

Abstract
Cells of Saccharomyces cerevisiae express two tryptophan permeases, Tat1 and Tat2, which have different characteristics in terms of their affinity for tryptophan and intracellular localization. Although the high-affinity permease Tat2 has been well documented in terms of its ubiquitin-dependent degradation, the low-affinity permease Tat1 has not yet been characterized fully. Here we show that a high hydrostatic pressure of 25 MPa triggers a degradation of Tat1 which depends on Rsp5 ubiquitin ligase and the EH domain-containing protein End3. Tat1 was resistant to a 3-h cycloheximide treatment, suggesting that it is highly stable under normal growth conditions. The ubiquitination of Tat1 most likely occurs at N-terminal lysines 29 and 31. Simultaneous substitution of arginine for the two lysines prevented Tat1 degradation, but substitution of either of them alone did not, indicating that the roles of lysines 29 and 31 are redundant. When cells were exposed to high pressure, Tat1-GFP was completely lost from the plasma membrane, while substantial amounts of Tat1K29R-K31R-GFP remained. The HPG1-1 (Rsp5P514T) and rsp5-ww3 mutations stabilized Tat1 under high pressure, but any one of the rsp5-ww1, rsp5-ww2, and bul1Δ bul2Δ mutations or single deletions of genes encoding arrestin-related trafficking adaptors did not. However, simultaneous loss of 9-arrestins and Bul1/Bul2 prevented Tat1 degradation at 25 MPa. The results suggest that multiple PPxY motif proteins share some essential roles in regulating Tat1 ubiquitination in response to high hydrostatic pressure.
INTRODUCTION
The genome of the yeast Saccharomyces cerevisiae encodes 24 amino acid permeases and their homologous proteins. These permeases consist of 12 transmembrane domains (TMDs) and cytoplasmic tails and have unique characteristics in terms of substrate specificity, affinity, capacity, and posttranslational modification (1–3). Tryptophan is hydrophobic and bulky and is one of the sparsest amino acids in nutrients and nature. This leads to the concept that the tryptophan uptake machinery can be specialized evolutionarily in heterotrophic organisms. In S. cerevisiae, tryptophan is imported by facilitated diffusion through Tat1 and Tat2, which are the low-affinity and high-affinity tryptophan permeases, respectively. TAT1 and TAT2 were originally identified as genes conferring resistance to the immunosuppressive agent tacrolimus (FK506) (4, 5). Because tryptophan uptake is highly sensitive to diverse environmental stresses, it becomes the limiting factor for cell growth in tryptophan-auxotrophic strains. Of the two permeases, Tat2 is relatively well characterized in terms of ubiquitin-dependent degradation and intracellular trafficking. In response to nutrient deprivation or rapamycin treatment, Tat2 undergoes ubiquitination in a manner dependent on Rsp5 ubiquitin ligase, followed by vacuolar degradation (6). Appropriate delivery of Tat2 to the cell surface requires ergosterol (7, 8) and glycosylphosphatidylinositol-anchored proteins (9).
We reported previously that hydrostatic pressure at nonlethal levels of 15 to 25 MPa (∼150 to 250 kg cm−2) or the low temperature of 15°C compromised the uptake of tryptophan and thereby inhibited growth of tryptophan-auxotrophic strains (10). Overexpression of TAT2 confers the ability of growth under these conditions (10). When cells were incubated under conditions of high pressure or low temperature, Tat1 and Tat2 underwent vacuolar degradation in a manner dependent on Rsp5 ubiquitin ligase (11, 12). High hydrostatic pressure has a profound impact on lipid membranes, primarily resulting in tighter packing and restriction of acyl chain motion, in a manner analogous to that with decreasing temperatures (13–15). With increasing pressure, the gel-to-liquid crystalline coexistence region is shifted toward higher temperatures by approximately 22°C/100 MPa (14). In this regard, tryptophan uptake is hypersensitive to reductions in membrane fluidity caused by either increasing pressure or decreasing temperature. Tryptophan uptake is also sensitive to some agents, such as 4-phenylbutyrate (16), isoflurane (17), and phytosphingosine (18), that potentially have disturbing effects on the membrane, and overexpression of TAT2 confers resistance to these agents. Accordingly, hydrostatic pressure in combination with temperature is a unique parameter to elucidate the function and regulation of tryptophan permeases by altering the membrane structure and lipid-protein interactions without introducing any components into the system (19, 20).
Although Tat1 and Tat2 are homologous, with a sequence identity of 39%, they are more different than might at first be surmised. In the presence of a normal or high concentration of tryptophan (20 to 40 μg ml−1 or 200 μg ml−1, respectively), Tat1 localizes predominantly to the plasma membrane, whereas Tat2 is more abundant in endosomes and multivesicular bodies (7, 11). Mutations rendering growth at 25 MPa in a tryptophan-auxotrophic strain have yielded HPG1 (high-pressure growth 1), a semidominant allele of RSP5 occurring as a mutation in the catalytic HECT (homologous to E6-AP C terminus) domain (11). Bul1 and Bul2 are PPxY motif proteins that bind to Rsp5 by interacting with the WW domain (21, 22). Deletion of BUL1 and BUL2 causes marked stabilization of Tat2, although it has no effect on Tat1 degradation (11). In a membrane flotation assay in which crude membrane extracts are treated with cold Triton X-100, Tat2 is associated with detergent-soluble membranes (7, 11), while Tat1 is associated with detergent-resistant membranes, i.e., so-called lipid rafts (11). Upon acquiring the HPG1 mutation, Tat2 becomes associated with lipid rafts, while Tat1 remains in rafts. When yeast cells are starved of nutrients, Tat2 degradation is initiated by covalent binding of ubiquitin to lysine residues in the N-terminal domain (6). Substitution of arginines for the five lysines prevents Tat2 from degrading. However, the ubiquitin binding site of Tat1 is still unknown.
