

Abstract
The mitochondrial DNA of trypanosomatid protozoa consists of a complex, intercatenated network of tens of maxicircles and thousands of minicircles. This structure, called kinetoplast DNA (kDNA), requires numerous proteins and multiprotein complexes for replication, segregation, and transcription. In this study, we used a proteomic approach to identify proteins that are associated with the kDNA network. We identified a novel protein encoded by Tb927.2.6100 that was present in a fraction enriched for kDNA and colocalized the protein with kDNA by fluorescence microscopy. RNA interference (RNAi) knockdown of its expression resulted in a growth defect and changes in the proportion of kinetoplasts and nuclei in the cell population. RNAi also resulted in shrinkage and loss of the kinetoplasts, loss of maxicircle and minicircle components of kDNA at similar rates, and (perhaps secondarily) loss of edited and pre-edited mRNA. These results indicate that the Tb927.2.6100 protein is essential for the maintenance of kDNA.
INTRODUCTION
The mitochondrial DNA of kinetoplastid protozoa consists of tens of maxicircles and thousands of minicircles, all of which are intercatenated into a single network that resides within the cell's large single mitochondrion across from the flagellar basal body (1–3). Early cytologists named the highly visible region in cells stained for DNA the kinetoplast by virtue of its association with the base of the motile flagellum. Subsequently, this mitochondrial DNA came to be called kinetoplast DNA (kDNA) (4).
The kDNA maxicircles encode two mitochondrial rRNAs and 17 proteins, of which most RNAs undergo posttranscriptional sequence remodeling called RNA editing (5–7). The minicircles encode small guide RNAs (gRNAs) that specify the edited sequences. Maxicircle and minicircle sizes vary among different kinetoplastids, but within each species their maxicircle sequences are homogeneous, while their minicircle sequences are heterogeneous (8, 9). The specific mRNAs that undergo editing and the extent of their editing vary among the different kinetoplastids, which is a reflection of the minicircle sequence diversity (10).
Transcription of both maxicircles and minicircles is polycistronic, and the primary transcripts are processed by cleavage (11, 12). In addition, at least some transcripts can undergo editing prior to the cleavage step (13). Both maxicircles and minicircles appear to be transcribed by a single mitochondrial RNA polymerase, which has the characteristics of a viral polymerase (14, 15). It is unclear if there are other RNA polymerases involved in mitochondrial transcription.
Although there are two classes of DNA molecules, there are six different mitochondrial DNA polymerases, the specific functions of which have yet to be completely elucidated (16–18). Replication of the kDNA network has been studied in detail, especially in the Englund laboratory (19). These studies revealed somewhat different mechanisms in different kinetoplastids. In Trypanosoma brucei, multiple proteins must functionally associate with kDNA to ensure its proper positioning and replication, and perhaps repair and segregation during cell division as well as its transcription. The processing of the primary transcripts may also be coordinated with transcription, as implied by the editing of preprocessed transcripts. Hence, there is likely a set of proteins that functionally interact with kDNA, directly or indirectly, to accomplish the replication, repair, segregation, and perhaps processing as well as suborganellar localization.
In this study, proteins in kDNA enriched fractions were identified by mass spectrometry (MS), and localization of candidate proteins was determined by immunofluorescence. One novel protein was demonstrated to colocalize with kDNA in vivo, and its functional role was assessed using RNA interference (RNAi). This protein, Tb927.2.6100, is found to be associated with kDNA and is essential for its maintenance.
MATERIALS AND METHODS
Trypanosome cell growth.
Procyclic-form (PF) T. brucei 164 clone IsTaR 1.7a was grown to densities of 1× 107 to 2 × 107 cells/ml in vitro at 27°C in SDM-79 medium containing hemin (7.5 mg/ml) and 10% (vol/vol) fetal bovine serum (FBS). PF T. brucei strain 29.13 (20), which contains integrated genes for T7 polymerase and the tetracycline repressor, was grown in the presence of G418 (15 μg/ml) and hygromycin (25 μg/ml). The cells were harvested by centrifugation at 6,000 × g for 10 min at 4°C.
Plasmid constructs, transfections, and induction.
To create constructs for the inducible expression of c-myc epitope-tagged proteins in T. brucei, the open reading frames (ORFs) of interest were PCR amplified from the genomic DNA of T. brucei strain Lister 427. A detailed list of all tagged proteins and primers used to amplify the selected ORFs is provided in the supplemental material. The PCR products were cloned into pGEM-T Easy vector (Promega), digested with BamHI or BglII and HindIII enzymes, and ligated into the pLEW79-MHTAP vector (21, 22). The plasmids were linearized with NotI enzyme and transfected into the PF T. brucei 29.13 cell line; phleomycin-resistant cell lines were selected and checked for tetracycline (Tet)-regulated expression. The transgenic PF cell lines expressing a tandem affinity purification (TAP)-tagged protein were supplemented with 2.5 μg/ml of phleomycin.
