PubMed] [Google Scholar]
- 16.Shaftel SS, Griffin WST, Kerry KM. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. Journal of Neuroinflammation. 2008;5, article 7 [Google Scholar]
- 17.Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(4):E197–E205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constam DB, Philipp J, Malipiero UV, Ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-β1, -β2, and -β3 by glioblastoma cells, astrocytes, and microglia. The Journal of Immunology. 1992;148(5):1404–1410. [PubMed] [Google Scholar]
- 19.Lodge PA, Sriram S. Regulation of microglial activation by TGF-β, IL-10, and CSF-1. Journal of Leukocyte Biology. 1996;60(4):502–508. doi: 10.1002/jlb.60.4.502. [DOI] [PubMed] [Google Scholar]
- 20.Williams K, Dooley N, Ulvestad E, Becher B, Antel JP. IL-10 production by adult human derived microglial cells. Neurochemistry International. 1996;29(1):55–64. doi: 10.1016/0197-0186(95)00138-7. [DOI] [PubMed] [Google Scholar]
- 21.Rivest S. Regulation of innate immune responses in the brain. Nature Reviews Immunology. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 22.Glezer I, Rivest S. Glucocorticoids: protectors of the brain during innate immune responses. Neuroscientist. 2004;10(6):538–552. doi: 10.1177/1073858404263494. [DOI] [PubMed] [Google Scholar]
- 23.Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. The Journal of Neuroscience. 2003;23(13):5536–5544. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64(1):33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 26.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β . Nature Immunology. 2008;9(8):857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, et al. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. Journal of Neuroinflammation. 2012;9:p. 73. doi: 10.1186/1742-2094-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster SD, Park M, Fonseca MI, Tenner AJ. Structural and functional evidence for microglial expression of C1qR(p), the C1q receptor that enhances phagocytosis. Journal of Leukocyte Biology. 2000;67(1):109–116. doi: 10.1002/jlb.67.1.109. [DOI] [PubMed] [Google Scholar]
- 29.Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Research Reviews. 2010;63(1-2):2–10. doi: 10.1016/j.brainresrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annual Review of Physiology. 2009;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010;463(7278):232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halassa MM, Florian C, Fellin T, et al. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61(2):213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki A, Stern SA, Bozdagi O, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144(5):810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in Immunology. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 36.Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(16):6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Neerven S, Nemes A, Imholz P, et al. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. Journal of Neuroimmunology. 2010;229(1-2):169–179. doi: 10.1016/j.jneuroim.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Johnstone M, Gearing AJH, Miller KM. A central role for astrocytes in the inflammatory response to β- amyloid; chemokines, cytokines and reactive oxygen species are produced. Journal of Neuroimmunology. 1999;93(1-2):182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- 39.Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death and Disease. 2011;2(6, article e167) doi: 10.1038/cddis.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barreto GE, Gonzalez J, Capani F, Morales L. Role of astrocytes in neurodegenerative diseases. In: Chang RCC, editor. Neurodegenerative Diseases—Processes, Prevention, Protection and Monitoring. InTech; 2011. http://www.intechopen.com/books/neurodegenerative-diseases-processes-prevention-protection-and-monitoring/role-of-astrocytes-in-neurodegenerative-diseases. [Google Scholar]
- 42.Kawai T, Akira S. Signaling to NF-κB by toll-like receptors. Trends in Molecular Medicine. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 44.O’Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Reviews Immunology. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 45.Ozinsky A, Underhill DM, Fontenot JD, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(25):13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oosting M, Ter Hofstede H, Sturm P, Adema GJ, Kullberg BJ, van der Meer JW, et al. TLR1/TLR2 heterodimers play an important role in the recognition of borrelia spirochetes. PLoS ONE. 2011;6(10) doi: 10.1371/journal.pone.0025998.e25998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leulier F, Lemaitre B. Toll-like receptors—taking an evolutionary approach. Nature Reviews Genetics. 2008;9(3):165–178. doi: 10.1038/nrg2303. [DOI] [PubMed] [Google Scholar]
- 48.Siednienko J, Gajanayake T, Fitzgerald KA, Moynagh P, Miggin SM. Absence of MyD88 results in enhanced TLR3-dependent phosphorylation of IRF3 and increased IFN-β and RANTES production. The Journal of Immunology. 2011;186(4):2514–2522. doi: 10.4049/jimmunol.1003093. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Zhan Z, Li D, et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nature Immunology. 2011;12(5):416–424. doi: 10.1038/ni.2015. [DOI] [PubMed] [Google Scholar]
- 50.Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by toll-like receptors. Nature Immunology. 2011;12(8):742–751. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- 51.Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nature Immunology. 2012 13(6):612–620. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(16):6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pais TF, Figueiredo C, Peixoto R, Braz MH, Chatterjee S. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. Journal of Neuroinflammation. 2008;5, article 43 doi: 10.1186/1742-2094-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolanle F, Yongshan M, Maria S, Modinat L, Hallenbeck J. Downstream toll-like receptor signaling mediates adaptor-specific cytokine expression following focal cerebral ischemia. Journal of Neuroinflammation. 2012;9:p. 174. doi: 10.1186/1742-2094-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. Journal of Neuroinflammation. 2012;9:p. 46. doi: 10.1186/1742-2094-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu S, Kielian T. MyD88 is pivotal for immune recognition of Citrobacter koseri and astrocyte activation during CNS infection. Journal of Neuroinflammation. 2011;8, article 35 doi: 10.1186/1742-2094-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drouin-Ouellet J, Gibrat C, Bousquet M, Calon F, Kriz J, Cicchetti F. The role of the MYD88-dependent pathway in MPTP-induced brain dopaminergic degeneration. Journal of Neuroinflammation. 2011;8:p. 137. doi: 10.1186/1742-2094-8-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng Z, Yuan R, Song M, Huo Y, Liu W, Cai X, et al. The toll-like receptor 4-mediated signaling pathway is activated following optic nerve injury in mice. Brain Research. 