

Abstract
To test the hypothesis that ablation of transient receptor potential vanilloid type 1 (TRPV1) channels leads to exacerbated inflammatory responses and organ damage during endotoxic shock, lipopolysaccharide (LPS; 5 million endotoxin units/kg of body weight) was injected intraperitoneally (i.p.) into wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. Mean arterial pressure and heart rate, determined by radiotelemetry, were severely depressed after LPS injection into WT and TRPV1−/− mice, with no distinction between the two strains. At 24 h after LPS injection, renal glomerular hypercellularity and hepatocellular injury were observed in both strains, accompanying further elevated serum levels of creatinine and alanine aminotransferase in TRPV1−/− mice compared to those in WT mice. At 6 or 24 h after LPS injection, neutrophil recruitment into kidneys and livers, serum cytokine (tumor necrosis factor alpha [TNF-α], interleukin 1β [IL-1β], IL-6) and renal chemokine (KC, macrophage inflammatory protein 2 [MIP-2]) levels, and renal VCAM-1 and ICAM-1 expression were greater in TRPV1−/− mice than WT mice. In addition, increased plasma calcitonin gene-related peptide (CGRP) levels observed in WT mice 6 h after LPS injection were absent in TRPV1−/− mice. Thus, TRPV1 ablation aggravates inflammatory responses, including neutrophil infiltration, proinflammatory cytokine production, and adhesion molecule expression, leading to intensified organ damage during endotoxic shock in the absence of worsened circulatory failure. The data indicate that TRPV1 activation may attenuate endotoxin-induced organ damage, possibly via its anti-inflammatory action rather than alteration of hemodynamics.
INTRODUCTION
The transient receptor potential vanilloid type 1 (TRPV1) channel, also known as vanilloid receptor type 1, is a ligand-gated nonselective ion channel (1). It functions as a molecular transducer to integrate multiple physical and chemical stimuli, including noxious heat, low pH, exogenous and endogenous vanilloid compounds, and lipid metabolites (2, 3). Immunohistochemical labeling studies have shown that, in addition to the central nervous system, TRPV1 resides predominantly in unmyelinated C fibers and thinly myelinated Aδ-afferent nerve fibers innervating the cardiovascular tissues, including the heart, blood vessel, and kidney (2). In addition to a function as afferent nerves sending information to the central nervous system, sensory nerves possess an efferent function by releasing a number of sensory neurotransmitters, commonly substance P and calcitonin gene-related peptide (CGRP) (2).
Endotoxic shock is caused mainly by an exaggerated systemic response to endotoxemia induced by Gram-negative bacteria and their characteristic cell wall component, lipopolysaccharide (LPS) (4). It is characterized by refractory hypotension, multiple organ failure, and high mortality (5). In addition to hypotension, abnormalities in renal, hepatic, pulmonary, and hematologic systems are common in the course of endotoxic shock. The development of multiple organ failure contributes to mortality associated with endotoxic shock (6). Beyond refractory hypotension, excessive inflammatory responses are believed to result in tissue and cellular injury during endotoxic shock. Inflammation-induced tissue damage may be attributed as a main cause of multiple organ failure and mortality in the course of endotoxic shock.
Increased production of an endocannabinoid, anandamide, by macrophages has been reported to contribute to endotoxin-induced hypotension (7, 8). Recently, evidence has shown that TRPV1 can be activated by anandamide, suggesting that TRPV1 may be involved in the pathogenesis of endotoxic shock (3, 9). Activation of TRPV1 expressed in sensory nerves may cause release of a number of sensory neuropeptides, including CGRP and substance P, which are potent vasodilators in various vascular beds (2). These studies suggest that activation of TRPV1 during endotoxic shock may contribute to decreased systemic blood pressure. However, the contrary has been shown, in which blockade or deletion of TRPV1 exaggerates LPS-induced hypotension and mortality (10, 11), suggesting that TRPV1 activation is essential in protecting vital organ perfusion and survival during endotoxic shock.
To further define the role of TRPV1 in pathogenesis of endotoxic shock, we evaluate LPS-induced hypotension and end-organ damage in TRPV1-null mutant (TRPV1−/−) mice compared to that in wild-type (WT) mice. This study was designed to determine (i) whether ablation of TRPV1 leads to aggravated hemodynamic failure manifested by more-depressed blood pressure and heart rates during endotoxic shock and (ii) whether ablation of TRPV1 leads to exaggerated inflammatory responses contributing to more-severe end-organ damage during endotoxic shock.
MATERIALS AND METHODS
Animals.
TRPV1−/− mice were kindly provided by David Julius (University of California, San Francisco, CA) and were generated by deleting an exon encoding part of the fifth and all of the sixth putative transmembrane domains of the channel, as described by Caterina et al. (1). TRPV1−/− mice were backcrossed to C57BL/6 wild-type (WT) mice for ≥6 generations and had been shown to have impaired nociception (1). The experiments were carried out in 10-week-old male TRPV1−/− and WT (C57BL/6) mice obtained from Charles River Laboratories (Wilmington, MA). The mice were allowed free access to regular mouse chow and water ad libitum. All of the experiments were approved by the institutional animal care and use committee.
Telemetry blood pressure measurement.
Mean arterial pressure (MAP) and heart rate (HR) were recorded using a telemetry system (Data Sciences International, St. Paul, MN) according to the manufacturer's instructions. In brief, the mice were anesthetized with ketamine and xylazine (80 and 4 mg/kg of body weight subcutaneously [s.c.], respectively), the catheter linked to the transmitter was inserted into the left carotid artery, and the body of the transmitter was placed subcutaneously in the lower right side of the abdomen. The mice were returned to their individual cages and allowed to recover for 3 days before radiotelemetric recording was started. MAP and HR were recorded over 10 s every 10 min. Data were averaged over 60-min interval periods.
Experimental protocol.
Three days after the radiotelemetric recording, WT and TRPV1−/− mice were given a single intraperitoneal injection of either vehicle (saline) or LPS (5 million endotoxin units/kg), derived from Escherichia coli (Sigma Chemical, St. Louis, MO). The mice were sacrificed 6 or 24 h after administration of LPS. Blood was collected for analysis of creatinine and alanine aminotransferase (ALT) activities and cytokine and CGRP levels. The kidney and liver were fixed in 4% phosphate-buffered paraformaldehyde for histological analysis or frozen in liquid nitrogen for chemokine assay.
Histology and immunohistochemistry.