To create the construct for RNAi of the Tb927.2.6100 transcript, a fragment of 439 bp was PCR amplified from genomic DNA of T. brucei strain Lister 427 using the sense primer 5′-ATACTCGAGGAGGCTCTAGCAGCAGAGGA-3′ and antisense primer 5′-ATAAAGCTTGAGTAACTGGGGCTGCTACG-3′. The resulting PCR product was cloned into pZJM plasmid (23, 24) via XhoI and HindIII restriction sites. RNAi cell lines were generated by transfection with 10 μg of NotI-linearized pZJM construct as described previously (25). Three independent cell lines were selected, and growth curves were generated in the absence or presence of 1 μg/ml of tetracycline, which induces expression of double-stranded RNA (dsRNA). The induced and noninduced cultures were maintained at between 2 × 106 and 2 × 107 cells/ml to maintain log-phase growth, and cell density was monitored daily using a particle counter (Beckman).
kDNA isolation.
Mitochondrial (mt) vesicles were isolated by hypotonic lysis and enriched by density gradient flotation in 20 to 35% linear Percoll gradients as described previously (26). The enriched vesicles were solubilized by adding n-dodecyl-β-d-maltoside (DDM) corresponding to a DDM/protein ratio of 2 (g/g) and incubated on ice for 30 min. Following centrifugation at 12,000 × g for 20 min at 4°C, the pellet was washed thrice with buffer containing 0.5 M sucrose, 20 mM Tris-HCl (pH 7.6), 0.1 mM EDTA, 0.8 mM spermidine, 1 μg/ml of pepstatin, 2 μg/ml of leupeptin, 1 mM Pefabloc, and 0.5% Nonidet P-40 (NP-40). The last centrifugation was at 15,000 × g for 20 min at 4°C, and the pellet was used as the kDNA fraction.
Tandem affinity purification.
Expression of TAP-tagged Tb927.2.6100 was induced with tetracycline (100 ng/ml of culture) for 48 h, and 500 ml of cells was harvested by centrifugation at a density of ∼9 × 106 cells/ml. Tagged complex was purified from cell lysate prepared by two-step lysis with Nonidet P-40 detergent as described previously (27, 28). Briefly, the cells were lysed with 0.25% Nonidet P-40 and cleared by low-speed centrifugation, and the supernatant was further lysed with 1.5% Nonidet P-40 and cleared by high-speed centrifugation. The tagged complexes were isolated by sequential IgG affinity and calmodulin affinity columns (29).
Protein identification by LC-MS-MS.
Proteins were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS-MS) as described previously (30). Briefly, the proteins in the enriched kDNA fraction were separated on 10% SDS-PAGE gels and visualized by SYPRO ruby staining. The entire gel lane was divided into 9 pieces, and the proteins were digested in-gel with sequencing-grade modified trypsin; the resulting peptides were extracted with 50% acetonitrile–5% formic acid and dried in a SpeedVac. For proteins isolated with TAP-tagged Tb927.2.6100, 200 μl of calmodulin eluate was digested in solution, and peptides were extracted as described above. The peptides were fractionated by nanoflow C18 liquid chromatography and analyzed using a LTQ linear ion trap mass spectrometer. The collision-induced dissociation spectra were compared with the Tb927, version 4.0, T. brucei protein database downloaded from GeneDB using TurboSEQUEST software, and protein matches were determined using PeptideProphet and ProteinProphet software (31, 32).
Sequence analysis of identified proteins.
The probable functions of the proteins were assigned based on GeneDB annotation, and for proteins with unknown function, possible motifs and/or domains were searched in InterPro (http://www.ebi.ac.uk/Tools/InterProScan/), Pfam (http://pfam.sanger.ac.uk/), and NCBI CDD (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) databases. Alignment of Tb927.2.6100 orthologs was performed using MUSCLE (version 3.8.31) (33). Mitochondrial targeting signal of Tb927.2.6100 was predicted using MitoProt II (version 1.101) (34).
Immunofluorescence microscopy.
Subcellular localizations of the expressed tagged proteins within the cell were determined by immunofluorescence analysis (IFA) using anti-c-myc antibody (Invitrogen) as described previously (27). Colocalization analysis was performed using monoclonal antibody (MAb) 78 (anti-mt heat shock protein 70) (27) coupled with Texas Red-X-conjugated secondary antibody. For 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) staining, ∼5 × 105 noninduced and RNAi-induced cells were pelleted by centrifugation, washed, fixed with 4% formaldehyde, and treated with 50 μl of 1-μg/ml DAPI to visualize DNA. Phase-contrast images of the cells and their fluorescence were captured with a Nikon fluorescence microscope equipped with a camera and appropriate filters.
RNA isolation and quantitative real-time PCR.
