

Abstract
Lipid peroxidation, a process known to induce oxidative damage to key cellular components, has been implicated in several diseases. Following three decades of explorations mainly on in vitro models reproducible in the laboratories, lipid peroxidation has become increasingly relevant for the interpretation of a wide range of pathophysiological mechanisms in the clinical setting. This cumulative effort has led to the identification of several lipid peroxidation end-products meeting the needs of the in vivo evaluation. Among these different molecules, isoprostanes and 4-hydroxy-2-nonenal protein adducts appear to be particularly interesting. This review shows how specific oxidation products, deriving from polyunsaturated fatty acids precursors, are strictly related to the clinical manifestations and the natural history of Rett syndrome, a genetically determined neurodevelopmental pathology, currently classified among the autism spectrum disorders. In our experience, Rett syndrome offers a unique setting for physicians, biologists, and chemists to explore the borders of the lipid mediators concept.
1. Introduction
Oxidative stress (O.S.), a biological condition determined by the imbalance between prooxidant and the antioxidant system, is involved in several conditions, including inflammation, carcinogenesis, neurodegeneration, and development. Lipid peroxidation is a critical component of O.S. In particular, free radicals and specifically reactive oxygen species (ROS) are able to attack polyunsaturated fatty acids (PUFAs) of cell membranes thus generating a family of α,β-unsaturated reactive aldehydes, such as 4-hydroxy-2-nonenal (4-HNE) and prostaglandin-like end-products termed isoprostanes (IsoPs). Emerging knowledge points out the key role of these molecules in generating oxidative-driven damage. Therefore, these compounds, rather than being considered simple biomarkers of disease, are to be considered as mediators. In the present review, we discuss the evidence on the issue by focusing our proof-of-concept reasoning on the unique O.S. model of disease represented by Rett syndrome (RTT), a genetically determined autism spectrum disorder.
2. Role of Oxidative Stress in Human Diseases
O.S., as defined as an imbalance between cellular antioxidant defenses and free radicals production, is implicated in a wide variety of disease processes. To this regard, a role of oxidative damage in cancer [1, 2], neurodegenerative diseases [3–6], and likely inflammatory bowel disease [7, 8] has been recently reported. In the matter of atherosclerosis, an oxidative damage, especially to lipids, is certain. However, as it concerns the size of its effects and underlying pathogenetic mechanisms there is likely much less certainty than a few years ago [9]. One method to quantify oxidative injury is to measure lipid peroxidation. However, a cause-effect relationship is often difficult, if not impossible, to prove.
3. Oxidative Stress: Shifting from the In Vitro to the In Vivo Concept
Until the ‘80s, lipid peroxidation was confined to specialized laboratories in which the phenomenon was mainly evaluated in vitro. At this stage, researchers were interested in reproducing lipid peroxidation as a macrophenomenon induced by xenobiotics of such an entity that would be quite unlikely to be present in human physiology and pathology. The meaning of those experiments and tests was mainly to evidence the phenomenon by measuring the products generated in the process. In this particular setting, oxidized aldehydes (chiefly malondialdehyde, MDA, and all the thiobarbituric acid reactive substances, TBARS) were considered to be reliable indicators of lipid peroxidation. Things changed dramatically when a number of laboratories around the world evidenced increased levels of lipid peroxidation markers in tissues and body fluids from patients as compared to those observed in healthy controls. Thus, the setting has progressively moved from in vitro to in vivo and has allowed to enlighten the role of lipid peroxidation within the mechanisms of several diseases and physiologic signaling pathways.
The entity of the phenomenon became dramatically downscaled so that more sensitive and reliable markers in vivo became an urgent necessity. At this further stage, several key markers of lipid peroxidation applicable in vivo have been discovered, including IsoPs and 4-HNE protein adducts (4-HNE PAs). The measurement of those markers has allowed a much deeper understanding of the key role of oxidative stress in health and disease. Likely, we are now at the stage in which we are asking whether these end-peroxidation products may have biological activity on their own, thus progressively shifting from markers to mediators.
4. Lipid Peroxidation End-Products: Shifting from Markers to Mediators
Lipid mediators are chemical messengers that are released in response to tissue injury and include prostaglandins, leukotrienes, lipoxins, and neuroprotectins, playing an essential role in the different phases of inflammation. While early in the inflammatory response, arachidonic acid (AA) metabolites (i.e., prostaglandins and leukotrienes) exert proinflammatory actions, by promoting chemotaxis of phagocytic leukocytes and inducing fever; the so-termed “lipid mediator class switching” is responsible for the production of specialized proresolving mediators, such as lipoxins, resolvins, maresins, and protectins, which exert specific roles in counterregulating inflammation and turning on resolution [10, 11]. Thus inflammation can be considered a self-limited process depending on lipid-derived mediators produced in the inflammatory exudates.
Emerging evidence indicates that biomarkers of lipid peroxidation can be interpreted as lipid mediators, acting as key factors in the regulation of the delicate balance between inflammation and the resolution process. One unanswered major question is concerning whether the end-products of peroxidation, such as IsoPs, are to be considered true markers or simple markers of O.S.; in particular whether different lipid mediators would determine different diseases; or whether the time course of production is a critical factor in physiology and disease.
5. Aldehyde Products
As mentioned earlier, the interaction of ROS and lipids can induce the peroxidation process, a chain reaction that produces multiple breakdown molecules, including many highly reactive aldehydes such as malondialdehyde, 4-HNE, 4-hydroxy-2-hexenal, and acrolein [12].
Among them, 4-HNE, a 4-hydroxyalkenal, is the most intensively studied aldehyde and it is one of the best recognized and most studied cytotoxic products derived from the lipid peroxidation processes.
4-HNE derives from the oxidation of ω-6 PUFAs, essentially arachidonic and linoleic acid, that is, the two most represented fatty acids in biomembranes. 4-HNE is an unusual compound containing three functional groups that in many cases act in concert explaining its high reactivity. There is, first of all, a conjugated system consisting of a C=C double bond and a C=O carbonyl group in 4-HNE. The hydroxyl group at carbon four contributes to the reactivity both by polarizing the C=C bond and by facilitating internal cyclisation reactions, such as thioacetal formation [13].
Because of its chemical properties, 4-HNE is an amphiphilic molecule (water soluble while exhibiting strong lipophilic properties). 4-HNE tends to concentrate in biomembranes, where phospholipids, like phosphatidylethanolamine, and proteins, such as transporters, ion channels and receptors, quickly react with it. In addition, since it is a highly electrophilic molecule, it easily reacts with low molecular weight compounds, such as glutathione, and at higher concentration with DNA [12]. Due to its electrophilic nature, 4-HNE can form adducts with cellular protein nucleophiles. Indeed, the reactivity of 4-HNE explains its potential involvement in the modulation of enzymes activity, signal transduction, and gene expression [13].