It has been shown that members of a family of arrestin-related trafficking adaptors (ARTs; referred to as arrestins in this work) mediate the ubiquitination and endocytosis of amino acid permeases, such as the lysine permease Lyp1 (23), the arginine permease Can1 (23, 24), the methionine permease Mup1 (23), the glutamate permease Dip5 (25), and Tat2 (26), in response to specific amino acids or environmental stresses (for a review, see references 27 and 28). The arrestins possess the PPxY motif and mediate Rsp5-dependent ubiquitination of these amino acid permeases. In the case of Lyp1, Art1 is required for lysine-induced endocytosis, while Art2 is required for cycloheximide-induced endocytosis (23). Art3 is required for the endocytosis of Dip5 in response to excess glutamate (25). Systematic analysis of 9 ART genes revealed that their simultaneous deletion blocks the endocytosis of Tat2 in response to excess tryptophan or cycloheximide (26). While either ART1 or ART2 restores the tryptophan-induced endocytosis of Tat2, ART1 does not restore cycloheximide-induced endocytosis (26). Accordingly, arrestins are unlikely to be general regulators of the degradation of amino acid permeases but rather participate in their signal-dependent endocytosis.
In this paper, we show that lysine residues 29 and 31 in the N-terminal domain are required for Tat1 ubiquitination, which depends on Rsp5, and that one of the two lysines is sufficient for degradation in response to increased hydrostatic pressure. We also show that pressure-induced Tat1 ubiquitination is mediated by functionally redundant multiple PPxY motif proteins, including the arrestins and Bul1/Bul2. For simplicity, “ART” is used as a synonym for yeast arrestin genes, except for 9-arrestin (ART10), to note simultaneous deletions of ART1, ART2, ART3, ART4, ART5, ART6, ART7, ART8, and ART10 (26).
MATERIALS AND METHODS
Yeast strains and culture conditions.
Strains used in this study are listed in Table 1. Strains EN60 and EN67 were kind gifts from H. R. B. Pelham of the MRC Laboratory of Molecular Biology (26). Cells were grown at 25°C with shaking in synthetic complete (SC) medium, with slight modifications (40 μg/ml tryptophan, 90 μg/ml leucine, and valine was eliminated) (11). High-pressure cultivation of the cells was carried out as described previously (10, 11). Other materials were laboratory stocks (29, 30).
Table 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| YPH499 (wild type) | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 | 29 |
| FAM1 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 end3Δ::URA3 | This study |
| FAY18A | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 HPG1-1 (Rsp5P514T) | 11 |
| FAJ75 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 bul1Δ::HIS3 bul2Δ::LEU2 | 11 |
| FAS626 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 rsp5-ww1 (Rsp5W257G) | This study |
| FAS630 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 rsp5-ww2 (Rsp5W359G) | This study |
| FAS634 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 rsp5-ww3 (Rsp5W451G) | 31 |
| FAS510 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art1Δ::KanMX | This study |
| FAS511 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art2Δ::KanMX | This study |
| FAS516 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art3Δ::KanMX | This study |
| FAS519 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art4Δ::KanMX | This study |
| FAS523 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art5Δ::KanMX | This study |
| FAS526 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art6Δ::KanMX | This study |
| FAS532 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art7Δ::KanMX | This study |
| FAS535 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 art8Δ::KanMX | This study |
| BY4742 (wild type) | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | EUROSCARF |
| EN60 (9-arrestin mutant) | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 art1Δ art2Δ art3Δ art4Δ art5Δ art6Δ art7Δ art8Δ art10Δ | 26 |
| EN67 (9-arrestin bul1 bul2 mutant) | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 art1Δ art2Δ art3Δ art4Δ art5Δ art6Δ art7Δ art8Δ art10Δ bul1Δ bul2Δ | 26 |
Construction of plasmids.
PCR-based site-directed mutagenesis was performed to create one or multiple lysine-to-arginine (K → R) substitutions in the Tat1 N-terminal domain, using a QuikChange II site-directed mutagenesis kit (Agilent Technologies Inc., Santa Clara, CA) or a PrimerSTAR mutagenesis basal kit (TaKaRa Bio Inc., Otsu, Shiga, Japan). Relevant primers and p3HA-TAT1c (three-hemagglutinin [3HA]-tagged TAT1 driven by its own promoter in YCplac33 [URA3 CEN4]) as a template were used for PCR amplification (11). The appropriate base substitution was confirmed by sequencing the entire region of the TAT1 open reading frame (ORF), using relevant sequence primers.
Plasmids containing TAT1-13c-Myc-GFP (simply denoted TAT1-GFP from now on) and its mutant forms driven by the TAT1 promoter were constructed as follows. pSCU709 (GAL1 promoter-13c-Myc-GFP-CYC1 terminator URA3 2μ; a kind gift from T. Ushimaru of Shizuoka University) was used to generate a plasmid containing TAT1-GFP. pSCU709 was digested with SacI and BamHI to remove the GAL1 promoter from the plasmid. The TAT1 ORF and its own promoter region were amplified by PCR, using pTAT1c as the template and the following primers: GGGAACAAAAGCTGGAGCTCCATCCTTGTTTAAGTAAACCATTTATGT and TTAATTAACCCGGGGGATCCGCACCAGAAATTGGTCATCCTCTTAAAA (sequences complementary to the ends of the digested plasmid are underlined). The linearized plasmid and the PCR product were introduced into strain YPH499 to generate pSCU709-TAT1-GFP in cells, based on homologous recombination. The plasmid was isolated, and the entire TAT1 ORF was sequenced. The KpnI sites are located at the 313th to 318th positions of the TAT1 ORF and its 3′ downstream region in YCplac33. pSCU709-TAT1-GFP was digested with KpnI to obtain a fragment containing TAT1-GFP lacking the first 315 nucleotides of the ORF. It was fused to the longer fragment from either p3HA-TAT1c or p3HA-TAT1-K29R-K31Rc, digested with KpnI, to give p3HA-TAT1-GFPc (3HA-tagged TAT1-GFP driven by its own promoter; URA3 CEN4) or p3HA-TAT1-K29R-K31R-GFPc (3HA-tagged TAT1-K29R-K31R-GFP driven by its own promoter; URA3 CEN4), respectively.