Total RNA was harvested from the cell lines using the TRIzol reagent according to the manufacturer's instructions. Ten micrograms of RNA was treated with DNase I by using a DNA-free kit (Ambion) and used as the templates for reverse transcription-PCR (RT-PCR) analysis. Real-time RT-PCR to measure the relative abundance of mRNAs was performed as previously described (35). The primer pairs used to amplify 18S rRNA and β-tubulin cDNAs, which were used as reference genes, A6 (pre-edited and edited), and cytochrome oxidase I (COI), 9S/ND8, and RPS12/ND5 have been described previously (35, 36). The sequences of primers for maxicircles are as follows: forward, GGTTCATAGAGGAGAATGGTTCAAG, and reverse, CCTCCACTTACACAATCTTTTGCTT. Those for minicircles are as follows: forward, GGCGTGCAAAAATACACATACAC, and reverse, GTACCTCGGACCTCAAAAATGC. The average of three cycle threshold (CT) values for each target was used in the calculations. The relative standard deviation of measured CT values in quantitative PCR (qPCR) assays was between 0.03% and 1.77%. Analysis was carried out using the Pfaffl method, with PCR efficiencies calculated by linear regression with LinRegPCR software (37, 38). Data were normalized to 18S rRNA and β-tubulin, and relative changes in mRNA abundance after RNAi induction were expressed as fold changes relative to noninduced control cells.
For the quantification of the minicircles and maxicircles, total DNA was isolated from cells collected during RNAi, and dyskinetoplastic EATRO164 blood form (DK164) cells (which lack kDNA) were used as a control (39, 40). The cells were harvested and washed once with phosphate-buffered saline (PBS) and then lysed in PBS containing 0.5% SDS and 0.2 mg/ml of proteinase K for 4 h at 56°C. The DNA was treated with 0.1 mg/ml of RNase at 37°C for 30 min and then extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated with ethanol. The pellets were resuspended in water and stored at −20°C until they were analyzed. For another control the mtDNA was isolated from wild-type (WT) PF 29.13 cells.
The quantitative PCR for minicircles and maxicircles was performed essentially as described above for cDNAs, with the differences being the use of EvaGreen (Bio-Rad) as the PCR mix and the experimental determination of the optimal concentration for each DNA preparation; 7.5 pg of DNA was used for samples in Fig. 3B and C. As reference genes, both the β-tubulin gene and the 18S rRNA gene were tested. The two reference genes gave very similar results, and thus, only the β-tubulin gene was used as a reference gene. The kDNA content was quantified relative to this gene using the PCR efficiency and the Pfaffl method as described above.
Fig 3.

Quantitative PCR of kDNA. (A) Threshold cycles using different amounts of DNA from DK164 cells (no mitochondrial DNA) and kDNA from PF 29.13 cells. Primers that target the maxicircle (max1, A6 pre) and minicircle (min2) DNA showed no significant amplification without mitochondrial DNA (DK164), whereas good amplification was obtained using kDNA as the template. Error bars represent standard deviations of measured CT replicates. (B) RNAi of Tb927.2.6100 results in decreases of both maxicircle and minicircle DNAs, with the maxicircle decreasing to a larger extent (∼20%) than the minicircle (∼30%). (C) Maxicircle and minicircle abundance is not altered by KREX2 repression by RNAi, a control that indicates that effects of Tb927.2.6100 RNAi are specific. β-Tubulin was used as the internal reference for relative comparisons.
RESULTS
Protein identification.
The kDNA was isolated from Percoll gradient-enriched mitochondria, and the proteins in this fraction were identified by mass spectrometry. A total of 2,573 unique peptides were identified by LC-MS-MS analyses. Five hundred twenty-three proteins were matched to these by two or more peptide hits, while 338 proteins were identified by a single peptide hit, of which 175 (52%) had a protein identification probability of ≥0.99, 109 (32%) had 0.98, and the rest had between 0.91 and 0.97; 510 (98%) of the proteins identified with two or more peptide matches had a protein identification probability of ≥0.99, 7 had 0.98, and the rest had between 0.93 and 0.97. The identified peptide sequences and associated probability values are presented in Table S1 in the supplemental material. Of the 861 proteins identified in this study, 44 had not been previously detected in our prior proteomic analyses of the T. brucei mitochondrion (30, 41, 42) (see Table S2 in the supplemental material).
Not all of the 861 different proteins identified in the kDNA fraction are mitochondrial, a finding which in part reflects the fact that the large, structurally complex mitochondrion is disrupted and reseals as vesicles during isolation, contributing to cross contamination. Based on available GeneDB annotation, keyword searches, and literature searches (although some genes may be misannotated), 424 proteins (∼49%) are mitochondrial and 105 proteins (∼12%) are likely contaminants of the enriched kDNA fraction, since they are assignable to non-mt locations, including glycosomes, endoplasmic reticulum, the nucleus, Golgi, and lysosomes (see Table S3 in the supplemental material). The remaining 332 proteins (∼39%) have no annotated localization; some are likely to be mitochondrial proteins whose location has not been demonstrated, while others are nonmitochondrial contaminants, as has been observed in previous analyses (30). As expected, several proteins known to be associated with kDNA were identified in this sample, including DNA polymerases (Tb927.5.2780 and Tb927.5.2790) (17, 43), DNA topoisomerase II (Tb09.160.4090) (44), kDNA-associated proteins (Tb10.6k15.1460 and Tb927.8.7260), and components of the HslVU protease complex (Tb11.01.2000, Tb11.01.4050, and Tb927.5.1520) (45).