Adduction to and modification of functional and/or signaling proteins most likely represents one of the main mechanisms by which 4-HNE and also the other α,β-unsaturated aldehydes can influence physiological as well as pathological processes.
Primary reactants for 4-HNE are the amino acids cysteine, histidine, and lysine, which—either free or protein-bound—undergo readily Michael addition reactions to the C=C double bond. Besides this type of reaction, a secondary reaction may occur involving the carbonyl and the hydroxyl groups to form a cyclic hemiacetal derivative. Amino groups (e.g., Lys) may alternatively react with the carbonyl group of 4-HNE to yield a Schiff base product [14].
Therefore, since proteins play an important role in normal structure and function of the cells, oxidative modifications by increased 4-HNE levels, as in O.S. conditions, may greatly alter their structure. These protein alterations may subsequently lead to loss of normal physiological cell functions and/or may lead to abnormal function of cell and eventually to cell death.
4-HNE adducts contribute to the pool of damaged enzymes, which increases in levels during aging and in several pathological states [15]. Furthermore, impaired protein clearance (i.e., ubiquitin proteosome system dysfunction) and/or the overwhelming production of abnormal proteins play an important role in the pathophysiology of disorders related to O.S., and a lot has been done in the last few decades on their role in neuro-related pathologies [16].
Based on this, 4-HNE represents one of the most useful biomarkers for the occurrence and/or the extent of O.S. [17].
6. Isoprostanes
IsoPs are a unique series of prostaglandin-like compounds generated, via a free radical-catalyzed mechanism, from a number of different PUFAs, including AA, eicosapentaenoic acid (EPA), adrenic acid (AdA), and docosahexaenoic acid (DHA).
Looking at the lipid composition of the tissues of the body [18], AA was found to be localized quite everywhere, whereas PUFAs such as DHA or AdA are mainly localized in nervous tissue and especially in grey and white matters. Thus the clinical relevance of the different classes of IsoPs hinges on the PUFA precursor anatomical distribution.
6.1. F2-Isoprostanes
The discovery of prostaglandin F2-like compounds, termed F2-isoprostanes (F2-IsoPs), generated by free radical-induced peroxidation of arachidonic acid, was for the first time reported by Morrow et al. [19].
Since F2-IsoPs, initially formed in situ on phospholipids [20], are released into the circulation and because these prostanoids are less reactive than other lipid peroxidation products (i.e., lipoperoxides and aldehydes), they can be detected more easily in plasma. Since the discovery of these molecules, F2-IsoPs have become the biomarker of choice for assessing endogenous OS, mainly due to their chemical stability and ubiquitousness in tissues and body fluids [21–23]. Elevated levels of plasma or urinary F2-IsoPs have been reported in several diseases [24–26]. For a correct O.S. damage quantification, Halliwell has suggested to use a combination of blood IsoPs and urinary IsoP metabolites determinations [27]. The 15-F2t-IsoP is the most represented isomer among the F2-IsoPs and is also referred to as 8-iso-prostaglandin F2α [28].
In addition to F2-IsoPs with F2-ring, a variety of IsoPs with different ring structures, E2-ring and D2-ring, have been so far identified. Central in the pathway of formation of IsoPs are PGH2-like endoperoxides. Just as cyclooxygenase-derived PGH2 are rearranged to PGD2 and PGE2, the H2-IsoP endoperoxides can be reduced to form F2-ring IsoPs [29] but can also undergo rearrangements to form E2- and D2-IsoPs. Biological effects for E2- and D2-IsoPs have been described [30]. Subsequent to E2- and D2-IsoPs dehydration [29], cyclopentenone IsoPs can be formed [31]; cyclopentenone IsoPs, A2/J2-IsoPs, are highly reactive; α,β-unsaturated carbonyl moieties are highly susceptible to Michael addition reactions [32, 33]. In particular, one of the A2-IsoPs, 15-A2t-IsoP is primarily metabolized by these cells via conjugation to glutathione [34].
6.2. F4-Neuroprostanes
A series of F2-IsoP-like molecules named F4-neuroprostanes (F4-NeuroPs) originate from the free radical catalyzed peroxidation of DHA, an essential constituent of nervous tissue, highly enriched in neurons, and highly susceptible to oxidation [35]. Quantification of these compounds appears to provide a very sensitive index of oxidative neuronal injury in contrast to the IsoPs, since the amounts of F4-NeuroPs formed from DHA oxidation exceed the levels of F2-IsoPs generated from AA by 3.4-fold [36]. Given the well-known role for free radicals in the pathogenesis of a number of neurodegenerative diseases, that is, Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis [20, 29, 37], the quantification of F4-NeuroPs appears to be a tool to evaluate a brain oxidative injury.
In experimental models of neurodegenerative phenomenon, F4-NeuroPs appear to be not related to excitotoxicity and epilepsy [38, 39], and a decrease of F4-NeuroPs levels, subsequent to treatment with antioxidant, was observed [39].
Furthermore, an increase of F4-NeuroPs has been reported in a neonatal hypoxic-ischemic encephalopathy [40]. The hypoxic-ischemia determines, in association with brain damage, formation of F4-NeuroPs, as well as F2-IsoPs. As F4-NeuroP levels are particularly elevated in ischemic areas, F4-NeuroPs may represent a specific marker of ischemic damage.
In Alzheimer's or Parkinson's diseases, F4-NeuroPs, as well as F2-IsoPs, are markedly increased in brain tissue and cerebrospinal fluid [41]. In particular, the F4-NeuroP levels were significantly higher as compared to those of F2-IsoPs, in the brain regions affected by the disease [42, 43]. Increased levels of F4-NeuroPs have been also reported in the cerebrospinal fluid of patients with subarachnoid hemorrhage from aneurysm. The generation of F4-NeuroPs in subarachnoid aneurysm hemorrhage and traumatic brain injury [44, 45] appears to be the consequence of a catastrophic central nervous system injury, and it can be considered an useful indicator of the pathological event. Regards to F4-NeuroPs determination, some researchers have used the chemically synthesized 17-F4c-NeuroP isomer [46], although interference with F2-dihomo-isoprostanes (F2-dihomo-IsoPs) cannot be ruled out [44].