2012;1489:90–97. doi: 10.1016/j.brainres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 59.Trudler D, Frenkel D, Farfara D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: towards future therapeutic application. Mediators of Inflammation. 2010;2010:12 pages. doi: 10.1155/2010/497987.497987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cole JE, Georgiou E, Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators of Inflammation. 2010;2010:18 pages. doi: 10.1155/2010/393946.393946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. Journal of Leukocyte Biology. 2004;76(3):514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 62.Beg AA. Endogenous ligands of Toll-like receptors: Implications for regulating inflammatory and immune responses. Trends in Immunology. 2002;23(11):509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- 63.Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathology. 1998;8(4):625–640. doi: 10.1111/j.1750-3639.1998.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quan N, Whiteside M, Kim L, Herkenham M. Induction of inhibitory factor κBα mRNA in the central nervous system after peripheral lipopolysaccharide administration: an in situ hybridization histochemistry study in the rat. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(20):10985–10990. doi: 10.1073/pnas.94.20.10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell. 2011;147(4):868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Glezer I, Chernomoretz A, David S, Plante MM, Rivest S. Genes involved in the balance between neuronal survival and death during inflammation. PLoS ONE. 2007;2(3, article e310) doi: 10.1371/journal.pone.0000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147(4):867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 68.Harms AS, Lee JK, Nguyen TA, Chang J, Ruhn KM, Trevino I, et al. Regulation of microglia effector functions by tumor necrosis factor signaling. Glia. 2012;60(2):189–202. doi: 10.1002/glia.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. The Journal of Immunology. 2005;175(7):4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 70.Packard AE, Leung PY, Vartanian KB, Stevens SL, Bahjat FR, Stenzel-Poore MP. TLR9 bone marrow chimeric mice define a role for cerebral TNF in neuroprotection induced by CpG preconditioning. Journal of Cerebral Blood Flow & Metabolism. 2012;32(12):2193–2200. doi: 10.1038/jcbfm.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hyakkoku K, Hamanaka J, Tsuruma K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171(1):258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 72.Rolls A, Shechter R, London A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nature Cell Biology. 2007;9(9):1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 73.Ma Y, Li J, Chiu I, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. The Journal of Cell Biology. 2006;175(2):209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okun E, Griffioen K, Barak B, et al. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(35):15625–15630. doi: 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaul D, Habbel P, Derkow K, Kruger C, Franzoni E, Wulczyn FG, et al. Expression of toll-like receptors in the developing brain. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037767.e37767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McKernan DP, Dennison U, Gaszner G, Cryan JF, Dinan TG. Enhanced peripheral toll-like receptor responses in psychosis: further evidence of a pro-inflammatory phenotype. Transl Psychiatry. 2011;1(8):p. e36. doi: 10.1038/tp.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zimmerman G, Shaltiel G, Barbash S, Cohen J, Gasho CJ, Shenhar-Tsarfaty S, et al. Post-traumatic anxiety associates with failure of the innate immune receptor TLR9 to evade the pro-inflammatory NFκB pathway. Transl Psychiatry. 2012;2:p. e78. doi: 10.1038/tp.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klein M, Obermaier B, Angele B, et al. Innate immunity to pneumococcal infection of the central nervous system depends on toll-like receptor (TLR) 2 and TLR4. The Journal of Infectious Diseases. 2008;198(7):1028–1036. doi: 10.1086/591626. [DOI] [PubMed] [Google Scholar]
- 79.Letiembre M, Echchannaoui H, Ferracin F, Rivest S, Landmann R. Toll-like receptor-2 deficiency is associated with enhanced brain TNF gene expression during pneumococcal meningitis. Journal of Neuroimmunology. 2005;168(1-2):21–33. doi: 10.1016/j.jneuroim.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 80.Dutta R, Krishnan A, Meng J, Das S, Ma J, Banerjee S, et al. Morphine modulation of toll-like receptors in microglial cells potentiates neuropathogenesis in a HIV-1 model of coinfection with pneumococcal pneumoniae. The Journal of Neuroscience. 2012;32(29):9917–9930. doi: 10.1523/JNEUROSCI.0870-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arpaia N, Godec J, Lau L, et al. TLR signaling is required for salmonella typhimurium virulence. Cell. 2011;144(5):675–688. doi: 10.1016/j.cell.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morrison LA. The Toll of herpes simplex virus infection. Trends in Microbiology. 2004;12(8):353–356. doi: 10.1016/j.tim.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 83.Sørensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. The Journal of Immunology. 2008;181(12):8604–8612. doi: 10.4049/jimmunol.181.12.8604. [DOI] [PubMed] [Google Scholar]
- 84.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 85.Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. The Journal of Clinical Investigation. 2012;122(4):1368–1376. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates west nile virus entry into the brain causing lethal encephalitis. Nature Medicine. 2004;10(12):1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 87.Mishra BB, Gundra UM, Teale JM. Toll-like receptors in CNS parasitic infections. Current Topics in Microbiology and Immunology. 2009;336(1):83–104. doi: 10.1007/978-3-642-00549-7_5. [DOI] [PubMed] [Google Scholar]
- 88.Greene JA, Sam-Agudu N, John CC, Opoka RO, Zimmerman PA, Kazura JW. Toll-like receptor polymorphisms and cerebral malaria: TLR2 Δ22 polymorphism is associated with protection from cerebral malaria in a case control study. Malaria Journal. 2012;11:p. 47. doi: 10.1186/1475-2875-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Franklin BS, Ishizaka ST, Lamphier M, et al. Therapeutical targeting of nucleic acid-sensing toll-like receptors prevents experimental cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu X, Pan Y, Li Y, Jiang Y, Shang H, Gowda DC, et al. Targeting toll-like receptors by chloroquine protects mice from experimental cerebral malaria. International Immunopharmacology. 2012;13(4):392–397. doi: 10.1016/j.intimp.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 91.Michaud JP, Richard KL, Rivest S. MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer’s disease. Molecular Neurodegeneration. 2011;6(1, article 5) doi: 10.1186/1750-1326-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Michaud JP, Richard KL, Rivest S. Hematopoietic MyD88-adaptor protein acts as a natural defense mechanism for cognitive deficits in Alzheimer's disease. Stem Cell Reviews and Reports. 2012;8(3):898–904. doi: 10.1007/s12015-012-9356-9. [DOI] [PubMed] [Google Scholar]