Paraformaldehyde-fixed kidney and liver were embedded in paraffin. From the paraffin-embedded tissue blocks, 4-μm sections were cut and stained with hematoxylin and eosin (H&E). Renal and hepatic damage was evaluated on H&E-stained sections.
For the immunohistochemical study of neutrophil infiltration in the kidney and liver, 4-μm sections of paraffin-embedded specimens were treated with 3% hydrogen peroxide for 5 min and blocked with 3% normal horse serum in phosphate buffer solution for 1 h. The sections were then incubated overnight at 4°C with rat anti-mouse monoclonal antibody to neutrophils (1:200; Sterotec, Oxford, United Kingdom). After being washed in phosphate buffer, the sections were incubated with 1:400-diluted horse anti-rat IgG conjugated with horseradish peroxidase (Vector Laboratories, Burlingame, CA) for 1 h at room temperature and visualized by incubation of the sections with the substrate vector fast red (Vector Laboratories, Burlingame, CA). Finally, the sections were counterstained in hematoxylin, mounted, and photographed. Negative-control experiments were performed with all the same conditions except that the primary antibody was omitted during incubation.
Neutrophils were identified by positive staining and morphology and were counted for at least 10 fields under ×400 magnification for each slide. The number of neutrophils was expressed as cells per square millimeter. The histological evaluation was performed in a blind fashion with regard to the treatment of animals.
Creatinine and ALT assay.
To further determine renal and hepatic injury, serum creatinine and ALT activities were assayed by the use of an improved Jaffe creatinine assay kit (BioAssay Systems, Hayward, CA) and a kinetic test kit (Biotron Diagnostics, Hernet, CA), respectively.
Cytokine/chemokine assay.
The levels of tumor necrosis factor alpha (TNF-α), interleukin 1β (IL-1β), and IL-6 in serum and the protein levels of KC and macrophage inflammatory protein 2 (MIP-2) in whole kidney tissues were determined using the corresponding enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN) by following the manufacturer's instructions. Fifty microliters or less of serum was used for the TNF-α, IL-1β, and IL-6 assay. For determination of levels of KC and MIP-2 in whole kidney tissues, kidneys were homogenized in ice-cold phosphate buffer containing protease inhibitor cocktail (Sigma Chemical, St. Louis, MO), and the total protein was extracted using NE-PER cytoplasmic extraction reagents (Pierce, Rockford, IL). The total protein concentration in tissue extract, determined by the use of a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA), was used for normalization of chemokine levels in kidney tissues.
Western blot analysis.
Frozen kidney samples were homogenized in ice-cold lysis buffer (20 mM HEPES, 75 mM NaCl, 2.5 mM MgCl2, 0.1 mM EDTA, 0.1% Triton X-100) containing a 1/10 volume of a protease inhibitor solution consisting of 25 μg/ml antipain, 1 μg/ml aprotinin, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, and 200 μM phenylmethylsulfonyl fluoride. The homogenate was centrifuged at 15,000 × g at 4°C for 20 min. Protein content of the supernatant was measured using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA). Samples (30 μg of protein) were loaded onto a 10% sodium dodecyl sulfate-polyacrylamide gel, followed by electrophoresis and blotting to a polyvinylidene difluoride membrane. The membranes were blocked for 1 h at room temperature in 5% milk solution (50 mM Tris-HCl, 100 mM NaCl, and 0.1% Tween 20 at pH 7.5). Subsequently, the membranes were incubated with goat anti-mouse ICAM-1 polyclonal antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) or goat anti-human VCAM-1 polyclonal antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) in blocking solution overnight at 4°C. After being washed, the membranes were incubated with bovine anti-goat horseradish peroxidase-conjugated antibody (1:3,000; Santa Cruz Biotechnology, Santa Cruz, CA) in blocking solution at room temperature for 1 h. The membranes were developed using the enhanced chemiluminescence ECL kit (GE Healthcare, Buckinghamshire, United Kingdom), and the signals were detected using Hyperfilm (Hyperfilm-ECL; GE Healthcare, Buckinghamshire, United Kingdom). Quantification was performed with Scion Image analysis software (Scion, Frederick, MD). β-Actin was used to normalize protein loaded on the membranes.
CGRP assay.
CGRP levels in plasma 6 h after LPS injection were assayed using the CGRP (rat, mouse) radioimmunoassay kit (Phoenix Pharmaceuticals, Belmont, CA) according to the manufacturer's protocol (12). This antibody has 100% cross-reactivity with α-CGRP and 79% cross-reactivity with β-CGRP. There is no cross-reactivity with amylin, calcitonin, somatostatin, or substance P.
Statistical analysis.
All values are expressed as means and standard errors (SE). The significance of differences among groups was analyzed using one-way analysis of variance (ANOVA), followed by Bonferroni's adjustment for multiple comparisons. Differences were considered statistically significant at a P value of <0.05.
RESULTS
WT and TRPV1−/− mice were implanted with telemetric probes, and their hemodynamic parameters (MAP and HR) were continuously measured before and after injection of LPS. These parameters were monitored for 24 h to study the changes in MAP and HR produced by LPS. As shown in Fig. 1, MAP was not different between WT and TRPV1−/− mice before LPS injection. MAP decreased immediately after LPS injection and reached the lowest point at about 8 to 12 h after LPS injection in both WT and TRPV1−/− mice, with no difference between the two strains. While LPS led to a transient increase followed by the prolonged decrease in HR, there was no difference between WT and TRPV1−/− mice.
Fig 1.
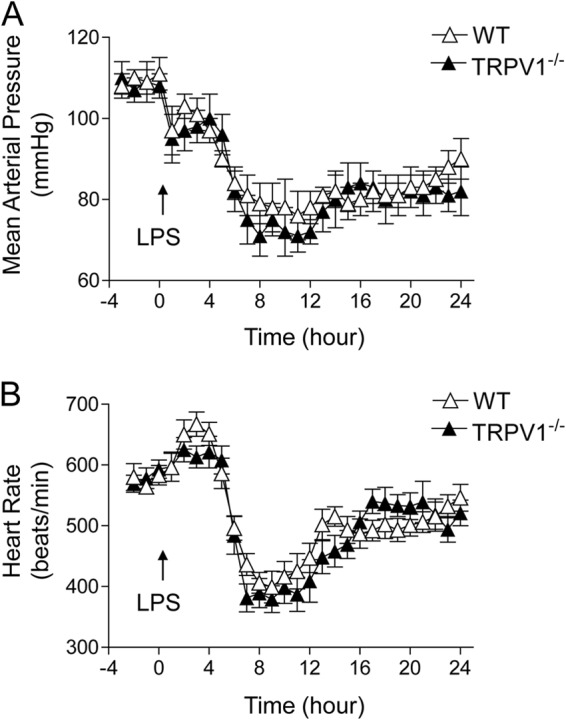
Effect of lipopolysaccharide (LPS) treatment on mean arterial pressure (A) and heart rate (B), as determined by radiotelemetry, in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. LPS (5 million endotoxin units/kg) was intraperitoneally injected at 0 min. Values are means ± SE (n = 6).