6.3. F2-Dihomo-Isoprostanes
F2-dihomo-IsoPs are specific markers for free radical-induced AdA peroxidation and have been characterized as potential markers of free radical damage to myelin in human brain [47]. To date, clinical applications for F2-dihomo-IsoPs are few and studies are reported for white brain matter [47], cerebrospinal fluid [44], and plasma [48]. For the first time, our studies have shown the possibility of F2-dihomo-IsoP evaluation in plasma.
7. IsoPs as Mediators of Disease
15-F2t-IsoP is previously also indicated as 8-epi-PGF2α or 8-iso-PGF2α, as it is one of the most abundantly F2-IsoP isomer produced in vivo and may exhibit biological activity [24].
F2-IsoPs seem activate receptors analogous or identical to those for the thromboxane A2 (TxA2) and induce platelet aggregation [49] and vasoconstriction of renal glomerular arterioles [50, 51]. Via activation of the TxA2 receptors, IsoPs inhibit angiogenesis [52]. Furthermore, stimulation of DNA synthesis and cell proliferation for F2-IsoPs on muscle vascular cells [51] and endothelial cells [53] is known, as well as the role of F2-IsoPs in the pulmonary pathophysiology [54]. In streptozotocin-induced diabetes, F2-IsoPs appear to mediate an increase of the transforming growth factor-β1 (TGF-β1) [55], while the 5-F2t-IsoPs and 5-epi-5-F2t-IsoPs regulate the [³H]d-aspartate release in isolated bovine retina [56].
The complex F2-IsoPs bioactivity has been summarized in recent reviews [57–59].
8. The Role of Chemical Synthesis in the Exploration of Lipid Mediators
Oxidative stress is evolved in neurodegeneration of grey matter (Alzheimer's diseases) [60] and white matter (multiple sclerosis or Rett syndrome).
As their isomers F2-IsoPs are known as the “gold standard” for systemic O.S. [61] and for their biological activities [62, 63], F4-NeuroPs and F2-dihomo-IsoPs are potential biomarkers in specific pathologies such as AD or RTT and may as well have biological activities.
In order to demonstrate theses various activities of PUFAs oxygenated metabolites, chemical synthesis is a need and chemists are the link between biochemists and biologists.
Thus, since the discovery of F2-IsoPs, chemists developed strategies to access to IsoPs [64–66] as well as NeuroPs [67, 68], dihomo-IsoPs [69, 70]. Among them, our laboratory, specialized in total synthesis of lipids metabolites (leukotrienes, isoprostanes, and resolvins), developed during the past twenty years three strategies allowing the access to F-type IsoPs [69, 71, 72] as well as E-type [69, 73, 74], D-type [74], and A-type (Bultel-Poncé et al., unpublished results). Those strategies permitted the syntheses of different series of oxygenated metabolites derived from α-linolenic acid (ALA), AA, DHA, EPA, and AdA (Scheme 1).
Scheme 1.
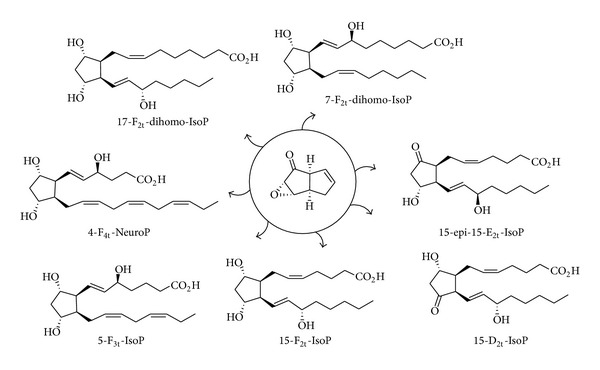
With those chemically pure metabolites in hands, biologists and clinicians have shown biomarkers activities [48] as well as biological activities [75, 76] of those oxidative stress-derived metabolites.
9. Rett Syndrome: A Genetic Model of Autism Spectrum Disorder
RTT (OMIM ID: 312750) occurs with a frequency of up to 1/10,000 live female births. Causative mutations in the X-linked methyl-CpG binding protein 2 gene (MECP2) are detectable in up to 95% of cases, although a wide genetical and phenotypical heterogeneity is well established [77]. Approximately 80% of RTT clinical cases show the so-called “typical” clinical picture; after an apparently normal development for 6–18 months, RTT girls lose their acquired cognitive, social, and motor skills in a typical 4-stage neurological regression and develop autistic behavior accompanied by stereotypic hand movements [78]. Autistic features are typically transient in RTT.
In addition to typical RTT, it has been recognized that some individuals present with many, but do not necessarily all, of the features of the disorder. New guidelines for the diagnosis of specific “variant” or “atypical” forms of RTT have been developed to identify the preserved speech, early seizure, and the congenital variant [79].
Although the genetic mechanisms of disease have been extraordinary explored in details in RTT, to date the biological mechanisms linking the gene mutation to the phenotypic expression of the disease including its wide heterogeneity are yet to be clarified. Several explanations have been proposed so far, including a key role of MECP2 in the neuronal maturation [80], maintenance of astroglia, immune dysfunction [81], and neurotransmission pathway abnormality [82]. However, recent discoveries, mainly by our team, concerning the emerging role of alteration of redox homeostasis offer an alternative explanations which is not mutually exclusive with the others previously proposed. Nevertheless, the whole history of RTT is cluttered with several apparently firm points that have been subsequently changed. Interestingly, one of the most firm points to date is that RTT is caused by a single-gene mutation either MECP2 or other more rarely affected (i.e., CDKL5 or FOXG1 genes) [79]. Recently the analysis of the full exome sequencing in two pairs of affected sisters, each with identical MECP2 gene mutation but discordant phenotype, indicates that several hundreds of gene mutations appear to be associated with the MECP2 gene mutation and therefore are to be considered previously unknown disease modifier [83].
Currently, no effective pharmacological therapies for RTT exist that can either halt progression, or reverse the neurological and cognitive abnormalities.
10. Lipid Peroxidation and Rett Syndrome
Mounting evidence indicates an emerging role for O.S. in genetically determined diseases [84]. Furthermore, the role of the redox alteration in the pathogenesis of the autism spectrum disorder is under debate [85, 86].
In 2001 indirect evidence for excessive lipid peroxidation, leading to increased plasma malondialdehyde levels, has been reported in RTT patients [87]. However, after that isolated report and before our subsequent series of specific studies in the field, the lipid oxidative damage in RTT had not been further investigated. Our studies were focused on the identification of different classes of IsoPs increased in RTT, thus allowing an inference on the individual oxidized fatty acids precursors relevant to the disease. On the other hand, our reports of increased 4-HNE PAs levels in RTT patients while further supporting the evidence of a lipid damage with formation of the aldehyde 4-HNE also detect the presence of a coexisting protein damage due to the formation of the adducts.