Construction of mutant strains.
PCR-based gene disruption was performed on the wild-type strain YPH499 to create an end3Δ::URA3 strain, using primers 5′-ATGCCCAAGTTGGAACAATTTGAAATAAAAAAATACTGGCAAATCTTCTCGGGTTTGAAAAATTCAATTCATCATTTTTT-3′ and 5′-TCAATTGATTTCTGCTTGTAATGCTTGCAATTCGTGTCTCTTATTGTTCAAGTAATTCTCTGGATTAGTCTCATCCTTCA-3′ (sequences complementary to YEplac195 are underlined), with YEplac195 as the template. It was also performed to create art1Δ::KanMX, art2Δ::KanMX, art3Δ::KanMX, art4Δ::KanMX, art5Δ::KanMX, art6Δ::KanMX, art7Δ::KanMX, and art8Δ::KanMX strains, using the relevant primers (20-mer oligonucleotides upstream and downstream of the individual open reading frames) and with genomic DNAs from the corresponding deletion strains of the EUROSCARF yeast deletion library (Invitrogen, Carlsbad, CA) as templates.
The rsp5-ww1 (W257G) and rsp5-ww2 (W359G) point mutations were introduced into the YPH499 genome as described previously for the rsp5-ww3 (W451G) mutant (31), by replacing RSP5 in the genome with RSP5 containing the rsp5-ww1 or rsp5-ww2 mutation. Each GGG → CCC base substitution was created in plasmid pFA499c, which contained RSP5 driven by its own promoter (11), using a QuikChange II site-directed mutagenesis kit and primers for rsp5-ww1 (5′-CACAAGGACTACCACTGGGAAACGTCCAACGC-3′ and 5′-GCGTTGGACGTTTCCCAGTGGTAGTCCTTGTG-3′; the mutation sites are underlined) or rsp5-ww2 (5′-CTAGAACAACCACTGGGGTGGATCCAAGGAGAC-3′ and 5′-GTCTCCTTGGATCCACCCCAGTGGTTGTTCTAG-3′; the mutation sites are underlined). The resulting rsp5-ww1 or rsp5-ww2 DNA fragment was introduced into plasmid YIplac211 (URA3) to transform strain YPH499. Multiple Ura3+ transformants were incubated in yeast extract-peptone-dextrose (YPD) medium overnight, and the cells were spread on synthetic dextrose (SD) plates containing uracil and 0.1% 5-fluoroorotic acid. Multiple colonies were isolated, and the rsp5-ww1 and rsp5-ww2 mutations of the clones were confirmed by DNA sequencing of the genome locus by use of the relevant primers.
Western blotting.
Exponentially growing cells (1.0 × 107 to 1.5 × 107 cells ml−1) were collected by centrifugation, and whole-cell extracts were prepared for Western blotting as described previously (11). To avoid the nonspecific removal of ubiquitin from ubiquitinated Tat1, PR-619, a nonselective cell-permeating inhibitor of ubiquitin isopeptidases (LifeSensors, Malvern, PA), was added to the cells at 20 μM during the preparation of the whole-cell extracts. The extracts were subjected to centrifugation at 13,000 × g for 10 min to yield the P13 membranes. The supernatants were subjected to centrifugation at 100,000 × g for 30 min to yield the P100 membranes. Antibodies were used to detect HA (16B12; BabCO, Richmond, CA), Adh1 (Rockland antibodies and assays; Rockland, Gilbertsville, PA), Pma1 (EnCor Biotechnology Inc., Gainesville, FL), and Dpm1 (5C5A7; Life Technologies Corp., Carlsbad, CA). Adh1 and Pma1 were used as loading controls for the whole-cell extracts and the P13 membranes, respectively. Dpm1 was used as a loading control for both P13 and P100 membranes. The signals were detected in an ImageQuant LAS4000 minisystem (GE Healthcare Life Sciences, Piscataway, NJ).
Fluorescence microscopy.
Cells expressing green fluorescent protein (GFP)-tagged Tat1 proteins were imaged on a model IX70 fluorescence microscope (Olympus Co. Ltd., Tokyo, Japan).
RESULTS
Tat1 undergoes endocytic degradation in response to high pressure.
Our previous study showed that Tat1 undergoes Rsp5-dependent degradation in response to high hydrostatic pressure (11). We first compared Tat1 degradation between cells with high-pressure treatment and those with cycloheximide treatment. Whole-cell extracts for Western blotting were prepared from cells after exposure to a pressure of 25 MPa or to cycloheximide (100 μg/ml at 0.1 MPa) for 3 h. We found that Tat1 was highly stable, without a significant decrease in its level, after 3 h of cycloheximide treatment. In contrast, Tat1 was decreased considerably after high-pressure treatment for 3 h. A control protein, Adh1, was not degraded after the cycloheximide or high-pressure treatment. The results suggest that high pressure promotes the degradation of Tat1 (Fig. 1A). Some higher-molecular-mass bands were detected above the main Tat1 band upon overexposure for signal detection (Fig. 1A, arrowheads; see below).
Fig 1.

Endocytotic degradation of Tat1 in response to high pressure. (A) Wild-type cells were exposed to cycloheximide (CHX; 100 μg/ml) or a pressure of 25 MPa (HP) for 3 h. Whole-cell extracts were prepared for Western blotting using an anti-HA antibody to detect the 3HA-Tat1 proteins. Amounts of 3HA-Tat1 relative to that of Adh1 were calculated from the signals detected in an ImageQuant LAS4000 minisystem. Arrowheads denote the higher-molecular-mass bands of 3HA-Tat1. (B) Wild-type and end3Δ cells were exposed to a pressure of 25 MPa for 3 h. The P13 and P100 membranes were collected from the whole-cell extracts (W) by differential centrifugation. Adh1 and Pma1 were used as loading controls for the whole-cell extracts and the P13 membranes, respectively. Dpm1 was used as a loading control for both P13 and P100 membranes.