- 93.Hao W, Liu Y, Liu S, et al. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain. 2011;134(1):278–292. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- 94.Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2008;28(22):5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. The Journal of Neuroscience. 2009;29(38):11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang SC, Lathia JD, Selvaraj PK, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Experimental Neurology. 2008;213(1):114–121. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song M, Jin J, Lim JE, et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of Neuroinflammation. 2011:p. 92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herber DL, Roth LM, Wilson D, et al. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Experimental Neurology. 2004;190(1):245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 99.Scholtzova H, Kascsak RJ, Bates KA, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. The Journal of Neuroscience. 2009;29(6):1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doi Y, Mizuno T, Maki Y, et al. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. American Journal of Pathology. 2009;175(5):2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iribarren P, Chen K, Hu J, et al. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid β 1-42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. The FASEB Journal. 2005;19(14):2032–2034. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- 102.Kang J, Rivest S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. The Journal of Cell Biology. 2007;179(6):1219–1230. doi: 10.1083/jcb.200705046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu Y, Hao W, Dawson A, Liu S, Fassbender K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. The Journal of Biological Chemistry. 2009;284(6):3691–3699. doi: 10.1074/jbc.M804446200. [DOI] [PubMed] [Google Scholar]
- 104.Zhao W, Beers DR, Henkel JS, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58(2):231–243. doi: 10.1002/glia.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Beraud D, Maguire-Zeiss KA. Misfolded alpha-synuclein and toll-like receptors: therapeutic targets for parkinson's disease. Parkinsonism & Related Disorders. 2012;(1):S17–S20. doi: 10.1016/S1353-8020(11)70008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spinner DS, In SC, Seung YP, et al. Accelerated prion disease pathogenesis in toll-like receptor 4 signaling-mutant mice. Journal of Virology. 2008;82(21):10701–10708. doi: 10.1128/JVI.00522-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sethi S, Lipford G, Wagner H, Kretzschmar H. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. The Lancet. 2002;360(9328):229–230. doi: 10.1016/S0140-6736(02)09513-2. [DOI] [PubMed] [Google Scholar]
- 108.Prinz M, Heikenwalder M, Schwarz P, Takeda K, Akira S, Aguzzi A. Prion pathogenesis in the absence of toll-like receptor signalling. The EMBO Reports. 2003;4(2):195–199. doi: 10.1038/sj.embor.embor731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chauhan VS, Sterka DG, Furr SR, Marriott I. NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia. 2009;57(4):414–423. doi: 10.1002/glia.20770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bajramovic JJ. Regulation of innate immune responses in the central nervous system. CNS & Neurological Disorders Drug Targets. 2011;10(1):4–24. doi: 10.2174/187152711794488610. [DOI] [PubMed] [Google Scholar]
- 111.Areschoug T, Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cellular Microbiology. 2009;11(8):1160–1169. doi: 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 112.Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. International Journal of Alzheimer's Disease. 2012;2012:10 pages. doi: 10.1155/2012/489456.489456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Furr SR, Chauhan V, Sterka D, Grdzelishvili V, Marriott I. Characterization of retinoic acid-inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus. Journal of NeuroVirology. 2008;14(6):503–513. doi: 10.1080/13550280802337217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crehan H, Hardy J, Pocock J. Microglia, alzheimer's disease, and complement. International Journal of Alzheimer's Disease. 2012;2012 doi: 10.1155/2012/983640.983640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. The Journal of Neuroscience Research. 2002;69(1):94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- 116.Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. Journal of Leukocyte Biology. 2009;85(3):352–370. doi: 10.1189/jlb.0608385. [DOI] [PubMed] [Google Scholar]
- 117.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 118.Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. The Journal of Immunology. 2002;169(1):10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 119.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 120.Takeuchi O, Kawai T, Mühlradt PF, et al. Discrimination of bacterial lipoproteins by Toll-like recepttor 6. International Immunology. 2001;13(7):933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 121.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. The Journal of Biological Chemistry. 1999;274(25):17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 122.Sato M, Sano H, Iwaki D, et al. Direct binding of toll-like receptor 2 to Zymosan, and Zymosan-induced NF-κB activation and TNF-α secretion are down-regulated by lung collectin surfactant protein A. The Journal of Immunology. 2003;171(1):417–425. doi: 10.4049/jimmunol.171.1.417. [DOI] [PubMed] [Google Scholar]
- 123.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 124.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 125.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis E Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 126.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 127.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 128.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. The Journal of Immunology. 2012;188(3):1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 130.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nature Immunology. 2010;11(2):155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Henning LN, Azad AK, Parsa KVL, Crowther JE, Tridandapani S, Schlesinger LS. Pulmonary surfactant protein a regulates TLR expression and activity in human macrophages. The Journal of Immunology. 2008;180(12):7847–7858. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Murakami S, Iwaki D, Mitsuzawa H, et al. Surfactant protein a inhibits peptidoglycan-induced tumor necrosis factor-α secretion in U937 cells and alveolar macrophages by direct interaction with toll-like receptor 2. The Journal of Biological Chemistry. 2002;277(9):6830–6837. doi: 10.1074/jbc.M106671200. [DOI] [PubMed] [Google Scholar]
- 133.Ohya M, Nishitani C, Sano H, et al. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry. 2006;45(28):8657–8664. doi: 10.1021/bi060176z. [DOI] [PubMed] [Google Scholar]