Therefore, our findings in RTT, a rare cause of genetically determined autism spectrum disorder, indicate that this pervasive development disorder can be considered a unique human model for chronic O.S. and could be considered a valuable testing ground for the link between lipid peroxidation byproducts and the mediation of disease processes.
10.1. Isoprostanes and Neuroprostanes in RTT
Our findings [48, 88–90] indicate that typical RTT is characterized by markedly increased levels of IsoPs deriving from the nonenzymatic oxidation of AA, DHA, and AdA at every clinical stage of the disease. IsoPs and NeuroPs levels appear to be closely interrelated to the RTT clinical presentation, suggesting that these lipid oxidation products could mediate the pathogenetic mechanisms underlying the syndrome.
Extremely high (i.e., two orders of magnitude) plasma levels of F2-dihomo-IsoPs are detectable in RTT girls in stage I of the disease. AdA, whatever the actual origin (brain white matter, adrenal gland, or kidney), is the PUFA that goes through the greatest degree of oxidation during the earliest stage of the typical form of the disease. An insult to AdA and the clinical onset of neuroregression occur at the same time [48]. Thus, during the first two years of the natural evolution of the disease, the peroxidation of AdA, a critical component of myelin [47] in the primate brain, is involved.
Oxidation of the AA appears to be another essential component in the pathogenesis of the first two stages of typical RTT, as can be deduced from the significantly high F2-IsoPs in the early stages as compared with the late natural progression of classic RTT. Nevertheless, F2-IsoPs increase not with the same marked raise of the F2-dihomo-IsoPs. Thus, F2-dihomo-IsoPs are the prominent lipid peroxidation end-product detectable at this stage of the disease. In RTT girls, plasma F2-IsoPs are always above the physiological range, and thus, considering the half-life of these prostanoids, lipid peroxidation is continuously carried out in the syndrome. Due to the systemic distribution of AA, free plasma F2-IsoPs can be considered an index of generalized lipid peroxidation, while in RTT, a specific site of peroxidation events has been identified in the erythrocyte membrane. In fact, esterified F2-IsoPs are increased in RTT erythrocyte membrane and their levels are correlated to altered red blood cells shape [91].
Patients with typical RTT had significantly higher F2-IsoPs than those with atypical phenotype and are correlated to RTT clinical severity, as well as F4-NeuroPs. In particular, F4-NeuroPs are related to clinical severity markers, including early regression, severe head growth deceleration, major motor impairment, hand use loss, and seizures. In our experience, MECP2 gene mutations located in critical regions that carry higher phenotype severity usually show a more severely shifted O.S. imbalance [89, 90]. Moreover, the demonstrated link between F4-NeuroPs and MECP2 genotype-phenotype correlation suggests that the degree of MeCP2 protein dysfunction is directly proportional to the O.S.-mediated neuronal damage, explaining ~90% of the expressed phenotype variability [90]. Also plasma F2-IsoPs concentrations are significantly related to MECP2 genotype; anyway, the strength of the relationship between plasma F2-IsoPs and MeCP2 phenotype appears to be far weaker than that between plasma F4-NeuroPs and MeCP2 phenotype [89, 90]. As F4-NeuroPs plasma levels mirror neurological severity, these molecules may provide evidence on neuronal damage, also considering the specific distribution of DHA in membrane neurons.
10.2. 4-HNE and RTT
In our recent work, we have shown that the levels of 4-HNE PAs change during the clinical progression of typical RTT [92]. Plasma 4-HNE PAs increase between stage I and stage II and to less extent in the following stages (i.e., III and IV).
Impairment of the GSH system and other detoxification enzymes involved in aldehyde metabolism or defect/dysfunction in the ubiquitin proteasome system, reported in the autism spectrum disorder [93–95], could be involved in the accumulation, in the early stages of RTT, of several 4-HNE plasma protein adducts. This, as a consequence, could play a role in the severe clinical features observed in the later stages of the disease.
As for the IsoPs, a relationship between 4-HNE PAs and phenotypical RTT presentation has been reported. In particular, with reference to the atypical RTT clinical presentation, CDKL5-related RTT patients have a significant increase in 4-HNE PAs levels, and on the contrary, FOXG1-related RTT patients are not different from the controls [92]. Although the possible cause for the observed difference in FOXG1-related RTT is not clear, the 4-HNE PAs, as well as the IsoPs, appear to be directly involved in the RTT pathogenesis. To date, it is not clear how the gene mutations can induce and increase O.S. in RTT patients. It is possible to speculate that it could be an indirect mechanism that might involve mitochondria respiration, modifying therefore the redox state of the cells.
It should be pointed out that the presence of 4-HNE PAs in plasma of RTT patients suggests two events. First, it is indicative of a generalized lipid peroxidation, indicating the occurrence of PUFAs oxidation in various organs and/or tissues. Second, by covalent modification of proteins, 4-HNE is able to cause long-lasting biological consequences. Therefore, plasma proteins can be considered a target of increased O.S. status in RTT. Hence, it is possible to speculate that 4-HNE PAs can contribute to the pathophysiology of RTT, both in the development and in the progression and complications of the disease. We can suggest that 4HNE PAs represent a potential biomarker of RTT as well as disease severity.
In future, to better understand the clinical consequences that the modified plasma proteins have on RTT patients, further studies are needed. It could be of extreme importance to be able to identifying the target proteins modified by 4-HNE in the plasma, as recently shown for mild cognitive impairment and Alzheimer's disease [96, 97].
A close relationship between levels of circulating lipid peroxidation markers in RTT patients and presence of the symptom is not a causative proofs but strongly supports the concept that lipid peroxidation plays a previously unrecognized key role in the pathogenesis of RTT due to the gene mutation. The increase of knowledge on nature of 4-HNE modified proteins in RTT, might lead to better prevention, diagnosis and treatments of the associated physiological processes altered in patients.
Finally, the involvement of lipid peroxidation in RTT was also confirmed in our recent study, where the morphology of erythrocytes in typical RTT patients has been evaluated [91]. Emerging evidence indicates that O.S. imbalance and hypoxemia can lead to erythrocyte shape abnormalities in chronic pulmonary disease [98, 99], and now it has been well proved that chronic hypoxia, impaired pulmonary gas exchange, and increased O.S. are all present in typical RTT [88]. In fact, our data show the presence of erythrocytes altered shape (mainly leptocytes) and membrane oxidative damage (i.e., 4-HNE PAs) in patients with clinical diagnosis of typical RTT [91]. Consequently, monitoring of erythrocytes morphology can be an important new diagnostic and prognostic tool in this particular form of autism spectrum disorder, in which the lung seems to represent an unexpectedly key organ for the disease pathogenesis.