To eliminate the possibility that the decrease in Tat1 under these conditions could be attributed to any nonphysiological causes, e.g., mechanical pinching off due to perturbation of the membrane by high pressure, we examined whether Tat1 was maintained in the endocytosis-deficient end3Δ mutant at 25 MPa. We also extended the analysis by fractionating the membranes. As demonstrated previously, Tat1 was localized predominantly with the P13 membranes, in which the plasma membrane was enriched. Small amounts of Tat1 in the P100 membranes were likely associated with endosomes. The end3Δ mutation reduced the amount of Tat1 in the P100 membranes. When a pressure of 25 MPa was applied to the end3Δ cells, substantial amounts of Tat1 remained in the P13 membranes, while it was reduced from the P13 membranes in the wild-type cells (Fig. 1B). A nonubiquitinated control protein, Pma1, remained unchanged in the wild-type and end3Δ cells upon high-pressure treatment. These results suggest that high pressure enhances the ubiquitination and/or endocytosis of Tat1. It is unlikely that high pressure generally promotes ubiquitination/endocytosis of membrane proteins, because the rate of Tat2 degradation is much more rapid with cycloheximide treatment (∼30 min) (31) than with high-pressure treatment (>2 h) (11). It is known that excess amounts of tryptophan in medium induce the degradation of Tat2 (7). However, the Tat1 level remained almost unchanged when cells were cultured with various concentrations of tryptophan (4 to 200 μg/ml) (our unpublished observation). Therefore, we assumed that the cells responded specifically to changes in hydrostatic pressure and downregulated Tat1 in a manner dependent on Rsp5-mediated ubiquitination and endocytosis.
Lysine residues 29 and 31 are targets of Tat1 ubiquitination.
We next determined the target lysine(s) for ubiquitination of Tat1 in its N-terminal domain. According to HMMTOP (http://www.enzim.hu/hmmtop/), a program for transmembrane helix prediction (32, 33), the cytoplasmic N-terminal domain of Tat1 is composed of 99 amino acids, among which there are 11 lysine residues. We created K → R substitutions in various combinations in the N-terminal domain to determine which lysine residues are prerequisites for degradation of Tat1. In the process of substituting arginines for all of the 11 lysines, we first obtained 16 plasmids expressing the Tat1 K → R variants (p3HA-TAT1-AS1c to -AS16c in Table 2; we use the AS numbers, i.e., AS1 to AS16, as a shorthand throughout the text). Whole-cell extracts were prepared from the plasmid-bearing cells for Western blot analysis. The quantities of variant Tat1 proteins were almost the same as that of wild-type Tat1 (Fig. 2A). We found that at least two higher-molecular-mass bands, with additional masses of 8 and 16 kDa, appeared above the main wild-type Tat1 band (Fig. 2A, arrowheads). They probably correspond to mono- and diubiquitinated forms of Tat1, respectively. These two higher-molecular-mass bands were much weaker in cells harboring the variant TAT1 plasmids AS5, AS6, AS7, AS8, AS9, AS10, and AS11 (Fig. 2A). Accordingly, two lysines among those at positions 10, 29, 31, 39, 56, and 71 could be the ubiquitination targets. We next created single and double K → R variants to determine ubiquitin acceptors in the N-terminal domain. Simultaneous K29R and K31R substitution (K29R-K31R) and 11K→R substitution resulted in the complete loss of the higher-molecular-mass bands, but K39R and K56R substitution (K39R-K56R) did not (Fig. 2B). In our preliminary experiment, we confirmed the ubiquitin conjugation at these two lysines by performing immunoprecipitation of Tat1 and its mutant forms in cells coexpressing c-Myc-tagged ubiquitin, followed by the detection of covalently bound ubiquitin molecules with c-Myc antibody. c-Myc ubiquitin was detected in Tat1 but not in Tat1K29R-K31R and Tat111K→R (data not shown). These results suggest that K29 and K31 are target lysines for Rsp5-dependent ubiquitination. In the process of this analysis, we noticed that the appearance of the higher-molecular-mass bands for the single K → R substitution mutants, i.e., the K10R, K29R, K31R, K39R, and K56R mutants, varied with experiments. This implies that ubiquitination of Tat1 potentially occurs on lysines other than K29 and K31. However, we show below that K29 and K31 are crucial for pressure-induced degradation of Tat1.
Table 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| YCplac33 | URA3 CEN4 | 30 |
| YCplac111 | LEU2 CEN4 | 30 |
| p3HA-TAT1c | 3HA-TAT1 driven by TAT1 promoter in YCplac33 | 11 |
| p3HA-TAT1c-LEU | 3HA-TAT1 driven by TAT1 promoter in YCplac111 | This study |
| p3HA-TAT1-AS1c | 3HA-TAT1-K10,39R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS2c | 3HA-TAT1-K10,39,71R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS3c | 3HA-TAT1-K10,39,56,71R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS4c | 3HA-TAT1-K10,39,56,71,87R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS5c | 3HA-TAT1-K10,29,31,39,56,71R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS6c | 3HA-TAT1-K10,29,31,39,56,71,87R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS7c | 3HA-TAT1-K10,29,31,39,56,71,87,95R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS8c | 3HA-TAT1-K10,29,31,39,56,62,64,71,87R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS9c | 3HA-TAT1-K10,29,31,39,56,71,87,92,95R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS10c | 3HA-TAT1-K10,29,31,39,56,64,71,87,92,95R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS11c | 3HA-TAT1-K10,29,31,39,56,62,64,71,87,92,95R (11K→R) in p3HA-TAT1c | This study |
| p3HA-TAT1-AS12c | 3HA-TAT1-K10R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS13c | 3HA-TAT1-K29R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS14c | 3HA-TAT1-K31R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS15c | 3HA-TAT1-K39R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS16c | 3HA-TAT1-K56R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS17c | 3HA-TAT1-K39R-K56R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS18c | 3HA-TAT1-K29R-K31R in p3HA-TAT1c | This study |
| p3HA-TAT1-AS19c | 3HA-TAT1-K10R-K29R-K31R in p3HA-TAT1c | This study |
| pSCU709 | 13c-Myc-GFP (URA3 2μ) | Provided by T. Ushimaru |
| p3HA-TAT1-GFPc | 3HA-TAT1-13c-Myc-GFP driven by TAT1 promoter in YCplac33 | This study |
| p3HA-TAT1-K29R-K31R-GFPc | 3HA-TAT1-K29R-K31R-13c-Myc-GFP driven by TAT1 promoter in YCplac33 | This study |
Fig 2.