Subcellular location of proteins.
In order to determine whether some of the identified proteins colocalize with the kDNA in T. brucei PF cells, several protein candidates were selected based on the following criteria: (i) identification of at least two tryptic peptides, (ii) absence of a known function (“hypothetical conserved” proteins in the T. brucei genome database), (iii) pI of >9.5, (iv) domains or motifs, and (v) mt localization sequence predicted by the available software MitoProt (http://ihg.gsf.de/ihg/mitoprot.html) and/or SignalP (http://www.cbs.dtu.dk/services/SignalP/). These criteria led to a set of 64 hypothetical proteins, of which 34 had already been assigned to an mt protein complex and/or mt membranes (30, 36, 41, 46). Of the remaining 30 proteins, 18 with a pI of >9.5 were TAP tagged, and their subcellular localization was assessed by immunofluorescence analysis (IFA) after induction of expression in PF T. brucei cells (Fig. 1; see also Fig. S1 in the supplemental material). However, it has also been shown that the protein p166, which has an acidic pI of 5.32, localizes to the kDNA (47). Therefore, in a second round of selection, nine protein candidates were selected with a pI of <9.5 (see Fig. S1).
Fig 1.
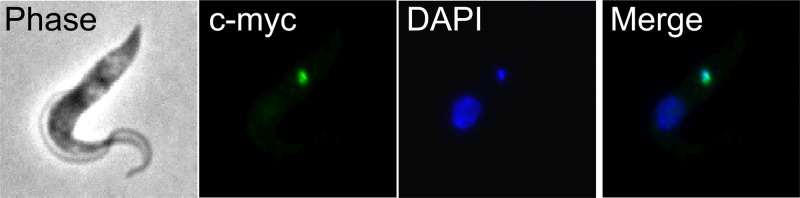
Tb927.2.6100 associates with the kDNA. The tagged protein was visualized by fluorescence microscopy using polyclonal anti-c myc antiserum coupled with fluorescein isothiocyanate (FITC)-conjugated secondary antibody. Panels show phase-contrast light microscopy of PF T. brucei cells, anti-c-myc antibody coupled with FITC-conjugated secondary antibody showing localization of the target protein, DAPI staining of nucleus and kDNA, and merge.
Immunofluorescence assay using anti-myc MAb (anti-tag) showed that among the 24 tagged and successfully expressed proteins (Table 1), Tb927.2.6100 is the only one that exclusively colocalized with the kDNA (Fig. 1). As for the remaining proteins, IFA showed that 15 of them were evenly distributed throughout the reticulated mitochondrion but did not intensely label the kinetoplast area, 3 localized to the nucleus, and 5 others also appeared to be nonmitochondrial (see Fig. S1 in the supplemental material). However, a definitive localization could not be assigned for the five last proteins due to indistinct speckled fluorescence; perhaps the expressed proteins were folded in a way that prevented the tag from being recognized by the MAb.
Table 1.
Subcellular localization of tagged proteins
| Protein namea | Theoretical pI/molecular mass (kDa) | Motif or domain | Previous mt assignment confidenceb | New assignment (IFA based) |
|---|---|---|---|---|
| Tb927.2.6100TAP233 | 10.77/53.28 | Low | mt-kDNA | |
| Tb927.10.2970TAP245 | 7.74/13.79 | Low | mt | |
| Tb927.5.4360TAP246 | 6.95/36.43 | S-Adenosylmethionine-dependent methyltransferase | Low | mt |
| Tb927.4.3080TAP247 | 8.58/18.44 | DUF167 | Low | mt |
| Tb09.211.0530TAP234 | 10.07/30.41 | Low | mt | |
| Tb927.3.1810TAP235 | 9.75/36.1 | Low | mt | |
| Tb927.10.1860 TAP273 | 9.61/39.1 | Low | Nuclear | |
| Tb11.01.3970TAP231 | 10.24/23.49 | Moderate | mt | |
| Tb927.5.740 TAP269 | 10.11/23.91 | Moderate | mt | |
| Tb11.01.8500 TAP270 | 9.91/25.87 | Moderate | mt | |
| Tb11.02.1860TAP268 | 5.52/51.88 | Putative transcriptional regulator | Moderate | mt |
| Tb11.01.2740TAP267 | 5.99/71.77 | AlkB, alkylated DNA repair protein | Moderate | mt |
| Tb09.160.2380TAP241 | 9.89/9.23 | Uncharacterized protein family (UPF0041) | Moderate | mt |
| Tb927.10.1660 TAP271 | 9.73/33.2 | Moderate | mt | |
| Tb927.5.1130 TAP272 | 9.59/34.3 | Moderate | Non-mt | |
| Tb927.3.1720TAP248 | 5.67/11.17 | Moderate | Non-mt | |
| Tb927.7.630 TAP274 | 10.73/28.19 | None | mt | |
| Tb927.4.1370TAP237 | 9.66/17.64 | None | mt | |
| Tb11.02.5230 TAP275 | 10.25/23.28 | None | Nuclear | |
| Tb09.211.1150 TAP277 | 10.38/20.69 | None | Nuclear | |
| Tb09.211.2620TAP238 | 10.31/22.75 | None | Non-mt | |
| Tb927.8.2490TAP250 | 8.52/35.23 | SET domain | Not detected | Non-mt |
GeneDB accession number (http://www.genedb.org) with superscript indicating TAP-tagged identifier.