LPS-induced damage of the kidney and liver was assessed in WT and TRPV1−/− mice after H&E staining. As shown in Fig. 2, renal and hepatic histology was not different between WT and TRPV1−/− mice before LPS challenge. Twenty-four hours after LPS injection, TRPV1−/− mice displayed manifested glomerular injury, including hypercellularity and parenchymal red blood cell congestion. Likewise, LPS-treated TRPV1−/− mice exhibited disrupted hepatic architecture manifested by extensive areas of hemorrhage and coagulative necrosis in the liver. On the other hand, mild tissue injury and hemorrhage were found in the kidney and liver of LPS-treated WT mice.
Fig 2.
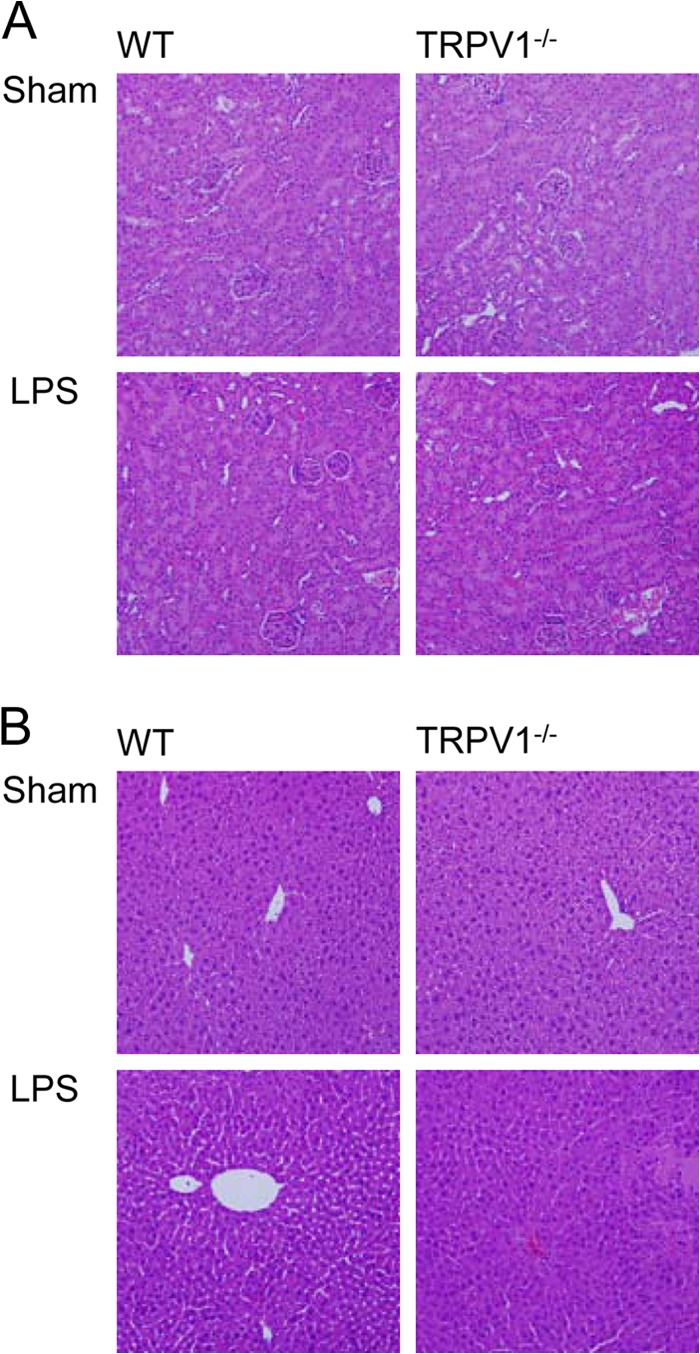
Effect of lipopolysaccharide (LPS) treatment on renal and hepatic morphology in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. H&E-stained kidney (A) and liver (B) sections showing morphological changes in WT and TRPV1−/− mice 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Magnification, ×200.
Renal and hepatic injury caused by LPS was confirmed by serum creatinine and ALT activity assays (Fig. 3). Serum creatinine levels were similar between WT and TRPV1−/− mice before LPS challenge but increased in WT and TRPV1−/− mice 24 h after LPS injection, with a greater effect in the latter. Similarly, there was no difference in baseline serum ALT levels between WT and TRPV1−/− mice. However, serum ALT levels increased 24 h after LPS injection in both strains, with a greater effect in TRPV1−/− mice than in WT mice. The results indicate that LPS induces exaggerated injury in kidney and liver when TRPV1 is ablated.
Fig 3.
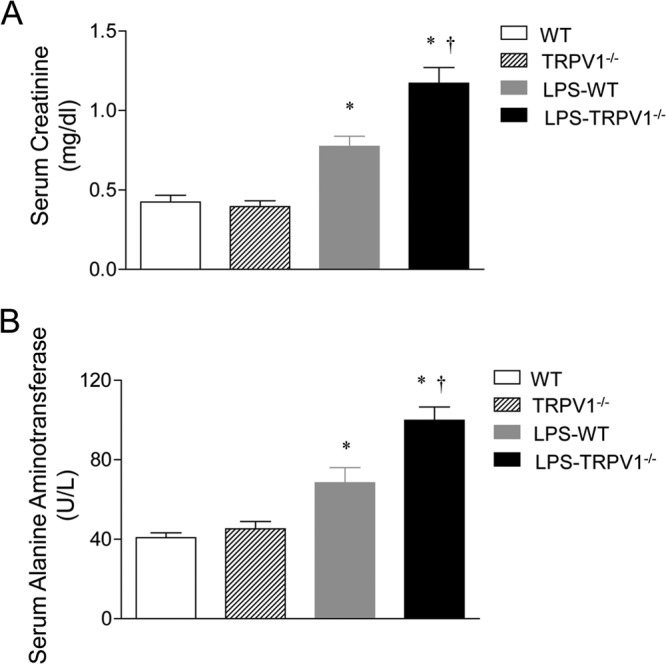
Effect of lipopolysaccharide (LPS) treatment on serum creatinine (A) and alanine aminotransferase (B) activities in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. Serum samples were collected at 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
As illustrated in Fig. 4 and 5, neutrophil infiltration was examined in the renal cortex and liver of WT and TRPV1−/− mice 6 or 24 h after LPS injection, given that neutrophils have been implicated in organ damage after LPS challenge. Immunohistochemical staining for neutrophils showed relatively few neutrophils in the kidney and liver of WT and TRPV1−/− mice before LPS injection. LPS treatment resulted in a marked increase in renal and hepatic neutrophil infiltration in WT mice, with a greater magnitude at 6 h than at 24 h after LPS challenge. The effects of LPS on neutrophil recruitment in the kidney and liver at 6 and 24 h were exaggerated in TRPV1−/− mice compared to WT mice.