Determination of lysine residues required for ubiquitin-dependent Tat1 degradation. 3HA-Tat1 proteins with multiple K → R substitutions (A) or single or double K → R substitutions (B) were visualized by Western blotting using an anti-HA antibody. The plasmids corresponding to the AS numbers are listed in Table 2. Asterisks denote plasmids containing either the K29R or K31R substitution (*) or both the K29R and K31R substitutions (**). Arrowheads denote the higher-molecular-mass bands of 3HA-Tat1. (C) Wild-type cells expressing the 3HA-Tat1 proteins with single or multiple K → R substitutions were exposed to a pressure of 25 MPa for 3 h. Whole-cell extracts were prepared for Western blotting to detect the variant 3HA-Tat1 proteins. Adh1 was used as a loading control.
Ubiquitination of either lysine 29 or 31 is sufficient for pressure-induced degradation of Tat1.
We next examined whether K29 and K31 were required for pressure-induced Tat1 degradation. While substantial decreases were observed in wild-type Tat1, Tat1K10R, Tat1K29R, Tat1K31R, Tat1K39R, Tat1K56R, and Tat1K39R-K56R, the amounts of Tat1K29R-K31R, Tat1K10R-K29R-K31R and Tat111K→R remained almost unchanged after the cells were exposed to high pressure for 3 h (Fig. 2C). The results indicate that either K29 or K31 is required for pressure-induced Tat1 degradation.
To examine whether the K29R-K31R substitution altered the intracellular localization of Tat1, GFP-tagged Tat1 was expressed in the cells. Tat1-GFP localized predominantly to the plasma membrane (Fig. 3). There was no marked difference in plasma membrane localization between Tat1-GFP and Tat1K29R-K31R-GFP at 0.1 MPa (Fig. 3). When cells were exposed to a pressure of 25 MPa for 3 h, Tat1-GFP was completely lost from the plasma membrane and transported to the vacuole for degradation. In contrast, substantial amounts of Tat1K29R-K31R-GFP remained in the plasma membrane under the same conditions (Fig. 3). Interestingly, the vacuoles were also GFP positive in cells expressing Tat1K29R-K31R-GFP. This result suggests that high hydrostatic pressure enhances the vacuolar sorting of newly synthesized Tat1 from the Golgi apparatus to the vacuole in a manner independent of K29- and K31-linked ubiquitination, as well as sorting of Tat1 that preexists in the plasma membrane in a manner dependent on K29 and K31.
Fig 3.
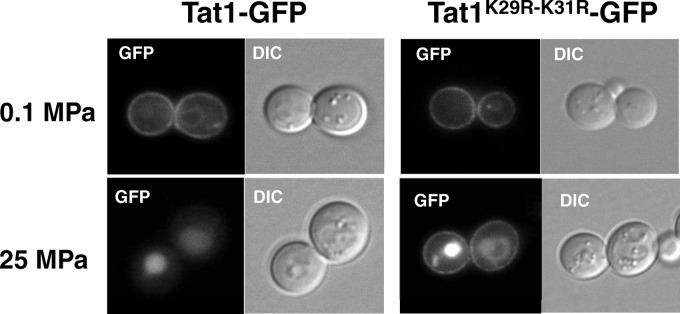
Intracellular localization of Tat1 and Tat1K29R-K31R under atmospheric pressure or high pressure. Wild-type cells were transformed with the centromeric plasmid p3HA-TAT1-GFPc or p3HA-TAT1-K29R-K31R-GFPc. The GFP-tagged Tat1 proteins were visualized by fluorescence microscopy after the cells were incubated at 0.1 MPa or 25 MPa for 3 h.
Pressure-induced endocytic degradation of Tat1 redundantly depends on PPxY motif proteins.
Pressure-induced Tat1 degradation is prevented by the HPG1 mutation (e.g., HPG1-1, encoding Rsp5P514T) but not by bul1Δ bul2Δ mutation (Fig. 4A) (11). The dispensability of Bul1 and Bul2 is in sharp contrast to the case for Tat2 degradation, where Bul1 is strictly required (7, 11). This implies that PPxY proteins other than Bul1 and Bul2 mediate Rsp5-dependent ubiquitination of Tat1. We first analyzed the effect of rsp5-ww mutations on pressure-induced Tat1 degradation. Base substitutions were introduced into the genome of strain YPH499 to create the rsp5-ww1 (W257G), rsp5-ww2 (W359G), and rsp5-ww3 (W451G) mutations. We found that Tat1 was degraded in the rsp5-ww1 and rsp5-ww2 mutants similarly to the case in the wild-type strain, although a somewhat larger amount remained in the rsp5-ww3 mutant (Fig. 4A). This suggests that other PPxY proteins mediate Tat1 ubiquitination through interaction with the Rsp5 WW3 domain, but probably partially redundantly with other Rsp5 WW domains, in response to high pressure. This prompted us to screen for yeast arrestins.