10.3. Lipid Peroxidation and ω-3 PUFAs Supplementation in RTT
Lipid peroxidation appears to be a peculiar characteristic of RTT, and the relationships between the described lipid peroxidation products and the clinical disease features are summarized in Figure 1.
Figure 1.
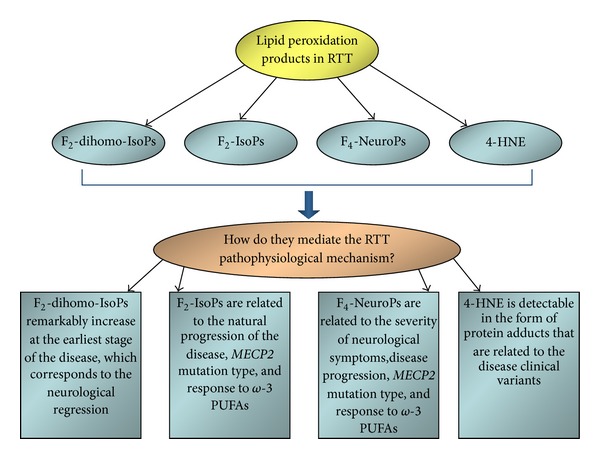
Different classes of isoprostanes (F2-dihomo-IsoPs, F2-IsoPs, and F4-NeuroPs), deriving from polyunsaturated fatty acids precursors (adrenic, arachidonic, and docosahexaenoic acids, resp.), and the 4-hydroxy-2-nonenal protein adducts are strictly related to the clinical manifestations and the natural history of Rett syndrome. The lipid peroxidation events and the disease pathogenic mechanisms are closely interrelated, as demonstrated by the dietary supplementation with ω-3 PUFAs. RTT: Rett syndrome; F2-dihomo-IsoPs: F2-dihomo-isoprostanes; F2-IsoPs: F2-isoprostanes; F4-NeuroPs: F4-neuroprostanes; 4-HNE: 4-hydroxy-2-nonenal; and PUFAs: polyunsaturated fatty acids.
A better comprehension of the lipid peroxidation involvement in the pathogenetic mechanisms of the disease derives from studies carried out with ω-3 PUFAs supplementation. Interestingly, the supplemented molecules are actually just the same category of molecules that undergo radical damage. Exogenous administration of ω-3 PUFAs, in disease stages I-IV, has been shown to moderately reduce clinical severity and significantly reduce the levels of IsoPs and 4-HNE PAs in RTT patients [89–91, 100]. Following ω-3 PUFAs supplementation, we might have expected an enhanced formation of the lipid oxidation products in plasma. Actually, the assumed fatty acids are not oxidized and the endogenous production is reduced. These results lead us to think that in RTT the lipid-oxidative damage is nonspecific, all fatty acids are not oxidatively damaged, but the oxidative insult regards specific biological targets.
As a consequence, these data, while confirming the involvement of lipid products in the intimate pathogenic mechanisms of the disease, indicate that the fatty acid oxidation is related to the clinical severity of the disease and is a reversible process.
11. Conclusion
Lipid peroxidation end-products appear to be associated with the modulation of RTT disease severity, thus suggesting their likely role as mediators. The identification of the involved classes of molecules (including metabolites) in the disease, the evaluation of their relationship with the disease natural history, and the chance to determine the effects of treatments (i.e., exogenous PUFAs supplementation) and of gene manipulation (i.e., reactivation of Mecp2 null mice) offer a unique setting for physicians, biologists, and chemists to explore the borders of the lipid mediators concept.
Authors' Contribution
Cinzia Signorini and Claudio De Felice contributed equally to the paper.
References
- 1.Halliwell B. Oxidative stress and cancer: have we moved forward? Biochemical Journal. 2007;401(1):1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson LR. Chronic inflammation and mutagenesis. Mutation Research. 2010;690(1-2):3–11. doi: 10.1016/j.mrfmmm.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B. Free radicals and antioxidants—quo vadis? Trends in Pharmacological Sciences. 2011;32(3):125–130. doi: 10.1016/j.tips.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 5.Praticò D. The neurobiology of isoprostanes and Alzheimer’s disease. Biochimica et Biophysica Acta. 2010;1801(8):930–933. doi: 10.1016/j.bbalip.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Aluise CD, Robinson RAS, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical alzheimer’s disease: insights into memory loss in MCI. Journal of Alzheimer’s Disease. 2011;23(2):257–269. doi: 10.3233/JAD-2010-101083. [DOI] [PubMed] [Google Scholar]
- 7.Halliwell B. Free Radicals in Biology and Medicine. 4th edition. Oxford, UK: Oxford University Press; 2007. [Google Scholar]
- 8.Brazil JC, Louis NA, Parkos CA. The role of polymorphonuclear leukocyte trafficking in the perpetuation of inflammation during inflammatory Bowel disease. Inflammatory Bowel Diseases. 2013;19(7):1556–1565. doi: 10.1097/MIB.0b013e318281f54e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halliwell B. Free radicals and antioxidants: updating a personal view. Nutrition Reviews. 2012;70(5):257–265. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- 10.Bannenberg GL. Therapeutic applicability of anti-inflammatory and proresolving polyunsaturated fatty acid-derived lipid mediators. The Scientific World Journal. 2010;10:676–712. doi: 10.1100/tsw.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? American Journal of Pathology. 2010;177(4):1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esterbauer H, Schaur RJ, Zollner H. Chemistry and Biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology and Medicine. 1991;11(1):81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-Hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Medicinal Research Reviews. 2008;28(4):569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 14.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radical Biology and Medicine. 2004;37(7):937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Progress in Lipid Research. 2003;42(4):318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 16.Grune T, Davies KJA. The proteasomal system and HNE-modified proteins. Molecular Aspects of Medicine. 2003;24(4-5):195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 17.Dubinina EE, Dadali VA. Role of 4-hydroxy-trans-2-nonenal in cell functions. Biochemistry. 2010;75(9):1069–1087. doi: 10.1134/s0006297910090014. [DOI] [PubMed] [Google Scholar]
- 18.Sastry PS. Lipids of nervous tissue: composition and metabolism. Progress in Lipid Research. 1985;24(2):69–176. doi: 10.1016/0163-7827(85)90011-6. [DOI] [PubMed] [Google Scholar]
- 19.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., II A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(23):9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., II Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrow JD, Harris TM, Roberts LJ., II Noncyclooxygenase oxidative formation of a series of novel prostaglandings: analytical ramifications for measurement of eicosanoids. Analytical Biochemistry. 1990;184(1):1–10. doi: 10.1016/0003-2697(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 22.Milne GL, Musiek ES, Morrow JD. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers. 2005;10(supplement 1):10–23. doi: 10.1080/13547500500216546. [DOI] [PubMed] [Google Scholar]