- 134.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nature Medicine. 2009;15(7):774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 135.Piccinini AM, Midwood KS. Endogenous control of immunity against infection: tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Reports. 2012;2(4):914–926. doi: 10.1016/j.celrep.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fcγ receptor. Arthritis and Rheumatism. 2011;63(1):53–62. doi: 10.1002/art.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hodgkinson CP, Patel K, Ye S. Functional Toll-like receptor 4 mutations modulate the response to fibrinogen. Thrombosis and Haemostasis. 2008;100(2):301–307. [PubMed] [Google Scholar]
- 138.Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, et al. An unconventional role for miRNA: let-7 activates toll-like receptor 7 and causes neurodegeneration. Nature Neuroscience. 2012;15(6):827–835. doi: 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- 139.Rosenstiel P, Schreiber S. NOD-like receptors—pivotal guardians of the immunological integrity of barrier organs. In: Kishore U, editor. Target Pattern Recognition in Innate Immunity. Austin, Tex, USA: Landes Bioscience; 2009. pp. 35–47. [DOI] [PubMed] [Google Scholar]
- 140.Langefeld T, Mohamed W, Ghai R, Chakraborty T. Toll-like receptors and NOD-like receptors: domain architecture and cellular signalling. In: Kishore U, editor. Target Pattern Recognition in Innate Immunity. Austin, Tex, USA: Landes Bioscience; 2009. pp. 48–57. [DOI] [PubMed] [Google Scholar]
- 141.Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. NOD-like receptors and the innate immune system: coping with danger, damage and death. Cytokine & Growth Factor Reviews. 2011;22(5-6):257–276. doi: 10.1016/j.cytogfr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 142.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nature Reviews Microbiology. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Medicine. 2010;16(1):90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 144.Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36(6):933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O'Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37(6):1009–1023. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annual Review of Cell and Developmental Biology. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 147.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annual Review of Immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 148.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 149.Wen H, Ting JP, O'Neill LA. A role for the NLRP3 inflammasome in metabolic diseases—did warburg miss inflammation? Nature Immunology. 2012;13(4):352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 151.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(49):19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shaw PJ, McDermott MF, Kanneganti TD. Inflammasomes and autoimmunity. Trends in Molecular Medicine. 2011;17(2):57–64. doi: 10.1016/j.molmed.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. American Journal of Physiology. 2012;303(8):L627–L633. doi: 10.1152/ajplung.00225.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nature Reviews Immunology. 2010;10(10):688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. The Journal of Immunology. 2011;187(2):596–602. doi: 10.4049/jimmunol.1100229. [DOI] [PubMed] [Google Scholar]
- 156.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β . Nature Immunology. 2008;9(8):857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jha S, Srivastava SY, Brickey WJ, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. The Journal of Neuroscience. 2010;30(47):15811–15820. doi: 10.1523/JNEUROSCI.4088-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(26):10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Science Signaling. 2012;5(225):p. ra38. doi: 10.1126/scisignal.2002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.De Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism. 2009;29(7):1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Abulafia DP, De Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. Journal of Cerebral Blood Flow and Metabolism. 2009;29(3):534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 162.Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, et al. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. The Journal of Immunology. 2011;187(10):5440–5451. doi: 10.4049/jimmunol.1100790. [DOI] [PubMed] [Google Scholar]
- 163.Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine japanese encephalitis. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0032270.e32270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zou J, Crews FT. Inflammasome-IL-1β signaling mediates ethanol inhibition of hippocampal neurogenesis. Frontiers in Neuroscience. 2012;6:p. 77. doi: 10.3389/fnins.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(1):333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Godoy B, Murgas P, Tichauer J, Von Bernhardi R. Scavenger receptor class a ligands induce secretion of IL1β and exert a modulatory effect on the inflammatory activation of astrocytes in culture. Journal of Neuroimmunology. 2012;251(1-2):6–13. doi: 10.1016/j.jneuroim.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mukhopadhyay S, Pluddemann A, Gordon S. Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. In: Kishore U, editor. Target Pattern Recognition in Innate Immunity. Austin, Tex, USA: Landes Bioscience; 2009. pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yamada Y, Doi T, Hamakubo T, Kodama T. Scavenger receptor family proteins: roles for atherosclerosis, host defence and disorders of the central nervous system. Cellular and Molecular Life Sciences. 1998;54(7):628–640. doi: 10.1007/s000180050191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. Journal of Lipid Research. 2009;50:S282–S286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wermeling F, Chen Y, Pikkarainen T, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. Journal of Experimental Medicine. 2007;204(10):2259–2265. doi: 10.1084/jem.20070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Oquendo P, Hundt E, Lawler J, Seed B. CD36 directly mediates cytoadherence of plasmodium falciparum parasitized erythrocytes. Cell. 1989;58(1):95–101. doi: 10.1016/0092-8674(89)90406-6. [DOI] [PubMed] [Google Scholar]
- 172.Urban BC, Ferguson DJP, Pain A, et al. Plasmodium falciparuminfected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400(6739):73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 173.Urban BC, Willcox N, Roberts DJ. A role for CD36 in the regulation of dendritic cell function. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8750–8755. doi: 10.1073/pnas.151028698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Stephen SL, Freestone K, Dunn S, Twigg MW, Homer-Vanniasinkam S, Walker JH, et al. Scavenger receptors and their potential as therapeutic targets in the treatment of cardiovascular disease. International Journal of Hypertension. 2010;2010:21 pages. doi: 10.4061/2010/646929.646929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thakur SA, Hamilton RF, Holian A. Role of scavenger receptor a family in lung inflammation from exposure to environmental particles. Journal of Immunotoxicology. 2008;5(2):151–157. doi: 10.1080/15476910802085863. [DOI] [PubMed] [Google Scholar]