Fig 4.

Roles of Rsp5 WW domains and multiple PPxY proteins in Tat1 degradation in response to high pressure. (A) Cells of the wild-type, HPG1-1, rsp5-ww1, rsp5-ww2, rsp5-ww3, and bul1Δ bul2Δ strains were exposed to a pressure of 25 MPa for 3 h. (B) Cells of the wild type and of art single deletion mutants were exposed to a pressure of 25 MPa for 3 h. (C) (Left) Cells of the wild-type (BY4742), 9-arrestin (EN60), and 9-arrestin bul1Δ bul2Δ (EN67) strains were exposed to a pressure of 25 MPa for 3 h. Whole-cell extracts were prepared for Western blotting to detect the variant 3HA-Tat1 proteins. Adh1 was used as a loading control. (Right) Amounts of 3HA-Tat1 relative to that of Adh1 were calculated from the signals detected in an ImageQuant LAS4000 minisystem. Data are shown as means ± standard deviations for three independent experiments.
Next, we created single deletions of ART1 to ART8 in the YPH499 genome. ART9 deletion was not performed because Art9 does not have the canonical PPxY motif. ART10 deletion in the YPH499 genome was not successful, for unknown reasons. We found that none of the single deletions of ART genes prevented Tat1 degradation at 25 MPa, suggesting that the roles of the arrestins are redundant (Fig. 4B). Simultaneous deletions of ART1, ART2, ART3, ART4, ART5, ART6, ART7, ART8, and ART10 (9-arrestin) (26) slightly prevented Tat1 degradation, and combined 9-arrestin and bul1Δ bul2Δ deletions substantially but not fully stabilized Tat1 at 25 MPa (Fig. 4C). This in turn suggests that 9-arrestins and Bul1/Bul2, and possibly other proteins, share some essential functions in regulating Tat1 ubiquitination through the interaction with Rsp5. However, we cannot rule out the possibility that the simultaneous loss of the 11 PPxY proteins indirectly caused the stabilization of Tat1 by exerting effects on any downstream proteins involved in the endocytic degradation of Tat1. Analysis of the 9-arrestin bul1Δ bul2Δ mutant with BUL1 or BUL2 on a plasmid would clarify which alternative is responsible for pressure-induced Tat1 degradation when Art proteins are absent. Unlike the observation by Nikko and Pelham (26), the 9-arrestin bul1Δ bul2Δ mutant did not exhibit slow growth under normal culture conditions in our modified SC medium (see Materials and Methods).
DISCUSSION
Endocytic degradation of Tat1 in response to high pressure.
The present study demonstrated that high hydrostatic pressure triggers the degradation of Tat1 in a manner dependent on ubiquitination and subsequent endocytosis. Under normal growth conditions, the low-affinity tryptophan permease Tat1 is highly stable, with a half-life much longer than the doubling time of proliferating cells (≫3 h), as opposed to the rapid turnover of the high-affinity tryptophan permease Tat2 (∼30 min) (31). Tat1 is insensitive to cycloheximide or various concentrations of tryptophan in culture medium (our unpublished observation), while Tat2 undergoes rapid degradation in response to excess tryptophan and cycloheximide (7, 26). Therefore, the ubiquitination machinery may specifically perform quality control of Tat1 when cells are exposed to nonlethal levels of hydrostatic pressure. Pressure-induced Tat1 degradation requires one of the N-terminal lysine residues 29 and 31, and simultaneous K29R-K31R substitution results in marked stabilization of Tat1. According to the protein mobility in SDS-PAGE gels, the two higher-molecular-mass bands appear to be attributed to mono- and diubiquitinated forms of Tat1, at positions 29 and/or 31.
Regulation of Tat1 degradation mediated by multiple PPxY motif proteins.
We showed that the WW3 domain of Rsp5 is important but probably redundant with other WW domains for pressure-induced degradation of Tat1. In fact, many proteins can bind to two of three WW domains of Rsp5 (34). In contrast to the requirement for specific arrestins for the degradation of Can1, Lyp1, Itr1, Dip5, or Jen1, 9-arrestins and Bul1/Bul2 redundantly regulate Tat1 degradation. Furthermore, the fact that Tat1 was not fully stabilized in the 9-arrestin bul1Δ bul2Δ mutant at high pressure suggests the presence of additional PPxY motif proteins that play a role in Tat1 degradation in response to high pressure. This is similar to the case for the uracil permease Fur4 with respect to the redundancy of 9-arrestins and Bul1 for vacuolar degradation after the addition of uracil or cycloheximide (26). It was reported previously that Art1 and Art2 effectively complement the 9-arrestin bul1Δ strain for Fur4 degradation, although Art8 shows weaker activity (26). The degradation of Tat2 is also blocked in the rsp5-ww3 mutant and the bul1Δ strain. Art1 and Art2 restore Tat2 degradation in the 9-arrestin mutant in the presence of excess tryptophan, whereas Art8 partially restores it with cycloheximide treatment (26). Nikko and Pelham mentioned a scheme of transporter regulation in which specificity is provided by arrestin interactions that promote endocytosis, and this is followed by further, relatively nonspecific ubiquitination in endosomes (26). It is still unclear how the functionally redundant PPxY motif proteins recognize Tat1 specifically after cells are exposed to high pressure. One possible explanation is that high pressure may perturb the structure of the raft domain, leading to the release of Tat1 from the rafts to nonrafts and thus enhancing the endocytosis of Tat1 for degradation. In the case of Can1, the addition of arginine releases Can1 from the raft domain, followed by endocytic degradation (35). In this regard, the structural instability of Tat1 within the unfavorable nonraft environment could provide an opportunity to expose the hydrophobic TMD peptides of Tat1 to the cytoplasm, and thereby a broad range of arrestins might become capable of interacting with the hypothetically denatured forms of Tat1 through hydrophobic interactions. In this sense, the ubiquitin system would differentiate dysfunctional membrane proteins from intact ones through the function of arrestins. It would be worthwhile to elucidate whether high pressure disrupts the raft structure mechanically or blocks delivery of raft components, such as ergosterol and sphingolipids, to the plasma membrane, and whether Tat1 can be released from the rafts.