From reference 30.
Based on our IFA results, seven of the proteins that were previously assigned to the mitochondrion with moderate confidence and six with low confidence (30) are clearly localized to the mitochondrion, whereas one protein (Tb927.10.1860) earlier assigned to the mitochondrion with low confidence localized to the nucleus (see Fig. S1 in the supplemental material).
Tandem affinity purification of tagged Tb927.2.6100.
To determine what proteins might associate with Tb927.2.6100, tandem affinity purification was performed from cells expressing Tb927.2.6100TAP, and purified proteins were trypsin digested and analyzed by mass spectrometry (see Table S4 in the supplemental material). Among the proteins identified by mass spectrometry, nine proteins previously identified as associating with the mitochondrial ribosome were found: Tb10.70.7960, Tb11.01.1840, Tb11.02.2250, Tb11.02.3180, Tb927.1.2990, Tb927.3.5610, Tb927.6.3930, Tb927.7.3430, and Tb927.7.3510 (42). Among these, both Tb11.02.3180 and Tb10.70.7960 have also previously been identified as being a part of the MRB1 complex, which is involved in mitochondrial RNA processing and is thought to mediate exchange of gRNAs during RNA editing (36, 48, 49).
Effect of Tb927.2.6100 RNAi on cell growth.
Tb927.2.6100 is encoded by a single-copy gene, and its predicted product has 487 amino acid residues and a pI of 10.77. Bioinformatic analyses of Tb927.2.6100 predict an N-terminal mitochondrial targeting signal but no known motif or domain. Homologous genes are present in related Trypanosoma species but not in Leishmania species. A sequence alignment with orthologs from T. brucei gambiense, T. cruzi, T. congolense, and T. vivax showed conservation of a putative mitochondrial import sequence and regions near the N and C termini but divergence between these regions (see Fig. S2 in the supplemental material). To evaluate whether Tb927.2.6100 is essential in T. brucei, its expression was knocked down in PF cells by RNAi. Growth inhibition became apparent after day 5 and lasted up to 10 days compared with that in noninduced cell lines (Fig. 2A). Induction of the RNAi expression resulted in ∼80% knockdown of Tb927.2.6100 mRNA abundance after 4 days compared to that in noninduced cells (Fig. 2B). Growth resumed after ∼10 days, indicating that the cells became increasingly resistant to RNAi knockdown, as previously reported for the T. brucei system (50). Tb927.2.6100 protein levels could not be directly assessed in the RNAi cells due to the lack of an antibody specific for Tb927.2.6100. The growth defects resulting from the repression of Tb927.2.6100 imply that it is an essential protein in PF T. brucei.
Fig 2.

Effects of Tb927.2.6100 RNAi. (A) Cell growth curves of a representative RNAi cell line of Tb927.2.6100 grown in the presence (open circles) or absence (filled squares) of Tet. The cumulative cell numbers were calculated by multiplying cell densities by the dilution factor. (B) qPCR analysis of total RNA isolated from cells, in which expression of Tb927.2.6100 was repressed for 4 days by RNAi. The relative change in Tb927.2.6100 mRNA abundance was determined by using either β-tubulin mRNA (right bar) or 18S rRNA (left bar) as an internal control and is presented in log scale. (C) Effect of RNAi on kinetoplast size. DAPI staining shows kinetoplasts from cells without RNAi or after 4 days of RNAi. n, nucleus; k, kinetoplast. (D) Quantitation of the different cell cycle stages during RNAi of Tb927.2.6100 showing that the proportion of 200 randomly chosen cells having one kinetoplast/one nucleus (1K1N) or two kinetoplasts/one nucleus (2K1N) is reduced during RNAi of Tb927.2.6100, whereas the proportion of these cells having no kinetoplast/one nucleus (0K/1N) is increased dramatically, as is the proportion having a reduced-size kinetoplast/one nucleus (1ksmall1N).
Effect of Tb927.2.6100 RNAi on mitochondrial DNA.
RNAi of proteins closely associated with the kDNA has been shown to cause kDNA loss (16, 47, 51–53), so maintenance of kDNA after Tb927.2.6100 RNAi was examined. DAPI staining revealed that Tb927.2.6100 RNAi also caused shrinking and disappearance of the kDNA network. In Fig. 2C, the top image shows DAPI-stained cells (noninduced) with a normal nucleus and kinetoplast. In contrast, the bottom image (4 days after RNAi induction) shows cells that have lost the majority of kinetoplast staining. In the RNAi cells that do retain kDNA, DAPI staining showed an obvious decrease in the size of these kinetoplasts, to the point of being barely detectable (Fig. 2C; see also Fig. S3 in the supplemental material).