Fig 4.
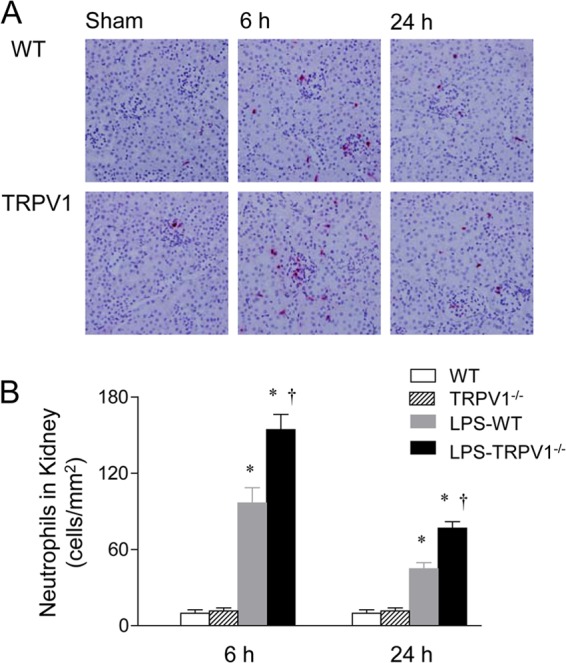
Effect of lipopolysaccharide (LPS) treatment on renal cortex neutrophil infiltration in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. (A) Immunohistochemically stained sections from the renal cortex demonstrating neutrophils in the renal cortex of control WT and TRPV1−/− mice and WT and TRPV1−/− mice treated with LPS. (B) Bar graph showing the number of renal cortical neutrophils, expressed as cells per square millimeter. Tissue samples were harvested at 6 or 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
Fig 5.
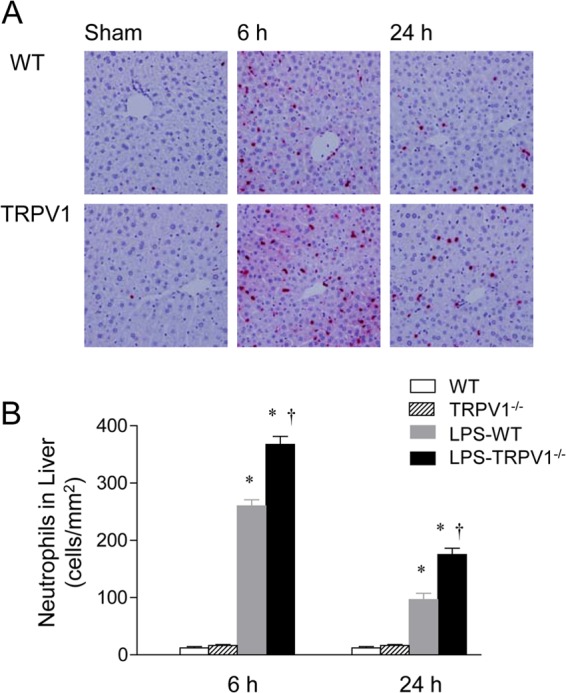
Effect of lipopolysaccharide (LPS) treatment on hepatic neutrophil infiltration in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. (A) Immunohistochemically stained sections from the liver demonstrating neutrophils in the liver of control WT and TRPV1−/− mice and WT and TRPV1−/− mice treated with LPS. (B) Bar graph showing the number of hepatic neutrophils, expressed as cells per square millimeter. Tissue samples were harvested at 6 or 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
To evaluate the systemic inflammatory response to LPS, serum concentrations of cytokines were determined 6 and 24 h after LPS treatment, and these data are summarized in Fig. 6. The concentrations of serum cytokines, including TNF-α, IL-1β, and IL-6, were not different in vehicle-treated WT and TRPV1−/− mice. In response to LPS treatment, TRPV1−/− mice exhibited significantly greater amounts of cytokines (TNF-α, IL-1β, and IL-6) 6 h after LPS injection than did WT mice. With the exception of IL-6, there was no difference in TNF-α and IL-1β levels between WT and TRPV1−/− mice 24 h after LPS injection.
Fig 6.
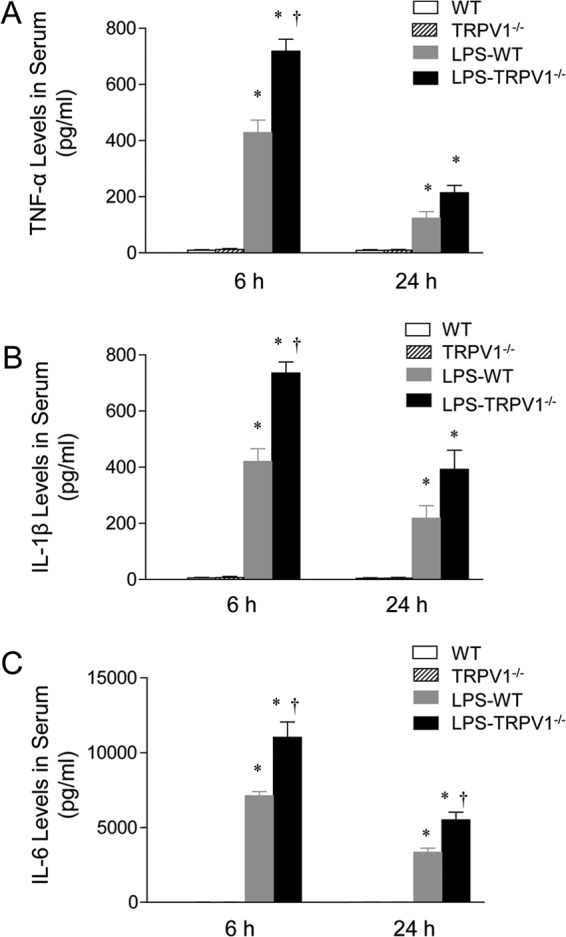
Effect of lipopolysaccharide (LPS) treatment on serum TNF-α (A), IL-1β (B), and IL-6 (C) levels in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. Serum samples were collected at 6 or 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
Chemokines, including KC and MIP-2, were assessed in the kidney of WT and TRPV1−/− mice 6 and 24 h after LPS injection (Fig. 7), given that KC and MIP-2 are potent chemokines for neutrophil recruitment. No difference was observed in KC and MIP-2 between vehicle-treated WT and TRPV1−/− mice. However, WT and TRPV1−/− mice had a marked increase in KC and MIP-2 6 h after LPS treatment, with a greater effect in TRPV1−/− mice than in WT mice. While still significantly elevated 24 h after LPS injection, there was no difference in KC and MIP-2 levels between WT and TRPV1−/− mice.