A recent key finding on the nutrient-sensing pathway is that TORC1 controls nutrient uptake by targeting the ubiquitin-mediated endocytosis of specific amino acid permeases. Endocytic downregulation of Can1 is promoted by cycloheximide, and this effect is abrogated by treatment of the cells with rapamycin, a TORC1 inhibitor (24). Cycloheximide also triggers the endocytosis of Mup1, and rapamycin abrogates the effect. However, rapamycin does not affect the methionine-induced endocytosis of Mup1. Thus, TORC1 signaling promotes the endocytosis of multiple cell surface proteins as a general mechanism to limit protein abundance at the plasma membrane. In the presence of nutrients, TORC1 kinase inactivates the downstream kinase Npr1 by phosphorylation. In this situation, the Rsp5-Art1 complex is translocated to the plasma membrane to ubiquitinate Can1 (24). Starvation or rapamycin treatment inhibits TORC1, which in turn activates Npr1 by dephosphorylation. The activated Npr1 protein is translocated to the plasma membrane and phosphorylates Art1. Rsp5-Art1 is then dissociated from the plasma membrane, and hence, Can1 ubiquitination does not occur (24).
TORC1 also controls nitrogen-induced Gap endocytosis in a manner dependent on Npr1 and the 14-3-3 proteins (36). Under nitrogen-poor conditions, Bul1 and Bul2 are phosphorylated in an Npr1-dependent manner and bind to the 14-3-3 proteins, which inhibit the capacity of Bul proteins to induce Gap1 ubiquitination. Upon the addition of ammonium ions, Bul proteins are dephosphorylated, concomitantly dissociated from the 14-3-3 proteins, and then ubiquitinated by Rsp5 (36). The PPxY and arrestin motifs of Bul proteins are required for ammonium-induced ubiquitination of Gap1. The role of Bul proteins in Gap1 ubiquitination is in sharp contrast to their dispensability in pressure-induced Tat1 degradation. It is known that the 14-3-3 proteins and Rod1/Art4 coordinately regulate ubiquitination of the lactate transporter Jen1 (37). Whether the pressure-induced Tat1 degradation also involves phosphorylation/dephosphorylation of any arrestins is unknown.
It is evident that there are considerable differences in the ways that plasma membrane transporters are regulated through the functions of PPxY motif proteins and Rsp5 in response to a broad range of environmental cues. A specific distinction that sets hydrostatic pressure apart from other cues, such as nutrients, salt, urea, or organic solvents, is that pressure merely changes the equilibrium among preexisting components, depending on the magnitude of volume changes (38, 39). Thus, hydrostatic pressure can bring minor elements out of the background noise and into the foreground for direct observation and study. In this sense, ubiquitination and/or endocytosis of Tat1 occurs at an extremely low rate under normal growth conditions at atmospheric pressure, but it becomes possible to analyze these processes at high hydrostatic pressures, probably because the rate-limiting step in the degradation of Tat1 is accompanied by a large negative volume change.
ACKNOWLEDGMENTS
We thank Hugh R. B. Pelham for providing yeast strains, Takashi Ushimaru for GFP plasmids, Yoichi Noda for valuable suggestions, and Hiroki Deguchi and Mai Nagata for technical assistance.
This work was supported by a grant from the Japan Society for the Promotion of Science (grant 22658031 to F. Abe).
Footnotes
Published ahead of print 10 May 2013
REFERENCES
- 1. Nelissen B, De Wachter R, Goffeau A. 1997. Classification of all putative permeases and other membrane plurispanners of the major facilitator superfamily encoded by the complete genome of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 21: 113– 134 [DOI] [PubMed] [Google Scholar]
- 2. Sophianopoulou V, Diallinas G. 1995. Amino acid transporters of lower eukaryotes: regulation, structure and topogenesis. FEMS Microbiol. Rev. 16: 53– 75 [DOI] [PubMed] [Google Scholar]
- 3. Lauwers E, Erpapazoglou Z, Haguenauer-Tsapis R, Andre B. 2010. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20: 196– 204 [DOI] [PubMed] [Google Scholar]
- 4. Heitman J, Koller A, Kunz J, Henriquez R, Schmidt A, Movva NR, Hall MN. 1993. The immunosuppressant FK506 inhibits amino acid import in Saccharomyces cerevisiae. Mol. Cell. Biol. 13: 5010– 5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmidt A, Beck T, Koller A, Kunz J, Hall MN. 1998. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17: 6924– 6931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck T, Schmidt A, Hall MN. 1999. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 146: 1227– 1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Umebayashi K, Nakano A. 2003. Ergosterol is required for targeting of tryptophan permease to the yeast plasma membrane. J. Cell Biol. 161: 1117– 1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daicho K, Makino N, Hiraki T, Ueno M, Uritani M, Abe F, Ushimaru T. 2009. Sorting defects of the tryptophan permease Tat2 in an erg2 yeast mutant. FEMS Microbiol. Lett. 298: 218– 227 [DOI] [PubMed] [Google Scholar]
- 9. Okamoto M, Yoko-o T, Umemura M, Nakayama K, Jigami Y. 2006. Glycosylphosphatidylinositol-anchored proteins are required for the transport of detergent-resistant microdomain-associated membrane proteins Tat2p and Fur4p. J. Biol. Chem. 281: 4013– 4023 [DOI] [PubMed] [Google Scholar]