- 176.Wright AKA, Rao S, Range S, et al. Pivotal advance: expansion of small sputum macrophages in CF: failure to express MARCO and mannose receptors. Journal of Leukocyte Biology. 2009;86(3):479–489. doi: 10.1189/jlb.1108699. [DOI] [PubMed] [Google Scholar]
- 177.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382(6593):716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 178.Christie RH, Freeman M, Hyman BT. Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. The American Journal of Pathology. 1996;148(2):399–403. [PMC free article] [PubMed] [Google Scholar]
- 179.Xu Y, Qian L, Zong G, Ma K, Zhu X, Zhang H, et al. Class A scavenger receptor promotes cerebral ischemic injury by pivoting microglia/macrophage polarization. Neuroscience. 2012;218:35–48. doi: 10.1016/j.neuroscience.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 180.Levy-Barazany H, Frenkel D. Expression of scavenger receptor A on antigen presenting cells is important for CD4+ T-cells proliferation in EAE mouse model. Journal of Neuroinflammation. 2012;9:120 pages. doi: 10.1186/1742-2094-9-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Huang F, Buttini M, Wyss-Coray T, et al. Elimination of the class a scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. The American Journal of Pathology. 1999;155(5):1741–1747. doi: 10.1016/S0002-9440(10)65489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Husemann J, Silverstein SC. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. The American Journal of Pathology. 2001;158(3):825–832. doi: 10.1016/S0002-9440(10)64030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Srivastava RAK, Jain JC. Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer’s disease brain. Journal of the Neurological Sciences. 2002;196(1-2):45–52. doi: 10.1016/s0022-510x(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 184.Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type i (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(48):20816–20821. doi: 10.1073/pnas.1005888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-β . Proceedings of the National Academy of Sciences of the United States of America. 2011;108(12):5063–5068. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(8):3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shi DY, Bierhaus A, Nawroth PP, Stern DM. RAGE and Alzheimer’s disease: a progression factor for amyloid-β- induced cellular perturbation? Journal of Alzheimer’s Disease. 2009;16(4):833–843. doi: 10.3233/JAD-2009-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fang, Lue LF, Yan S, et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. The FASEB Journal. 2010;24(4):1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Origlia N, Righi M, Capsoni S, et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. The Journal of Neuroscience. 2008;28(13):3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Takuma K, Fang F, Zhang W, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Porter RR, Reid KBM. Activation of the complement system by antibody-antigen complexes: the classical pathway. Advances in Protein Chemistry. 1979;33:1–71. doi: 10.1016/s0065-3233(08)60458-1. [DOI] [PubMed] [Google Scholar]
- 192.Dahl MR, Thiel S, Matsushita M, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15(1):127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 193.Thiel S, Vorup-Jensen T, Stover CM, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386(6624):506–510. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 194.Carroll MV, Sim RB. Complement in health and disease. Advanced Drug Delivery Reviews. 2011;63(12):965–975. doi: 10.1016/j.addr.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 195.Kang YH, Tan LA, Carroll MV, Gentle ME, Sim RB. Target pattern recognition by complement proteins of the classical and alternative pathways. Advances in Experimental Medicine and Biology. 2009;653:117–128. doi: 10.1007/978-1-4419-0901-5_8. [DOI] [PubMed] [Google Scholar]
- 196.Thiel S, Gadjeva M. Humoral pattern recognition molecules: mannan-binding lectin and ficolins. Advances in Experimental Medicine and Biology. 2009;653:58–73. doi: 10.1007/978-1-4419-0901-5_5. [DOI] [PubMed] [Google Scholar]
- 197.Reid KB, Porter RR. The proteolytic activation systems of complement. Annual Review of Biochemistry. 1981;50:433–464. doi: 10.1146/annurev.bi.50.070181.002245. [DOI] [PubMed] [Google Scholar]
- 198.Hajishengallis G, Lambris JD. Crosstalk pathways between toll-like receptors and the complement system. Trends in Immunology. 2010;31(4):154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37(2):199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Petersen SV, Thiel S, Jensen L, Vorup-Jensen T, Koch C, Jensenius JC. Control of the classical and the MBL pathway of complement activation. Molecular Immunology. 2001;37(14):803–811. doi: 10.1016/s0161-5890(01)00004-9. [DOI] [PubMed] [Google Scholar]
- 201.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nature Reviews Immunology. 2009;9(10):729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 202.Kishore U, Sim RB. Factor H as a regulator of the classical pathway activation. Immunobiology. 2012;217(2):162–168. doi: 10.1016/j.imbio.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 203.Tan LA, Yang AC, Kishore U, Sim RB. Interactions of complement proteins C1q and factor H with lipid A and Escherichia coli: further evidence that factor H regulates the classical complement pathway. Protein & Cell . 2011;2(4):320–332. doi: 10.1007/s13238-011-1029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tan LA, Yu B, Sim FCJ, Kishore U, Sim RB. Complement activation by phospholipids: the interplay of factor H and C1q. Protein & Cell. 2010;1(11):1033–1049. doi: 10.1007/s13238-010-0125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kang YH, Urban BC, Sim RB, Kishore U. Human complement factor H modulates C1q-mediated phagocytosis of apoptotic cells. Immunobiology. 2012;217(4):455–464. doi: 10.1016/j.imbio.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 206.Walker DG, McGeer PL. Complement gene expression in neuroblastoma and astrocytoma cell lines of human origin. Neuroscience Letters. 1993;157(1):99–102. doi: 10.1016/0304-3940(93)90652-2. [DOI] [PubMed] [Google Scholar]
- 207.Walker DG, Yasuhara O, Patston PA, McGeer EG, McGeer PL. Complement C1 inhibitor is produced by brain tissue and is cleaved in Alzheimer disease. Brain Research. 1995;675(1-2):75–82. doi: 10.1016/0006-8993(95)00041-n. [DOI] [PubMed] [Google Scholar]
- 208.Veerhuis R, Janssen I, Hoozemans JJM, De Groot CJA, Hack CE, Eikelenboom P. Complement C1-inhibitor expression in Alzheimer’s disease. Acta Neuropathologica. 1998;96(3):287–296. doi: 10.1007/s004010050896. [DOI] [PubMed] [Google Scholar]
- 209.Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. International Immunology. 2000;12(7):1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- 210.Barnum SR, Jones JL, Benveniste EN. Interleukin-1 and tumor necrosis factor-mediated regulation of C3 gene expression in human astroglioma cells. Glia. 1993;7(3):225–236. doi: 10.1002/glia.440070306. [DOI] [PubMed] [Google Scholar]