The effects on cell division and kinetoplast segregation were also monitored by DAPI staining (Fig. 2D). The cells in G1 and S phases would have one nucleus and one kinetoplast (1N1K). Since mt division precedes nuclear division, cells in G2/M phase have one nucleus and two kinetoplasts (1N2K), and cells undergoing cytokinesis have two nuclei and two kinetoplasts (2N2K). We evaluated the proportion of cells with nuclear and mitochondrial DNA in 200 cells per sample that were randomly chosen. In the control Tb927.2.6100 cell line (noninduced cells), ∼90% of the cells were found to have a normal-size single nucleus and a single kinetoplast (1N1K); the rest of the cells (∼10%) were either 1N2K or 2N2K. However, in RNAi-induced cells, a reduction in the proportion of cells in G1/S phase (1N1K) and a proportional increase in cells lacking the kinetoplast (1N0K) were observed. The percentage of 1N0K cells increased over time: 55% at day 2, 60% at day 4, and 75% at day 6 (Fig. 2D). Moreover, the percentage of 1N1K cells harboring a smaller kinetoplast remained constant over time, ∼20%. These results suggest that Tb927.2.6100 could be involved in controlling replication and/or segregation of the kDNA network.
To assess if the decrease of total kDNA was due to a specific reduction of minicircle and/or the maxicircle component of the mitochondrial DNA, quantitative PCR (qPCR) was performed using primers binding to either the maxicircle or the minicircle. DNA from DK164, which does not contain mitochondrial DNA, was used as a control, testing 7.5 pg of DNA per reaction; the mitochondrial DNA was tested in 3 dilutions of 75, 7.5, and 0.75 pg per reaction. The analysis of the threshold cycle shows that there was no amplification when using the DK164 DNA as a template (CT > 30) for the maxicircle primer pairs. This shows that there is no nonspecific binding of these primers to nuclear DNA, making them suitable to quantify the mitochondrial DNA (Fig. 3A). The qPCR of serial dilutions of mitochondrial DNA produced CT values reflecting robust amplification (14 to 19 over the concentration range), demonstrating the specific amplification of the mitochondrial targets. The linearity of the CT value when plotted over the DNA concentration (R2 > 0.9 for all of the primer pairs) shows that these primers can be used to quantify the mitochondrial DNA. To further ensure that these primers amplify only their intended target, qPCR amplicons were ligated into the pGEM-T Easy vector and sequenced, which confirmed the specificity of the primers.
Quantitative PCR was performed using total DNA purified from cells during RNAi of Tb927.2.6100 and from noninduced cells as controls at the same time points. The DNA was isolated every day of the time course of 6 days, as 6 days corresponds well with the development of the phenotype (Fig. 2A); after that point, growth stalls due to the depletion of Tb927.2.6100. The qPCR shows that in the first 4 days the levels of maxicircle DNA changed only slightly (to a maximum of an increase of 1.5-fold), but on days 5 and 6 of RNAi the maxicircle DNA decreased dramatically, to eventually 20% compared to the noninduced cells (Fig. 3B). Using two different primer pairs targeting different parts of the maxicircle gave very similar results, further confirming that the measured reduction is correct. The qPCR of minicircle DNA showed a similar pattern: for the first 4 days the changes were small, even though the variation was higher than the maxicircle, with a 2.4-fold increase on day 4 (Fig. 3B). But on days 5 and 6, the minicircle DNA also decreased to about 30% of the levels in noninduced cells. This shows that the decrease of kDNA during RNAi of Tb927.2.6100 was due to a concurrent loss of maxicircles and minicircles.
As an additional control, mitochondrial DNA was isolated on days 2, 4, and 6 of growth from Trypanosoma brucei PF 29.13 cells where the editing exonuclease KREX2 has been repressed by RNAi (54). Because KREX2 is not essential (55), these cells are a suitable control in which a mitochondrial protein is also lacking, but a decrease in the maxicircle and minicircle DNA is not expected. The qPCR showed almost no change in the abundance of the mitochondrial DNA, other than a small increase, of less than 2-fold, for the minicircle after 6 days (Fig. 3C). This shows that the observed decrease of mitochondrial DNA during RNAi of Tb927.2.6100 was not due to a general reaction of the cell to the depletion of a mitochondrial protein but a specific response. Southern blot analysis also showed that both minicircles and maxicircles diminished during the course of the RNAi knockdown in Tb927.2.6100 (results not shown).