Fig 7.
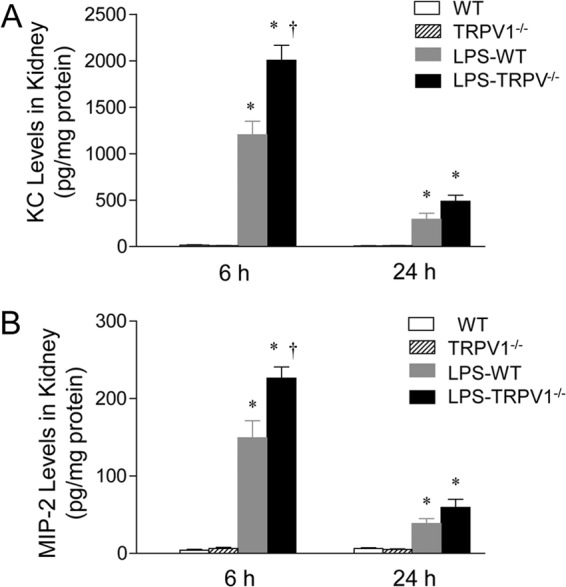
Effect of lipopolysaccharide (LPS) treatment on renal chemokine KC (A) and MIP-2 (B) levels in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. Tissue samples were collected at 6 or 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
In addition to proinflammatory chemokines, enhanced expression of adhesion molecules may also contribute to neutrophil extravasation caused by LPS. As shown in Fig. 8, the effect of LPS on expression of key adhesion molecules, ICAM-1 and VCAM-1, in the kidney was determined using Western blot. ICAM-1 expression in the kidney increased in WT and TRPV1−/− mice 6 h after LPS injection, with no difference between the two strains. ICAM-1 expression further increased in WT and TRPV1−/− mice 24 h after LPS injection, with a greater magnitude in the latter. In addition, the induction of VCAM-1 expression caused by LPS was enhanced 6 h after LPS injection, with substantial levels remaining for 24 h in both WT and TRPV1−/− mice. Moreover, a greater increase in VCAM-1 expression was observed in TRPV1−/− mice than in WT mice at 6 but not 24 h after LPS treatment.
Fig 8.
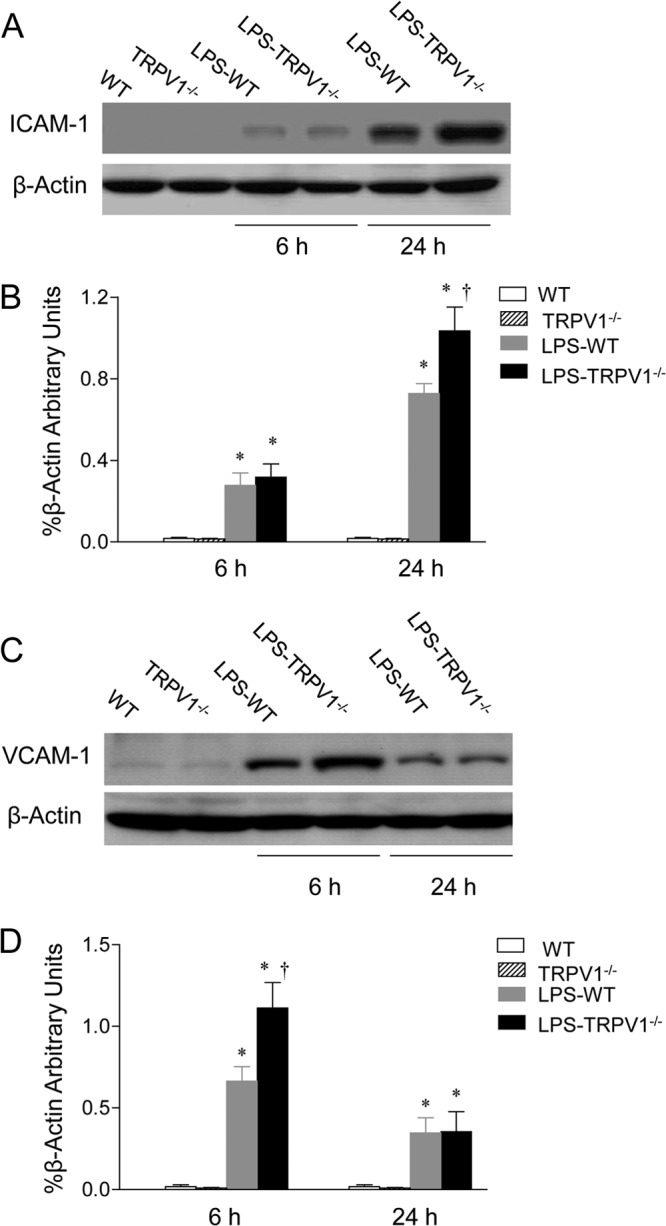
Effect of lipopolysaccharide (LPS) treatment on renal ICAM-1 and VCAM-1 protein expression in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. (A and C) Representative Western blots of ICAM-1 (A) and VCAM-1 (C) in the kidney of control WT and TRPV1−/− mice and WT and TRPV1−/− mice treated with and without LPS. (B and D) Bar graphs showing the relative optical density values for ICAM-1 (B) and VCAM-1 (D) in WT and TRPV1−/− mice with and without LPS. Tissue samples were collected at 6 or 24 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Protein expression levels are normalized with the housekeeping protein β-actin. Values are means ± SE (n = 4 to 5). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
Plasma CGRP levels were determined 6 h after LPS administration. As shown in Fig. 9, we found that intraperitoneal injection of LPS significantly increased plasma CGRP levels in WT mice. The enhanced plasma CGRP levels were abolished in TRPV1−/− mice.