- 211.Gasque P, Chan P, Mauger C, et al. Identification and characterization of complement C3 receptors on human astrocytes. The Journal of Immunology. 1996;156(6):2247–2255. [PubMed] [Google Scholar]
- 212.Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglia derived from postmortem human brains. The Journal of Neuroscience Research. 1995;40(4):478–493. doi: 10.1002/jnr.490400407. [DOI] [PubMed] [Google Scholar]
- 213.Veerhuis R, Janssen I, De Groot CJA, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer’s disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Experimental Neurology. 1999;160(1):289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- 214.Gasque P, Singhrao SK, Neal JW, et al. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. The Journal of Immunology. 1998;160(7):3543–3554. [PubMed] [Google Scholar]
- 215.Rahpeymai Y, Hietala MA, Wilhelmsson U, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. The EMBO Journal. 2006;25(6):1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Moriyama M, Fukuhara T, Britschgi M, et al. Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. The Journal of Neuroscience. 2011;31(11):3981–3989. doi: 10.1523/JNEUROSCI.3617-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annual Review of Neuroscience. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 218.Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64(1):93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 219.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 220.Nayak A, Ferluga J, Tsolaki AG, Kishore U. The non-classical functions of the classical complement pathway recognition subcomponent C1q. Immunology Letters. 2010;131(2):139–150. doi: 10.1016/j.imlet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 221.Chu Y, Jin X, Parada I, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(17):7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. The Journal of Neuroscience. 2011;31(9):3459–3469. doi: 10.1523/JNEUROSCI.3932-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bénard M, Raoult E, Vaudry D, et al. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Molecular Immunology. 2008;45(14):3767–3774. doi: 10.1016/j.molimm.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 224.Van Beek J, Nicole O, Ali C, et al. Complement anaphylatoxin c3a is selectively protective against nmda-induced neuronal cell death. NeuroReport. 2001;12(2):289–293. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]
- 225.Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. Journal of Cellular Biochemistry. 1999;73(3):303–311. [PubMed] [Google Scholar]
- 226.Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. Journal of Neurochemistry. 2001;77(1):43–49. doi: 10.1046/j.1471-4159.2001.00167.x. [DOI] [PubMed] [Google Scholar]
- 227.Mukherjee P, Thomas S, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. Journal of Neuroinflammation. 2008;5, article 5 doi: 10.1186/1742-2094-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clinical Microbiology Reviews. 2010;23(4):740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kugelberg E, Gollan B, Tang CM. Mechanisms in Neisseria meningitidis for resistance against complement-mediated killing. Vaccine. 2008;26(8):I34–I39. doi: 10.1016/j.vaccine.2008.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Schneider MC, Exley RM, Chan H, et al. Functional significance of factor H binding to Neisseria meningitidis . The Journal of Immunology. 2006;176(12):7566–7575. doi: 10.4049/jimmunol.176.12.7566. [DOI] [PubMed] [Google Scholar]
- 231.Schneider MC, Prosser BE, Caesar JJE, et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature. 2009;458(7240):890–893. doi: 10.1038/nature07769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bathum L, Hansen H, Teisner B, et al. Association between combined properdin and mannose-binding lectin deficiency and infection with Neisseria meningitidis . Molecular Immunology. 2006;43(5):473–479. doi: 10.1016/j.molimm.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 233.Rupprecht TA, Angele B, Klein M, et al. Complement C1q and C3 are critical for the innate immune response to Streptococcus pneumoniae in the central nervous system. The Journal of Immunology. 2007;178(3):1861–1869. doi: 10.4049/jimmunol.178.3.1861. [DOI] [PubMed] [Google Scholar]
- 234.Wooster DG, Maruvada R, Blom AM, Prasadarao NV. Logarithmic phase Escherichia coli K1 efficiently avoids serum killing by promoting C4bp-mediated C3b and C4b degradation. Immunology. 2006;117(4):482–493. doi: 10.1111/j.1365-2567.2006.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. 2011;411(2):362–373. doi: 10.1016/j.virol.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kapadia SB, Levine B, Speck SH, Virgin HW. Critical role of complement and viral evasion of complement in acute, persistent, and latent γ-herpesvirus infection. Immunity. 2002;17(2):143–155. doi: 10.1016/s1074-7613(02)00369-2. [DOI] [PubMed] [Google Scholar]
- 237.Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS. Complement activation is required for induction of a protective antibody response against west nile virus infection. Journal of Virology. 2005;79(12):7466–7477. doi: 10.1128/JVI.79.12.7466-7477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fuchs A, Pinto AK, Schwaeble WJ, Diamond MS. The lectin pathway of complement activation contributes to protection from west nile virus infection. Virology. 2011;412(1):101–109. doi: 10.1016/j.virol.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Libbey JE, Kirkman NJ, Wilcox KS, White HS, Fujinami RS. Role for complement in the development of seizures following acute viral infection. Journal of Virology. 2010;84(13):6452–6460. doi: 10.1128/JVI.00422-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Singh KK, Nathamu S, Adame A, Alire TU, Dumaop W, Gouaux B, et al. Expression of mannose binding lectin in HIV-1-infected brain: implications for HIV-related neuronal damage and neuroAIDS. Neurobehavioral HIV Medicine. 2011;3:41–52. doi: 10.2147/NBHIV.S19969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rambach G, Maier H, Vago G, et al. Complement induction and complement evasion in patients with cerebral aspergillosis. Microbes and Infection. 2008;10(14-15):1567–1576. doi: 10.1016/j.micinf.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 242.Rambach G, Dum D, Mohsenipour I, et al. Secretion of a fungal protease represents a complement evasion mechanism in cerebral aspergillosis. Molecular Immunology. 2010;47(7-8):1438–1449. doi: 10.1016/j.molimm.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 243.Speth C, Rambach G. Complement attack against aspergillus and corresponding evasion mechanisms. Interdisciplinary Perspectives on Infectious Diseases. 2012;2012:9 pages. doi: 10.1155/2012/463794.463794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lackner P, Hametner C, Beer R, et al. Complement factors C1q, C3 and C5 in brain and serum of mice with cerebral malaria. Malaria Journal. 2008;7, article 207 doi: 10.1186/1475-2875-7-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ramos TN, Darley MM, Hu X, et al. Cutting edge: the membrane attack complex of complement is required for the development of murine experimental cerebral malaria. The Journal of Immunology. 2011;186(12):6657–6660. doi: 10.4049/jimmunol.1100603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mitchell DA, Kirby L, Paulin SM, Villiers CL, Sim RB. Prion protein activates and fixes complement directly via the classical pathway: implications for the mechanism of scrapie agent propagation in lymphoid tissue. Molecular Immunology. 2007;44(11):2997–3004. doi: 10.1016/j.molimm.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 247.Sim RB, Kishore U, Villiers CL, Marche PN, Mitchell DA. C1q binding and complement activation by prions and amyloids. Immunobiology. 2007;212(4-5):355–362. doi: 10.1016/j.imbio.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 248.Ishii T, Haga S, Yagishita S, Tateishi J. The presence of complements in amyloid plaques of creutzfeldt-Jakob disease and gerstmann-straussler-scheinker disease. Applied Pathology. 1984;2(6):370–379. [PubMed] [Google Scholar]