We found that the decrease in mitochondrial DNA was accompanied by a reduction in the abundance of both pre-edited and edited kDNA transcripts. This was assessed using qPCR of RNA isolated from cells in which RNAi for Tb927.2.6100 was induced. The transcripts examined were pre-edited and edited A6, COI that does not get edited, and regions of 9S/ND8 and RPS12/ND5 that span their site that is cleaved during the processing of the polycistronic primary transcript. All of these transcripts showed a slight reduction after 2 days and a dramatic reduction, of about 90% (apart from edited A6, which had a reduction of 75%), compared to the levels in the noninduced cells at day 6 (Fig. 4). These results were expected and serve as an additional control to show that depletion of Tb927.2.6100 leads to a massive decrease in mitochondrial DNA and subsequently in mitochondrial transcripts.
Fig 4.

Quantitative PCR of mtRNA. RNAi of Tb927.2.6100 results in the strong decrease of the examined mt transcripts over the course of 6 days. RNAs examined included both pre-edited and edited ATPase subunit 6 (A6 pre and A6 ed, respectively), never-edited COI, and the polycistronic junction regions of primary transcripts corresponding to RPS12 and ND5 or 9S and ND8.
Together, these results confirmed the loss of the kinetoplast upon inhibition of Tb927.2.6100 and showed that maxicircle and minicircle DNA disappear at comparable rates during the same time period as a result of RNAi knockdown. Thus, it is not possible to determine if the protein is involved in the replication and/or maintenance of one over the other; the results rather point to Tb927.2.6100 having a more general function in the regulation of the kinetoplast copy numbers.
DISCUSSION
A multitude of proteins are required for the replication, segregation, and transcription of the complex trypanosome mitochondrial DNA, also known as kDNA, that consists of intercatenated network of tens of maxicircles and thousands of minicircles (19). We identified a novel protein, Tb927.2.6100, by MS analysis of proteins that are associated with enriched kDNA. Although sequence analysis did not identify any known motifs, alignment with orthologs from other trypanosomes revealed regions of sequence conservation near the N and C termini that may be involved in protein function. We showed that this protein localizes exclusively to the kDNA area of the mitochondrion. Furthermore, we demonstrated by RNAi that its presence is essential for kDNA maintenance and perhaps replication. Hence, the association with kDNA is functional as well as physical. The results from these RNAi studies indicate that the function of the protein is not exclusive to either the maxicircle or minicircle components, since knockdown resulted in the loss of both molecules at similar rates.
Loss of kDNA following RNAi was observed by both the loss of DAPI staining and loss of kDNA sequence by qPCR. However, the loss of kDNA by DAPI staining and the loss of maxicircle transcripts by qPCR occurred by day 2, while the loss of kDNA by qPCR occurred at day 5. This difference probably reflects the fact that maxicircle DNA abundance was measured by qPCR, which does not require that the maxicircles remain intact but only that the template DNA is present. However, kDNA transcripts are rapidly turned over upon disruption of transcription, and DAPI staining is affected by kDNA structure and supercoiling.
Immunofluorescence analysis of 13 additional TAP-tagged proteins that were also enriched in the kDNA fraction showed that they localize to the mitochondria but are not restricted to the kDNA region. This study also identified three proteins that localize to the nucleus. It is not surprising that there are many proteins associated with kDNA, which contains tens of identical ∼20-kb maxicircles and hundreds of very heterogeneous ∼1-kb minicircles all intercatenated in a single complex DNA network that is located within the mitochondrion across from the flagellar basal body. These proteins include DNA polymerases, DNA helicases, topoisomerases, and DNA ligases that function in the replication and reassembly into a network of the kDNA, many of which were observed in the kDNA preparation isolated in this work. They also include p166, which links kDNA to the flagellar basal body and functions analogously to a centriole in segregating the kDNA at cell division. In addition, at least one protease functions in regulating the level of kDNA. Furthermore, several proteins function in the polygenic transcription of kDNA as well as the subsequent RNA processing, which includes transcript cleavage, addition of 3′ oligo(U) tails to gRNAs and rRNA, and the extensive editing of the mRNAs. The apparent functions of these proteins are consistent with their being localized with kDNA.
Our previous proteomics studies identified a total of 3,043 T. brucei proteins and assigned 979 of those to the mitochondrion with progressively diminishing stringent criteria for 459 high-confidence, 218 moderate-confidence, and 302 low-confidence proteins (30, 41, 42). This study identified 44 additional proteins that had not previously been detected by MS. Analysis of TAP-tagged proteins identified 2 additional mitochondrial proteins, enhanced the confidence for a mitochondrial location for 12 proteins, and demonstrated nuclear localization for 3 others (Table 1). These results therefore update the list to 978 proteins assigned to the mitoproteome, with 474 high-confidence, 209 moderate-confidence, and 295 low-confidence mitochondrial proteins (see Table S5 in the supplemental material). These probably do not represent all mitochondrial proteins, and some may have dual localization (i.e., isoleucyl-tRNA synthetase) (56).