Fig 9.
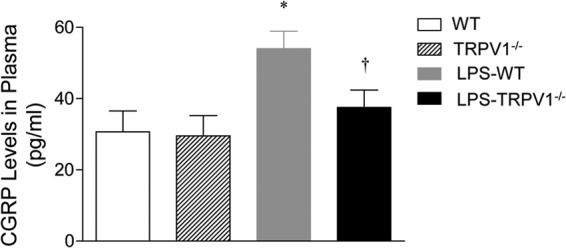
Effects of lipopolysaccharide (LPS) treatment on plasma calcitonin gene-related peptide (CGRP) levels in wild-type (WT) and TRPV1-null mutant (TRPV1−/−) mice. Plasma samples were collected at 6 h after intraperitoneal injection of vehicle or LPS (5 million endotoxin units/kg). Values are means ± SE (n = 6 to 8). *, P value of <0.05 compared with control WT or TRPV1−/− mice; †, P value of <0.05 compared with LPS-treated WT mice.
DISCUSSION
The development of multiple organ failure has been shown to be a key predictor of the outcome of septic shock (13). We therefore examined the role of TRPV1 in LPS-induced end-organ damage. The results demonstrate that deletion of TRPV1 did not affect LPS-induced circulatory failure in terms of telemetry-determined MAP and HR but markedly accelerated renal and hepatic injury revealed by histological and biochemical analysis, including further elevated serum creatinine and ALT levels after LPS injection. Moreover, the exaggerated organ damage in TRPV1-ablated mice injected with LPS is accompanied by heightened inflammatory responses, as evidenced by the enhancement in neutrophil infiltration, proinflammatory cytokine and chemokine production, and adhesion molecule expression. The results suggest that LPS-induced aggravated organ damage in TRPV1−/− mice may be attributed to exaggerated inflammatory responses rather than worsened circulatory failure.
In light of the fact that LPS-induced organ damage is more severe when TRPV1 is ablated, the data suggest that TRPV1 may be a key target for LPS or LPS-induced signaling pathways. It has been well established that TRPV1 can be activated by various stimuli, including low pH and prostanoids (2, 3). In addition, it has been reported that the endocannabinoid anandamide, produced by macrophages, contributes to endotoxin-induced hypotension (7, 8). While activation of cannabinoid receptor 1 (CB1) by anandamide elicits hypotension (14, 15), Zygmunt et al. (3) have shown that activation of TRPV1 receptors acts as a predominant mechanism for anandamide-induced relaxation in the rat mesenteric arteries, indicating that TRPV1 plays a key role in anandamide-induced action. The notion is further supported by previous in vivo studies showing that the depressor response to methanandamide, a metabolically stable analog of anandamide, is attenuated by blockade of TRPV1 with its antagonist, capsazepine (9). Given that endotoxemia often leads to tissue acidification and increased production of various eicosanoids, including anandamide and arachidonic acid metabolites (7, 8, 16, 17), it is likely that TRPV1 receptors are activated during endotoxemia.
The pathophysiology of LPS-induced organ damage is complex, involving altered hemodynamics (18, 19), inflammation (20, 21), and/or cytotoxic injury (22). Previous studies by Clark et al. using a tail-cuff technique have shown that LPS-induced hypotension is accelerated in TRPV1−/− mice (10). Although the tail-cuff method is noninvasive, direct and continuing measurement of arterial pressure with radiotelemetry provides a more accurate and reliable assessment that is recommended for blood pressure detection under conscious conditions, especially in mice. The data from the present study show that blood pressure determined with the use of radiotelemetry decreased in response to LPS injection; however, it was not different between WT and TRPV1−/− mice. These results suggest that TRPV1 ablation is insignificant in terms of affecting LPS-induced hypotension, and therefore, aggravated organ damage observed in TRPV1-ablated mice is likely attributed to mechanisms other than altered hemodynamics.
In general, in the case of bacterial infection or sepsis in animal or human hosts, the hosts remove pathogens primarily by increasing neutrophil recruitment to the afflicted sites. Although neutrophil recruitment to the affected organs is part of the natural defense mechanism, overwhelming accumulation of neutrophils usually causes further damage of the affected organs, thus contributing significantly to the development of multiple organ failure (23, 24). Neutrophils may produce several proinflammatory mediators, including oxygen-free radicals, neutrophil-specific proteases, and products of lipid peroxidation, many of which are injurious to cells and may lead to irreversible organ damage (25). The fact that LPS-induced neutrophil infiltration in the kidney and liver is worsened when TRPV1 is ablated indicates that activation of TRPV1 in WT mice during endotoxic shock may prevent neutrophil infiltration, an action that constitutes an anti-inflammatory property of TRPV1.
In response to LPS administration, a number of cytokines (i.e., TNF-α, IL-1, and IL-6) and chemokines (KC and MCP-1) will be produced and released (26). Our results show that heightened neutrophil infiltration in the kidney and liver of TRPV1−/− mice after LPS injection was accompanied by amplified cytokine and chemokine responses in circulation and the kidney, respectively. These results indicate that TRPV1 protects against LPS-induced organ damage via inhibition of the production of proinflammatory cytokines and chemokines and are consistent with the reports showing that TRPV1 plays a counterregulatory role against acute lung inflammatory injury induced by intratracheal administration of LPS (27). This notion is supported by most recent studies showing that TRPV1 protects against the transition from a local to a systemic inflammatory state in a cecal ligation and puncture (CLP) model of sepsis (28). In addition, it has been shown that the proinflammatory cytokines stimulate expression of adhesion molecules that lead to neutrophil-dependent organ damage (29). The adhesion molecules, including ICAM-1 and VCAM-1, are essential for the regulation of leukocyte trafficking across the vascular endothelium and are critically involved in vascular inflammatory responses (30, 31). Heightened expression of these adhesion molecules may lead to enhanced leukocyte attachment to endothelial cells and, consequently, tissue infiltration by leukocytes.