- 249.Kovacs GG, Gasque P, Ströbel T, et al. Complement activation in human prion disease. Neurobiology of Disease. 2004;15(1):21–28. doi: 10.1016/j.nbd.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 250.Bellander BM, Olafsson IH, Ghatan PH, et al. Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochirurgica. 2011;153(1):90–100. doi: 10.1007/s00701-010-0737-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. Journal of Neurotrauma. 2001;18(12):1295–1311. doi: 10.1089/08977150152725605. [DOI] [PubMed] [Google Scholar]
- 252.Stahel PF, Morganti-Kossmann MC, Perez D, et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. Journal of Neurotrauma. 2001;18(8):773–781. doi: 10.1089/089771501316919139. [DOI] [PubMed] [Google Scholar]
- 253.Stahel PF, Flierl MA, Morgan BP, et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. Journal of Neuroinflammation. 2009;6, article 2 doi: 10.1186/1742-2094-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Rancan M, Morganti-Kossmann MC, Barnum SR, et al. Central nervous system-targeted complement inhibition mediates neuroprotection after closed head injury in transgenic mice. Journal of Cerebral Blood Flow and Metabolism. 2003;23(9):1070–1074. doi: 10.1097/01.WCB.0000084250.20114.2C. [DOI] [PubMed] [Google Scholar]
- 255.Yanamadala V, Friedlander RM. Complement in neuroprotection and neurodegeneration. Trends in Molecular Medicine. 2010;16(2):69–76. doi: 10.1016/j.molmed.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Komotar RJ, Kim GH, Otten ML, et al. The role of complement in stroke therapy. Advances in Experimental Medicine and Biology. 2008;632:23–33. [PubMed] [Google Scholar]
- 257.Di Napoli M. Early inflammatory response in ischemic stroke. Thrombosis Research. 2001;103(3):261–264. doi: 10.1016/s0049-3848(01)00290-0. [DOI] [PubMed] [Google Scholar]
- 258.Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The role of the complement system in ischemia-reperfusion injury. Shock. 2004;21(5):401–409. doi: 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 259.Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. The FASEB Journal. 2012;26(9):3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- 260.Cervera A, Planas AM, Justicia C, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0008433.e8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pedersen ED, Løberg EM, Vege E, Daha MR, Mæhlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scandinavian Journal of Immunology. 2009;69(6):555–562. doi: 10.1111/j.1365-3083.2009.02253.x. [DOI] [PubMed] [Google Scholar]
- 262.Arumugam TV, Tang SC, Lathia JD, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(35):14104–14109. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mocco J, Mack WJ, Ducruet AF, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circulation Research. 2006;99(2):209–217. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 264.Heimann A, Takeshima T, Horstick G, Kempski O. C1-esterase inhibitor reduces infarct volume after cortical vein occlusion. Brain Research. 1999;838(1-2):210–213. doi: 10.1016/s0006-8993(99)01740-0. [DOI] [PubMed] [Google Scholar]
- 265.Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2009;158(3):1074–1089. doi: 10.1016/j.neuroscience.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Molecular Immunology. 2007;44(5):999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 267.Rogers J, Cooper NR, Webster S, et al. Complement activation by β-amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(21):10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Eikelenboom P, Hack CE, Rozemuller JM, Stam FC. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Archiv Abteilung B Cell Pathology. 1989;56(4):259–262. doi: 10.1007/BF02890024. [DOI] [PubMed] [Google Scholar]
- 269.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neuroscience Letters. 1989;107(1–3):341–346. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- 270.Webster S, Bonnell B, Rogers J. Charge-based binding of complement component C1q to the Alzheimer amyloid β-peptide. The American Journal of Pathology. 1997;150(5):1531–1536. [PMC free article] [PubMed] [Google Scholar]
- 271.Kishore U, Gupta SK, Perdikoulis MV, Kojouharova MS, Urban BC, Reid KBM. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, and C chains. The Journal of Immunology. 2003;171(2):812–820. doi: 10.4049/jimmunol.171.2.812. [DOI] [PubMed] [Google Scholar]
- 272.Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Aβ by microglia. Experimental Neurology. 2000;161(1):127–138. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- 273.Fan R, Tenner AJ. Complement C1q expression induced by Aβ in rat hippocampal organotypic slice cultures. Experimental Neurology. 2004;185(2):241–253. doi: 10.1016/j.expneurol.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 274.Sarvari M, Vago I, Weber CS, Nagy J, Gal P, Mak M, et al. Inhibition of C1q-beta-amyloid binding protects hippocampal cells against complement mediated toxicity. Journal of Neuroimmunology. 2003;137(1-2):12–18. doi: 10.1016/s0165-5728(03)00040-7. [DOI] [PubMed] [Google Scholar]
- 275.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. The Journal of Neuroscience. 2004;24(29):6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Strohmeyer R, Shen Y, Rogers J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Molecular Brain Research. 2000;81(1-2):7–18. doi: 10.1016/s0169-328x(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 277.Strohmeyer R, Ramirez M, Cole GJ, Mueller K, Rogers J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. Journal of Neuroimmunology. 2002;131(1-2):135–146. doi: 10.1016/s0165-5728(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 278.Trouw LA, Nielsen HM, Minthon L, et al. C4b-binding protein in Alzheimer’s disease: binding to Aβ1-42 and to dead cells. Molecular Immunology. 2008;45(13):3649–3660. doi: 10.1016/j.molimm.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 279.Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathologica. 1989;77(6):569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- 280.Korotzer AR, Watt J, Cribbs D, et al. Cultured rat microglia express C1q and receptor for C1q: implications for amyloid effects on microglia. Experimental Neurology. 1995;134(2):214–221. doi: 10.1006/exnr.1995.1051. [DOI] [PubMed] [Google Scholar]