- 10. Abe F, Horikoshi K. 2000. Tryptophan permease gene TAT2 confers high-pressure growth in Saccharomyces cerevisiae. Mol. Cell. Biol. 20: 8093– 8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abe F, Iida H. 2003. Pressure-induced differential regulation of the two tryptophan permeases Tat1 and Tat2 by ubiquitin ligase Rsp5 and its binding proteins, Bul1 and Bul2. Mol. Cell. Biol. 23: 7566– 7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagayama A, Kato C, Abe F. 2004. The N- and C-terminal mutations in tryptophan permease Tat2 confer cell growth in Saccharomyces cerevisiae under high-pressure and low-temperature conditions. Extremophiles 8: 143– 149 [DOI] [PubMed] [Google Scholar]
- 13. Winter R. 2002. Synchrotron X-ray and neutron small-angle scattering of lyotropic lipid mesophases, model biomembranes and proteins in solution at high pressure. Biochim. Biophys. Acta 1595: 160– 184 [DOI] [PubMed] [Google Scholar]
- 14. Winter R, Dzwolak W. 2005. Exploring the temperature-pressure configurational landscape of biomolecules: from lipid membranes to proteins. Philos. Trans. A Math. Phys. Eng. Sci. 363: 537– 562 [DOI] [PubMed] [Google Scholar]
- 15. Matsuki H, Goto M, Tada K, Tamai N. 2013. Thermotropic and barotropic phase behavior of phosphatidylcholine bilayers. Int. J. Mol. Sci. 14: 2282– 2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu M, Brusilow WS, Needleman R. 2004. Activity of the yeast Tat2p tryptophan permease is sensitive to the anti-tumor agent 4-phenylbutyrate. Curr. Genet. 46: 256– 268 [DOI] [PubMed] [Google Scholar]
- 17. Palmer LK, Wolfe D, Keeley JL, Keil RL. 2002. Volatile anesthetics affect nutrient availability in yeast. Genetics 161: 563– 574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skrzypek MS, Nagiec MM, Lester RL, Dickson RC. 1998. Inhibition of amino acid transport by sphingoid long chain bases in Saccharomyces cerevisiae. J. Biol. Chem. 273: 2829– 2834 [DOI] [PubMed] [Google Scholar]
- 19. Abe F. 2004. Piezophysiology of yeast: occurrence and significance. Cell. Mol. Biol. (Noisy-le-Grand) 50: 437– 445 [PubMed] [Google Scholar]
- 20. Abe F. 2007. Exploration of the effects of high hydrostatic pressure on microbial growth, physiology and survival: perspectives from piezophysiology. Biosci. Biotechnol. Biochem. 71: 2347– 2357 [DOI] [PubMed] [Google Scholar]
- 21. Yashiroda H, Oguchi T, Yasuda Y, Toh-e A, Kikuchi Y. 1996. Bul1, a new protein that binds to the Rsp5 ubiquitin ligase in Saccharomyces cerevisiae. Mol. Cell. Biol. 16: 3255– 3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yashiroda H, Kaida D, Toh-e A, Kikuchi Y. 1998. The PY-motif of Bul1 protein is essential for growth of Saccharomyces cerevisiae under various stress conditions. Gene 225: 39– 46 [DOI] [PubMed] [Google Scholar]
- 23. Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. 2008. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135: 714– 725 [DOI] [PubMed] [Google Scholar]
- 24. MacGurn JA, Hsu PC, Smolka MB, Emr SD. 2011. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147: 1104– 1117 [DOI] [PubMed] [Google Scholar]
- 25. Hatakeyama R, Kamiya M, Takahara T, Maeda T. 2010. Endocytosis of the aspartic acid/glutamic acid transporter Dip5 is triggered by substrate-dependent recruitment of the Rsp5 ubiquitin ligase via the arrestin-like protein Aly2. Mol. Cell. Biol. 30: 5598– 5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nikko E, Pelham HR. 2009. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 10: 1856– 1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leon S, Haguenauer-Tsapis R. 2009. Ubiquitin ligase adaptors: regulators of ubiquitylation and endocytosis of plasma membrane proteins. Exp. Cell Res. 315: 1574– 1583 [DOI] [PubMed] [Google Scholar]
- 28. Becuwe M, Herrador A, Haguenauer-Tsapis R, Vincent O, Leon S. 2012. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012: 242764. 10.1155/2012/242764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19– 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527– 534 [DOI] [PubMed] [Google Scholar]
- 31. Hiraki T, Abe F. 2010. Overexpression of Sna3 stabilizes tryptophan permease Tat2, potentially competing for the WW domain of Rsp5 ubiquitin ligase with its binding protein Bul1. FEBS Lett. 584: 55– 60 [DOI] [PubMed] [Google Scholar]
- 32. Tusnady GE, Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283: 489– 506 [DOI] [PubMed] [Google Scholar]
- 33. Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849– 850 [DOI] [PubMed] [Google Scholar]
- 34. Hesselberth JR, Miller JP, Golob A, Stajich JE, Michaud GA, Fields S. 2006. Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteins. Genome Biol. 7: R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grossmann G, Malinsky J, Stahlschmidt W, Loibl M, Weig-Meckl I, Frommer WB, Opekarova M, Tanner W. 2008. Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 183: 1075– 1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merhi A, Andre B. 2012. Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul arrestin-like adaptors. Mol. Cell. Biol. 32: 4510– 4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Becuwe M, Vieira N, Lara D, Gomes-Rezende J, Soares-Cunha C, Casal M, Haguenauer-Tsapis R, Vincent O, Paiva S, Leon S. 2012. A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 196: 247– 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balny C, Masson P, Heremans K. 2002. High pressure effects on biological macromolecules: from structural changes to alteration of cellular processes. Biochim. Biophys. Acta 1595: 3– 10 [DOI] [PubMed] [Google Scholar]
- 39. Lassalle MW, Akasaka K. 2007. The use of high-pressure nuclear magnetic resonance to study protein folding. Methods Mol. Biol. 350: 21– 38 [DOI] [PubMed] [Google Scholar]