Interestingly, we found that the time course of ICAM-1 expression did not correlate with neutrophil recruitment in the kidney. These data suggest that ICAM-1 may not be the key factor for neutrophil migration into tissues in TRPV1−/− mice. In contrast, we found that upregulation of VCAM-1 protein in the kidneys of TRPV1−/− mice occurred as early as 6 h after exposure to LPS, suggesting that VCAM-1 may facilitate egress of neutrophils into tissues, as reported by others in hosts with high cytokine levels, although VCAM-1 was not originally recognized as an important mediator of neutrophil infiltration (32).
Given the complexity of inflammatory responses, mechanisms underlying TRPV1 anti-inflammatory effects may involve several pathways. TRPV1 is known to be expressed mainly in primary sensory nerves. Pretreatment with capsaicin, a selective TRPV1 agonist, in a rodent model of allergic inflammation leads to increased TNF-α levels, suggesting that loss of TRPV1-positive sensory nerves enhances TNF-α production (33). Consistent with the results, we found that TRPV1 ablation aggravated inflammatory responses, including increasing proinflammatory cytokine production. In addition, most recent studies by us have shown that TRPV1 is activated during the early phase of sepsis (11). One of the consequences of TRPV1 activation is the release of sensory neurotransmitters, including CGRP. In addition to its well-recognized cardiovascular effects, CGRP has been shown to modulate antigen presentation, phagocytosis, and cytokine production induced by endotoxin (34–36). Indeed, inflammatory responses are worsened in CGRP-null mutant mice (37). In the present study, we found that ablation of TRPV1 abolished the enhanced levels of plasma CGRP during endotoxic shock. Consistent with the previous studies (37), our results suggest that the exaggerated inflammatory response may be attributed to the loss of CGRP release in TRPV1−/− mice suffering from endotoxic shock. Moreover, evidence has shown that TRPV1 receptors are expressed in the vascular endothelium and that TRPV1 activation increases nitric oxide (NO) production via activation of endothelial NO synthase (eNOS) (38). While excessive NO production can be cytotoxic, NO produced by eNOS in the case of TRPV1 activation serves a beneficial role as a messenger and host defense molecule against inflammation (39, 40). Although several mechanisms may contribute to the anti-inflammatory effects of TRPV1, further studies are required to clarify the detailed molecular mechanisms.
In summary, we found that deletion of TRPV1 exacerbates organ damage during endotoxic shock despite the lack of worsened circulatory failure. The aggravated organ damage is associated with the enhancement of neutrophil infiltration, cytokine and chemokine production, and adhesion molecule expression in tissues. These data suggest that the enhancement in organ damage may be derived primarily from the loss of TRPV1-induced anti-inflammatory effects rather than affecting blood pressure.
ACKNOWLEDGMENTS
This work was supported, in part, by grants from the National Institutes of Health (HL-57853, HL-73287, and DK67620) and Michigan Economic Development Corporation to D. H. Wang. This work was also supported, in part, by grants from the National Natural Science Foundation of China (no. 81170243) and Henan Provincial Science and Technology Innovative Program for Outstanding Scholarship (no. 124200510007) to Y. Wang.
We declare no conflicts of interest.
Footnotes
Published ahead of print 1 May 2013
REFERENCES
- 1. Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. 2000. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313 [DOI] [PubMed] [Google Scholar]
- 2. Szallasi A, Blumberg PM. 1999. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol. Rev. 51:159–212 [PubMed] [Google Scholar]
- 3. Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, DiMarzo V, Julius D, Hogestatt ED. 1999. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457 [DOI] [PubMed] [Google Scholar]
- 4. Glauser MP, Zanetti G, Baumgartner JD, Cohen J. 1991. Septic shock: pathogenesis. Lancet 338:732–736 [DOI] [PubMed] [Google Scholar]
- 5. Parrillo JE. 1993. Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 328:1471–1477 [DOI] [PubMed] [Google Scholar]
- 6. Wheeler AP, Bernard GR. 1999. Treating patients with severe sepsis. N. Engl. J. Med. 340:207–214 [DOI] [PubMed] [Google Scholar]
- 7. Bátkai S, Pacher P, Járai Z, Wagner JA, Kunos G. 2004. Cannabinoid antagonist SR-141716 inhibits endotoxic hypotension by a cardiac mechanism not involving CB1 or CB2 receptors. Am. J. Physiol. Heart Circ. Physiol. 287:H595–H600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Varga K, Wagner JA, Bridgen DT, Kunos G. 1998. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 12:1035–1044 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Kaminski NE, Wang DH. 2005. VR1-mediated depressor effects during high-salt intake: role of anandamide. Hypertension 46:986–991 [DOI] [PubMed] [Google Scholar]
- 10. Clark N, Keeble J, Fernandes ES, Starr A, Liang L, Sugden D, de Winter P, Brain SD. 2007. The transient receptor potential vanilloid 1 (TRPV1) receptor protects against the onset of sepsis after endotoxin. FASEB J. 21:3747–3755 [DOI] [PubMed] [Google Scholar]
- 11. Wang Y, Novotný M, Quaiserová-Mocko V, Swain GM, Wang DH. 2008. TRPV1-mediated protection against endotoxin-induced hypotension and mortality in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294:R1517–R1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y, Kaminski NE, Wang DH. 2007. Endocannabinoid regulates blood pressure via activation of the transient receptor potential vanilloid type 1 in Wistar rats fed a high-salt diet. J. Pharmacol. Exp. Ther. 321:763–769 [DOI] [PubMed] [Google Scholar]
- 13. Hebert PC, Drummond AJ, Singer J, Bernard GR, Russell JA. 1993. A simple multiple system organ failure scoring system predicts mortality of patients who have sepsis syndrome. Chest 104:230–235 [DOI] [PubMed] [Google Scholar]