- 23.Kadiiska MB, Gladen BC, Baird DD, et al. Biomarkers of oxidative stress study II. Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radical Biology and Medicine. 2005;38(6):698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Montuschi P, Barnes PJ, Roberts LJ., II Isoprostanes: markers and mediators of oxidative stress. FASEB Journal. 2004;18(15):1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 25.Basu S. Fatty acid oxidation and isoprostanes: oxidative strain and oxidative stress. Prostaglandins Leukotrienes and Essential Fatty Acids. 2010;82(4–6):219–225. doi: 10.1016/j.plefa.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 26.Basu S. F2-isoprostanes in human health and diseases: from molecular mechanisms to clinical implications. Antioxidants and Redox Signaling. 2008;10(8):1405–1434. doi: 10.1089/ars.2007.1956. [DOI] [PubMed] [Google Scholar]
- 27.Halliwell B, Lee CYJ. Using isoprostanes as biomarkers of oxidative stress: some rarely considered issues. Antioxidants and Redox Signaling. 2010;13(2):145–156. doi: 10.1089/ars.2009.2934. [DOI] [PubMed] [Google Scholar]
- 28.Morrow JD, Zackert WE, Yang JP, et al. Quantification of the major urinary metabolite of 15-F(2T)-isoprostane (8-iso-PGF(2α)) by a stable isotope dilution mass spectrometric assay. Analytical Biochemistry. 1999;269(2):326–331. doi: 10.1006/abio.1999.4008. [DOI] [PubMed] [Google Scholar]
- 29.Roberts LJ, II, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cellular and Molecular Life Sciences. 2002;59(5):808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morrow JD, Minton TA, Mukundan CR, et al. Free radical-induced generation of isoprostanes in vivo. Evidence for the formation of D-ring and E-ring isoprostanes. Journal of Biological Chemistry. 1994;269(6):4317–4326. [PubMed] [Google Scholar]
- 31.Chen Y, Morrow JD, Roberts LJ., II Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. Journal of Biological Chemistry. 1999;274(16):10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- 32.Boyland E, Chasseaud LF. Enzymes catalysing conjugations of glutathione with alpha-beta-unsaturated carbonyl compounds. Biochemical Journal. 1968;109(4):651–661. doi: 10.1042/bj1090651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atsmon J, Sweetman BJ, Baertschi SW, Harris TM, Roberts LJ., II Formation of thiol conjugates of 9-deoxy-Δ9,Δ12(E)-prostaglandin D2 and Δ12(E)-prostaglandin D2 . Biochemistry. 1990;29(15):3760–3765. doi: 10.1021/bi00467a023. [DOI] [PubMed] [Google Scholar]
- 34.Milne GL, Zanoni G, Porta A, et al. The Cyclopentenone Product of Lipid Peroxidation, 15-A 2t-Isoprostane, Is Efficiently Metabolized by HepG2 Cells via Conjugation with Glutathione. Chemical Research in Toxicology. 2004;17(1):17–25. doi: 10.1021/tx034213o. [DOI] [PubMed] [Google Scholar]
- 35.Nourooz-Zadeh J, Liu EHC, Yhlen B, Änggård EE, Halliwell B. F4-isoprostanes as specific marker of docosahexaenoic acid peroxidation in Alzheimer’s disease. Journal of Neurochemistry. 1999;72(2):734–740. doi: 10.1046/j.1471-4159.1999.0720734.x. [DOI] [PubMed] [Google Scholar]
- 36.Roberts LJ, II, Montine TJ, Markesbery WR, et al. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. Journal of Biological Chemistry. 1998;273(22):13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- 37.Roberts LJ, II, Morrow JD. Measurement of F2-isoprostanes as an index of oxidative stress in vivo . Free Radical Biology and Medicine. 2000;28(4):505–513. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- 38.Zaja-Milatovic S, Gupta RC, Aschner M, Montine TJ, Milatovic D. Pharmacologic suppression of oxidative damage and dendritic degeneration following kainic acid-induced excitotoxicity in mouse cerebrum. NeuroToxicology. 2008;29(4):621–627. doi: 10.1016/j.neuro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaja-Milatovic S, Gupta RC, Aschner M, Milatovic D. Protection of DFP-induced oxidative damage and neurodegeneration by antioxidants and NMDA receptor antagonist. Toxicology and Applied Pharmacology. 2009;240(2):124–131. doi: 10.1016/j.taap.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Signorini C, Ciccoli L, Leoncini S, et al. Free iron, total F2-isoprostanes and total F4- neuroprostanes in a model of neonatal hypoxic-ischemic encephalopathy: neuroprotective effect of melatonin. Journal of Pineal Research. 2009;46(2):148–154. doi: 10.1111/j.1600-079X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 41.Montine TJ, Quinn JF, Milatovic D, et al. Peripheral F2-isoprostanes and F4-neuroprostanes are not increased in Alzheimer’s disease. Annals of Neurology. 2002;52(2):175–179. doi: 10.1002/ana.10272. [DOI] [PubMed] [Google Scholar]
- 42.Montine KS, Quinn JF, Zhang J, et al. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chemistry and Physics of Lipids. 2004;128(1-2):117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 43.Nourooz-Zadeh J, Liu EHC, Yhlen B, Änggård EE, Halliwell B. F4-isoprostanes as specific marker of docosahexaenoic acid peroxidation in Alzheimer’s disease. Journal of Neurochemistry. 1999;72(2):734–740. doi: 10.1046/j.1471-4159.1999.0720734.x. [DOI] [PubMed] [Google Scholar]
- 44.Hsieh Y-P, Lin C-L, Shiue A-L, et al. Correlation of F4-neuroprostanes levels in cerebrospinal fluid with outcome of aneurysmal subarachnoid hemorrhage in humans. Free Radical Biology and Medicine. 2009;47(6):814–824. doi: 10.1016/j.freeradbiomed.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 45.Corcoran TB, Mas E, Barden AE, et al. Are isofurans and neuroprostanes increased after subarachnoid hemorrhage and traumatic brain injury? Antioxidants and Redox Signaling. 2011;15(10):2663–2667. doi: 10.1089/ars.2011.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quan LG, Cha JK. Preparation of isoprostanes and neuroprostanes. Journal of the American Chemical Society. 2002;124(42):12424–12425. doi: 10.1021/ja027451z. [DOI] [PubMed] [Google Scholar]
- 47.Van Rollins M, Woltjer RL, Yin H, Morrow JD, Montine TJ. F2-Dihomo-isoprostanes arise from free radical attack on adrenic acid. Journal of Lipid Research. 2008;49(5):995–1005. doi: 10.1194/jlr.M700503-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Felice C, Signorini C, Durand T, et al. F2-dihomo-isoprostanes as potential early biomarkers of lipid oxidative damage in Rett syndrome. Journal of Lipid Research. 2011;52(12):2287–2297. doi: 10.1194/jlr.P017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khasawneh FT, Huang J-S, Mir F, Srinivasan S, Tiruppathi C, Le Breton GC. Characterization of isoprostane signaling: evidence for a unique coordination profile of 8-iso-PGF2α with the thromboxane A2 receptor, and activation of a separate cAMP-dependent inhibitory pathway in human platelets. Biochemical Pharmacology. 2008;75(12):2301–2315. doi: 10.1016/j.bcp.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi K, Nammour TM, Fukunaga M, et al. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi- prostaglandin F(2α), in the rat. Evidence for interaction with thromboxane A2 receptors. Journal of Clinical Investigation. 1992;90(1):136–141. doi: 10.1172/JCI115826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukunaga M, Makita N, Roberts LJ, II, Morrow JD, Takahashi K, Badr KF. Evidence for the existence of F2-isoprostane receptors on rat vascular smooth muscle cells. American Journal of Physiology. 1993;264(6):C1619–C1624. doi: 10.1152/ajpcell.1993.264.6.C1619. [DOI] [PubMed] [Google Scholar]