- 281.McGeer PL, McGeer EG. The possible role of complement activation in Alzheimer disease. Trends in Molecular Medicine. 2002;8(11):519–523. doi: 10.1016/s1471-4914(02)02422-x. [DOI] [PubMed] [Google Scholar]
- 282.Sokolowski JD, Mandell JW. Phagocytic clearance in neurodegeneration. The American Journal of Pathology. 2011;178(4):1416–1428. doi: 10.1016/j.ajpath.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2008;28(25):6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wyss-Coray T, Yan F, Lin AHT, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. Journal of Neurochemistry. 2008;106(5):2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Britschgi M, Takeda-Uchimura Y, Rockenstein E, Johns H, Masliah E, Wyss-Coray T. Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. Journal of Neuroinflammation. 2012;9:220 pages. doi: 10.1186/1742-2094-9-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 288.Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN. Common micro RNAs (miRNAs) target complement factor H, (CFH) regulation in alzheimer's disease (AD) and in age-related macular degeneration (AMD) International Journal of Biochemistry and Molecular Biology. 2012;3(1):105–116. [PMC free article] [PubMed] [Google Scholar]
- 289.Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clinical and Experimental Immunology. 2009;155(2):128–139. doi: 10.1111/j.1365-2249.2008.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Barnett MH, Parratt JDE, Cho ES, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Annals of Neurology. 2009;65(1):32–46. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- 291.Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, et al. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(3):965–970. doi: 10.1073/pnas.1111924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ingram G, Hakobyan S, Hirst CL, et al. Complement regulator factor H as a serum biomarker of multiple sclerosis disease state. Brain. 2010;133(6):1602–1611. doi: 10.1093/brain/awq085. [DOI] [PubMed] [Google Scholar]
- 293.Compston DAS, Morgen BP, Campbell AK, et al. Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathology and Applied Neurobiology. 1989;15(4):307–316. doi: 10.1111/j.1365-2990.1989.tb01231.x. [DOI] [PubMed] [Google Scholar]
- 294.Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Annals of Neurology. 2001;50(5):646–657. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- 295.Brink BP, Veerhuis R, Breij ECW, Van Der Valk P, Dijkstra CD, Bö L. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. Journal of Neuropathology and Experimental Neurology. 2005;64(2):147–155. doi: 10.1093/jnen/64.2.147. [DOI] [PubMed] [Google Scholar]
- 296.Breij ECW, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Annals of Neurology. 2008;63(1):16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 297.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 298.McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism and Related Disorders. 2004;10(1):S3–S7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 299.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathologica. 1992;84(1):100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 300.Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. Journal of Neuroinflammation. 2006;3, article 29 doi: 10.1186/1742-2094-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wang Y, Hancock AM, Bradner J, et al. Complement 3 and factor H in human cerebrospinal fluid in Parkinson’s disease, Alzheimer’s disease, and multiple-system atrophy. The American Journal of Pathology. 2011;178(4):1509–1516. doi: 10.1016/j.ajpath.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature. 1988;334(6180):345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 303.Zhang W, Phillips K, Wielgus AR, et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of parkinson’s disease. Neurotoxicity Research. 2011;19(1):63–72. doi: 10.1007/s12640-009-9140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Depboylu C, Schäfer MKH, Arias-Carrión O, Oertel WH, Weihe E, Höglinger GU. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. Journal of Neuropathology and Experimental Neurology. 2011;70(2):125–132. doi: 10.1097/NEN.0b013e31820805b9. [DOI] [PubMed] [Google Scholar]
- 305.MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 306.Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Experimental Neurology. 1999;159(2):362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- 307.Hakobyan S, Boyajyan A, Sim RB. Classical pathway complement activity in schizophrenia. Neuroscience Letters. 2005;374(1):35–37. doi: 10.1016/j.neulet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 308.Arakelyan A, Zakharyan R, Khoyetsyan A, Poghosyan D, Aroutiounian R, Mrazek F, et al. Functional characterization of the complement receptor type 1 and its circulating ligands in patients with schizophrenia. BMC Clinical Pathology. 11:p. 10. doi: 10.1186/1472-6890-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Mayilyan KR, Dodds A, Boyajyan AS, Soghoyan AF, Sim RB. Complement C4B protein in schizophrenia. World Journal of Biological Psychiatry. 2008;9(3):225–230. doi: 10.1080/15622970701227803. [DOI] [PubMed] [Google Scholar]
- 310.Boyajyan A, Khoyetsyan A, Chavushyan A. Alternative complement pathway in schizophrenia. Neurochemical Research. 2010;35(6):894–898. doi: 10.1007/s11064-010-0126-2. [DOI] [PubMed] [Google Scholar]
- 311.Mayilyan KR, Arnold JN, Presanis JS, Soghoyan AF, Sim RB. Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neuroscience Letters. 2006;404(3):336–341. doi: 10.1016/j.neulet.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 312.Mayilyan KR, Weinberger DR, Sim RB. The complement system in schizophrenia. Drug News and Perspectives. 2008;21(4):200–210. doi: 10.1358/dnp.2008.21.4.1213349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zakharyan R, Khoyetsyan A, Arakelyan A, Boyajyan A, Gevorgyan A, Stahelova A, et al. Association of C1QB gene polymorphism with schizophrenia in armenian population. BMC Medical Genetics. 2011;12:p. 126. doi: 10.1186/1471-2350-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Håvik B, Le Hellard S, Rietschel M, et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biological Psychiatry. 2011;70(1):35–42. doi: 10.1016/j.biopsych.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 315.Rudduck C, Beckman L, Franzen G, Lindstrom L. C3 and C6 complement types in schizophrenia. Human Heredity. 1985;35(4):255–258. doi: 10.1159/000153555. [DOI] [PubMed] [Google Scholar]
- 316.Rudduck C, Beckman L, Franzen G, Lindstrom L. C3 and C6 complement types in schizophrenia. Human Heredity. 1985;35(4):255–258. doi: 10.1159/000153555. [DOI] [PubMed] [Google Scholar]
- 317.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Shastri A, Bonifati DM, Kishore U. Other Dementias. In: Kishore U, editor. Neurodegenerative Diseases. InTech; 2013. http://www.intechopen.com/books/neurodegenerative-diseases/other-dementias. [Google Scholar]