- 14. Lake KD, Martin BR, Kunos G, Varga K. 1997. Cardiovascular effects of anandamide in anesthetized and conscious normotensive and hypertensive rats. Hypertension 29:1204–1210 [DOI] [PubMed] [Google Scholar]
- 15. Varga K, Lake KD, Huangfu D, Guyenet PG, Kunos G. 1996. Mechanism of the hypotensive action of anandamide in anesthetized rats. Hypertension 28:682–686 [DOI] [PubMed] [Google Scholar]
- 16. Morrison DC, Ulevitch RJ. 1978. The effects of bacterial endotoxins on host mediation systems. A review. Am. J. Pathol. 93:526–617 [PMC free article] [PubMed] [Google Scholar]
- 17. West MA, Wilson C. 1996. Hypoxic alterations in cellular signal transduction in shock and sepsis. New Horiz. 4:168–178 [PubMed] [Google Scholar]
- 18. Wang W, Zolty E, Falk S, Basava V, Reznikov L, Schrier R. 2006. Pentoxifylline protects against endotoxin-induced acute renal failure in mice. Am. J. Physiol. Renal Physiol. 291:F1090–F1095 [DOI] [PubMed] [Google Scholar]
- 19. Wu L, Tiwari MM, Messer KJ, Holthoff JH, Gokden N, Brock RW, Mayeux PR. 2007. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipoplysaccharide administration in mice. Am. J. Physiol. Renal Physiol. 292:F261–F268 [DOI] [PubMed] [Google Scholar]
- 20. Miyaji T, Hu X, Yuen PST, Muramatsu Y, Iyer S, Hewitt SM, Star RA. 2003. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 64:1620–1631 [DOI] [PubMed] [Google Scholar]
- 21. Wang W, Faubel S, Ljubanovic D, Mitra A, Falk SA, Kim J, Tao Y, Soloviev A, Reznikov LL, Dinarello CA, Schrier RW, Edelstein CL. 2005. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am. J. Physiol. Renal Physiol. 288:F997–F1004 [DOI] [PubMed] [Google Scholar]
- 22. Wang W, Jittikanont S, Falk SA, Li P, Feng L, Gengaro PE, Poole BD, Bowler RP, Day BJ, Crapo JD, Schrier RW. 2003. Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am. J. Physiol. Renal Physiol. 284:F532–F537 [DOI] [PubMed] [Google Scholar]
- 23. He M, Horuk R, Moochhala SM, Bhatia M. 2007. Treatment with BX471, a CC chemokine receptor 1 antagonist, attenuates systemic inflammatory response during sepsis. Am. J. Physiol. Gastrointest. Liver Physiol. 292:G1173–G1180 [DOI] [PubMed] [Google Scholar]
- 24. Macdonald J, Galley HF, Webster NR. 2003. Oxidative stress and gene expression in sepsis. Br. J. Anaesth. 90:221–232 [DOI] [PubMed] [Google Scholar]
- 25. Fitzpatrick MM, Shah V, Filler G, Dillon MJ, Barratt TM. 1992. Neutrophil activation in the hemolytic uremic syndrome: free and complexed elastase in plasma. Pediatr. Nephrol. 6:50–53 [DOI] [PubMed] [Google Scholar]
- 26. Hack CE, Aarden LA, Thijs LG. 1997. Role of cytokines in sepsis. Adv. Immunol. 66:101–195 [DOI] [PubMed] [Google Scholar]
- 27. Helyes Z, Elekes K, Németh J, Pozsgai G, Sándor K, Kereskai L, Börzsei R, Pintér E, Szabó Á Szolcsányi J. 2007. Role of transient receptor potential vanilloid 1 receptors in endotoxin-induced airway inflammation in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L1173–L1181 [DOI] [PubMed] [Google Scholar]
- 28. Fernandes ES, Liang L, Smillie SJ, Kaiser F, Purcell R, Rivett DW, Alam S, Howat S, Collins H, Thompson SJ, Keeble JE, Riffo-Vasquez Y, Bruce KD, Brain SD. 2012. TRPV1 deletion enhances local inflammation and accelerates the onset of systemic inflammatory response syndrome. J. Immunol. 188:5741–5751 [DOI] [PubMed] [Google Scholar]
- 29. Horgan MJ, Palace GP, Everitt JE, Malik AB. 1993. TNF-alpha release in endotoxemia contributes to neutrophil-dependent pulmonary edema. Am. J. Physiol. Heart Circ. Physiol. 264:H1161–H1165 [DOI] [PubMed] [Google Scholar]
- 30. Albelda SM, Smith CW, Ward PA. 1994. Adhesion molecules and inflammatory injury. FASEB J. 8:504–512 [PubMed] [Google Scholar]
- 31. Zhou MY, Lo SK, Bergenfeldt M, Tiruppathi C, Jaffe A, Xu N, Malik AB. 1998. In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by a beta2 integrin-dependent mechanism. J. Clin. Invest. 101:2427–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ibbotson GC, Doig C, Kaur J, Gill V, Ostrovsky L, Fairhead T, Kubes P. 2001. Functional alpha4-integrin: a newly identified pathway of neutrophil recruitment in critically ill septic patients. Nat. Med. 7:465–470 [DOI] [PubMed] [Google Scholar]
- 33. Franco-Penteado CF, De Souza IA, Camargo EA, Teixeira SA, Muscara MN, De Nucci G, Antunes E. 2005. Mechanisms involved in the enhancement of allergic airways neutrophil influx by permanent C-fiber degeneration in rats. J. Pharmacol. Exp. Ther. 313:440–448 [DOI] [PubMed] [Google Scholar]
- 34. Asahina A, Moro O, Hosoi J, Lerner EA, Xu S, Takashima A, Granstein RD. 1995. Specific induction of cAMP in Langerhans cells by calcitonin gene-related peptide: relevance to functional effects. Proc. Natl. Acad. Sci. U. S. A. 92:8323–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ichinose M, Sawada M. 1996. Enhancement of phagocytosis by calcitonin gene-related peptide (CGRP) in cultured mouse peritoneal macrophages. Peptides 17:1405–1414 [DOI] [PubMed] [Google Scholar]
- 36. Monneret G, Pachot A, Laroche B, Picollet J, Bienvenu J. 2000. Procalcitonin and calcitonin gene-related peptide decrease LPS-induced TNF production by human circulating blood cells. Cytokine 12:762–764 [DOI] [PubMed] [Google Scholar]
- 37. Bowers MC, Katki KA, Rao A, Koehler M, Patel P, Spiekerman A, DiPette DJ, Supowit SC. 2005. Role of calcitonin gene-related peptide in hypertension-induced renal damage. Hypertension 46:51–57 [DOI] [PubMed] [Google Scholar]
- 38. Poblete IM, Orliac ML, Briones R, Adler-Graschinsky E, Huidobro-Toro JP. 2005. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J. Physiol. 568:539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang W, Mitra A, Poole B, Falk S, Lucia S, Tayal S, Schrier R. 2004. Endothelial nitric oxide synthase-deficient mice exhibit increased susceptibility to endotoxin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 287:F1044–F1048 [DOI] [PubMed] [Google Scholar]
- 40. Yamashita T, Kawashima S, Ohashi Y, Ozaki M, Ueyama T, Ishida T, Inoue N, Hirata KI, Akita H, Yokoyama M. 2000. Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation 101:931–937 [DOI] [PubMed] [Google Scholar]