- 52.Benndorf RA, Schwedhelm E, Gnann A, et al. Isoprostanes inhibit vascular endothelial growth factor-induced endothelial cell migration, tube formation, and cardiac vessel sprouting in vitro, As well as angiogenesis in vivo via activation of the thromboxane A2 receptor: a potential link between oxidative stress and impaired angiogenesis. Circulation Research. 2008;103(9):1037–1046. doi: 10.1161/CIRCRESAHA.108.184036. [DOI] [PubMed] [Google Scholar]
- 53.Yura T, Fukunaga M, Khan R, Nassar GN, Badr KF, Montero A. Free-radical-generated F2-isoprostane stimulates cell proliferation and endothelin-1 expression on endothelial cells. Kidney International. 1999;56(2):471–478. doi: 10.1046/j.1523-1755.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 54.Janssen LJ. Isoprostanes: an overview and putative roles in pulmonary pathophysiology. American Journal of Physiology. 2001;280(6):L1067–L1082. doi: 10.1152/ajplung.2001.280.6.L1067. [DOI] [PubMed] [Google Scholar]
- 55.Montero A, Munger KA, Khan RZ, et al. F2-isoprostanes mediate high glucose-induced TGF-β synthesis and glomerular proteinuria in experimental type I diabetes. Kidney International. 2000;58(5):1963–1972. doi: 10.1111/j.1523-1755.2000.00368.x. [DOI] [PubMed] [Google Scholar]
- 56.Jamil J, Wright A, Harrison N, et al. Regulation of [3H]D-aspartate release by the 5-F2t-isoprostane and its 5-epimer in isolated bovine retina. Neurochemical Research. 2012;37(3):574–582. doi: 10.1007/s11064-011-0645-5. [DOI] [PubMed] [Google Scholar]
- 57.Basu S. Bioactive eicosanoids: role of prostaglandin F2α and F2-isoprostanes in inflammation and oxidative stress related pathology. Molecules and Cells. 2010;30(5):383–391. doi: 10.1007/s10059-010-0157-1. [DOI] [PubMed] [Google Scholar]
- 58.Ting HJ, Khasawneh FT. Platelet function and Isoprostane biology. Should Isoprostanes be the newest member of the Orphan-ligand family? Journal of Biomedical Science. 2010;17(1, article 24) doi: 10.1186/1423-0127-17-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ. Isoprostane generation and function. Chemical Reviews. 2011;111(10):5973–5996. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kepp KP. Bioinorganic chemistry of Alzheimer's disease. Chemical Reviews. 2012;112:5193–5239. doi: 10.1021/cr300009x. [DOI] [PubMed] [Google Scholar]
- 61.Kadiiska MB, Gladen BC, Baird DD, et al. Biomarkers of oxidative stress study III. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in CCl4 poisoning. Free Radical Biology and Medicine. 2005;38(6):711–718. doi: 10.1016/j.freeradbiomed.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 62.Jahn U, Galano J-M, Durand T. Beyond prostaglandins—chemistry and biology of cyclic oxygenated metabolites formed by free-radical pathways from polyunsaturated fatty acids. Angewandte Chemie. 2008;47(32):5894–5955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- 63.Brooks JD, Musiek ES, Koestner TR, et al. The fatty acid oxidation product 15-A3t-Isoprostane is a potent inhibitor of NFκB transcription and macrophage transformation. Journal of Neurochemistry. 2011;119(3):604–616. doi: 10.1111/j.1471-4159.2011.07422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zanoni G, Porta A, Vidari G. First total synthesis of A2 isoprostane. Journal of Organic Chemistry. 2002;67(12):4346–4351. doi: 10.1021/jo025652f. [DOI] [PubMed] [Google Scholar]
- 65.Hwang SW, Adiyaman M, Khanapure S, Schio L, Rockach J. Total synthesis of 8-epi-PGF(2α). A novel strategy for the synthesis of isoprostanes. Journal of the American Chemical Society. 1994;116(23):10829–10830. [Google Scholar]
- 66.Schrader TO, Snapper ML. Stereodivergent synthesis of all 15-F2 isoprostanes. Journal of the American Chemical Society. 2002;124(37):10998–11000. doi: 10.1021/ja027154u. [DOI] [PubMed] [Google Scholar]
- 67.Taber DF, Reddy PG, Arneson KO. A potential route to neuroprostanes and isoprostanes: preparation of the four enantiomerically pure diastereomers of 13-F4t-NeuroP. Journal of Organic Chemistry. 2008;73(9):3467–3474. doi: 10.1021/jo702600v. [DOI] [PubMed] [Google Scholar]
- 68.Quan LG, Cha JK. Preparation of isoprostanes and neuroprostanes. Journal of the American Chemical Society. 2002;124(42):12424–12425. doi: 10.1021/ja027451z. [DOI] [PubMed] [Google Scholar]
- 69.Oger C, Brinkmann Y, Bouazzaoui S, Durand T, Galano J-M. Stereocontrolled access to isoprostanes via a bicyclo[3.3.0]octene framework. Organic Letters. 2008;10(21):5087–5090. doi: 10.1021/ol802104z. [DOI] [PubMed] [Google Scholar]
- 70.Oger C, Bultel-Poncé V, Guy A, Durand T, Galano J-M. Total synthesis of isoprostanes derived from adrenic acid and EPA. European Journal of Organic Chemistry. 2012;(13):2621–2634. [Google Scholar]
- 71.Rondot B, Durand T, Vidal J-P, Girard J-P, Rossi J-C. Synthesis and determination of cis or trans isoprostane precursors by a 1H NMR NOE study. Journal of the Chemical Society, Perkin Transactions 2. 1995;(8):1589–1594. [Google Scholar]
- 72.El Fangour S, Guy A, Vidal J-P, Rossi J-C, Durand T. A flexible synthesis of the phytoprostanes B1 type I and II. Journal of Organic Chemistry. 2005;70(3):989–997. doi: 10.1021/jo048179+. [DOI] [PubMed] [Google Scholar]
- 73.Pinot E, Guy A, Fournial A, Balas L, Rossi J-C, Durand T. Total synthesis of the four enantiomerically pure diasteroisomers of the phytoprostanes E1type II and of the 15-E2t-isoprostanes. Journal of Organic Chemistry. 2008;73(8):3063–3069. doi: 10.1021/jo702455g. [DOI] [PubMed] [Google Scholar]
- 74.Brinkmann Y, Oger C, Guy A, Durand T, Galano J-M. Total synthesis of 15-D2t- and 15-epi-15-E2t-isoprostanes. Journal of Organic Chemistry. 2010;75(7):2411–2414. doi: 10.1021/jo1000274. [DOI] [PubMed] [Google Scholar]
- 75.Le Guennec J-Y, Galano J-M, Oger C, et al. Methods and pharmaceutical composition for the treatment and prevention of cardiac arrhytmias. European Patent, EP12306519. 3, 2012.
- 76.Galano J-M, Mas E, Barden A, et al. Isoprostanes and neuroprostanes: Total synthesis, biological activity and biomarkers of oxidative stress in humans. Prostaglandins & Other Lipid Mediators. 2013 doi: 10.1016/j.prostaglandins.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 77.Amir RE, Van Den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nature Genetics. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 78.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: rett’s syndrome. Report of 35 cases. Annals of Neurology. 1983;14(4):471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 79.Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Annals of Neurology. 2010;68(6):944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annual Review of Cell and Developmental Biology. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 81.Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484(7392):105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chao H-T, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grillo E, Lo Rizzo C, Bianciardi L, et al. Revealing the complexity of a monogenic disease: rett syndrome exome sequencing. PLoS One. 2013;8(2) doi: 10.1371/journal.pone.0056599.e56599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pagano G, Castello G. Oxidative stress and mitochondrial dysfunction in down syndrome. Advances in Experimental Medicine and Biology. 2012;724:291–299. doi: 10.1007/978-1-4614-0653-2_22. [DOI] [PubMed] [Google Scholar]
- 85.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13(3):171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 86.Meguid NA, Dardir AA, Abdel-Raouf ER, Hashish A. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biological Trace Element Research. 2011;143(1):58–65. doi: 10.1007/s12011-010-8840-9. [DOI] [PubMed] [Google Scholar]
- 87.Sierra C, Vilaseca MA, Brandi N, et al. Oxidative stress in Rett syndrome. Brain and Development. 2001;23(1):S236–S239. doi: 10.1016/s0387-7604(01)00369-2. [DOI] [PubMed] [Google Scholar]
- 88.De Felice C, Ciccoli L, Leoncini S, et al. Systemic oxidative stress in classic Rett syndrome. Free Radical Biology and Medicine. 2009;47(4):440–448. doi: 10.1016/j.freeradbiomed.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 89.Leoncini S, de Felice C, Signorini C, et al. Oxidative stress in Rett syndrome: natural history, genotype, and variants. Redox Report. 2011;16(4):145–153. doi: 10.1179/1351000211Y.0000000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Signorini C, De Felice C, Leoncini S, et al. F4-neuroprostanes mediate neurological severity in Rett syndrome. Clinica Chimica Acta. 2011;412(15-16):1399–1406. doi: 10.1016/j.cca.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 91.Ciccoli L, De Felice C, Paccagnini E, et al. Morphological changes and oxidative damage in Rett Syndrome erythrocytes. Biochimica et Biophysica Acta. 2012;1820(4):511–520. doi: 10.1016/j.bbagen.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 92.Pecorelli A, Ciccoli L, Signorini C, et al. Increased levels of 4HNE-protein plasma adducts in Rett syndrome. Clinical Biochemistry. 2011;44(5-6):368–371. doi: 10.1016/j.clinbiochem.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 93.Al-Yafee YA, Al- Ayadhi LY, Haq SH, El-Ansary AK. Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurology. 2011;11(article 139) doi: 10.1186/1471-2377-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bowers K, Li Q, Bressler J, Avramopoulos D, Newschaffer C, Fallin MD. Glutathione pathway gene variation and risk of autism spectrum disorders. Journal of Neurodevelopmental Disorders. 2011;3(2):132–143. doi: 10.1007/s11689-011-9077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathologica. 2009;118(3):329–347. doi: 10.1007/s00401-009-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reed T, Perluigi M, Sultana R, et al. Redox proteomic identification of 4-Hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiology of Disease. 2008;30(1):107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 97.Perluigi M, Sultana R, Cenini G, et al. Redox proteomics identification of 4-hydroxynonenalmodified brain proteins in Alzheimer’s disease: role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics. 2009;3(6):682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lucantoni G, Pietraforte D, Matarrese P, et al. The red blood cell as a biosensor for monitoring oxidative imbalance in chronic obstructive pulmonary disease: an ex vivo and in vitro study. Antioxidants and Redox Signaling. 2006;8(7-8):1171–1182. doi: 10.1089/ars.2006.8.1171. [DOI] [PubMed] [Google Scholar]
- 99.Minetti M, Leto TL, Malorni W. Radical generation and alterations of erythrocyte integrity as bioindicators of diagnostic or prognostic value in COPD? Antioxidants and Redox Signaling. 2008;10(4):829–836. doi: 10.1089/ars.2007.1864. [DOI] [PubMed] [Google Scholar]
- 100.De Felice C, Signorini C, Durand T, et al. Partial rescue of Rett syndrome by ω-3 polyunsaturated fatty acids (PUFAs) oil. Genes and Nutrition. 2012;7(3):447–458. doi: 10.1007/s12263-012-0285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]