Analysis of proteins associated with TAP-tagged Tb927.2.6100 suggests that it may interact directly or indirectly with mitochondrial ribosomes. The mass spectrometry coverage of tagged Tb927.2.6100 itself consisted of only two identified peptides, which may be a consequence of the location and abundance of its lysine and arginine residues that may have affected susceptibility to protease and the generation of optimally sized peptides for mass spectrometry. Coverage of individual proteins of the mitochondrial ribosome was also limited, with only two ribosomal proteins observed by two or more peptides. However, the observation of nine distinct ribosomal proteins implies an association between Tb927.2.6100 and mitochondrial ribosomes. Two of these proteins have also been found in some isolations of the MRB1 complex, which is involved in some aspect of the processing of kDNA transcripts. Immunofluorescence studies have revealed that ribosomal proteins and proteins that function in RNA editing are located proximal to kDNA (57; L. Simpson, personal communication). Since Tb927.2.6100 is associated with kDNA and is essential for its maintenance, a simple explanation is that the proteins that function in the maintenance of kDNA, its transcription and RNA processing and translation, are generally colocalized in the region within which kDNA resides, i.e., the kinetoplast.
The specific role of Tb927.2.6100 is uncertain. It does not appear to function directly in replication or to be specific to either maxicircle or minicircle DNA, since these molecules are lost at similar rates. Its restriction to the submitochondrial location where kDNA and the proteins involved in kDNA replication, transcription, and RNA processing and translation are located and the apparent loss of kDNA structure and its transcripts may suggest a role associated with the structural organization of the kDNA or the integration of these macromolecular processes. The absence of the protein in Leishmania but not T. cruzi implies that its role may have been assumed by another protein, perhaps the histone-like protein or the hypothetical protein that is encoded at the syntenic position in Leishmania. Alternatively, this absence may reflect functional differences between trypanosomes and Leishmania that could be associated with the expression or segregation of kDNA (19).
The many proteins that are associated with kDNA reflect the complexity of this unusual mitochondrial genome, and the differences between kinetoplastid species reflect its evolutionary diversification. Differences in kDNA replication and segregation that have evolved between these species (9, 19, 58) may also reflect how the genetic information is differentially utilized and regulated in diverse kinetoplastid species, with substantial differences in their host target tissues, their vectors, and their metabolic (and other) processes.
Supplementary Material
ACKNOWLEDGMENTS
This work was primarily supported by National Institutes of Health grant AI065935 and received some support from grant AI014102.
We thank Yuko Ogata for mass spectrometry analysis.
Footnotes
Published ahead of print 6 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00352-12.
REFERENCES
- 1.Riou G, Delain E. 1969. Electron microscopy of the circular kinetoplastic DNA from Trypanosoma cruzi: occurrence of catenated forms. Proc. Natl. Acad. Sci. U. S. A. 62:210–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleisen CM, Weislogel PO, Fonk K, Borst P. 1976. The structure of kinetoplast DNA. 2. Characterization of a novel component of high complexity present in the kinetoplast DNA network of Crithidia luciliae. Eur. J. Biochem. 64:153–160 [DOI] [PubMed] [Google Scholar]
- 3.Shapiro TA, Englund PT. 1995. The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol. 49:117–143 [DOI] [PubMed] [Google Scholar]
- 4.Vickerman K. 1976. The diversity of the kinetoplastid flagellates, p 1–34 Lumsden WHR, Evans DA. (ed), Biology of kinetoplastida. Academic Press, London, United Kingdom [Google Scholar]
- 5.Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. 2005. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 30:97–105 [DOI] [PubMed] [Google Scholar]
- 6.Aphasizhev R, Aphasizheva I. 2011. Mitochondrial RNA processing in trypanosomes. Res. Microbiol. 162:655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hajduk S, Ochsenreiter T. 2010. RNA editing in kinetoplastids. RNA Biol. 7:229–236 [DOI] [PubMed] [Google Scholar]
- 8.Kleisen CM, Borst P, Weijers PJ. 1976. The structure of kinetoplast DNA. 1. The mini-circles of Crithidia luciliae are heterogeneous in base sequence. Eur. J. Biochem. 64:141–151 [DOI] [PubMed] [Google Scholar]
- 9.Lukes J, Guilbride DL, Votypka J, Zikova A, Benne R, Englund PT. 2002. Kinetoplast DNA network: evolution of an improbable structure. Eukaryot. Cell 1:495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson L, Maslov DA. 1999. Evolution of the U-insertion/deletion RNA editing in mitochondria of kinetoplastid protozoa. Ann. N. Y. Acad. Sci. 870:190–205 [DOI] [PubMed] [Google Scholar]
- 11.Grams J, McManus MT, Hajduk SL. 2000. Processing of polycistronic guide RNAs is associated with RNA editing complexes in Trypanosoma brucei. EMBO J. 19:5525–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madina BR, Kuppan G, Vashisht AA, Liang YH, Downey KM, Wohlschlegel JA, Ji X, Sze SH, Sacchettini JC, Read LK, Cruz-Reyes J. 2011. Guide RNA biogenesis involves a novel RNase III family endoribonuclease in Trypanosoma brucei. RNA 17:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koslowsky DJ, Yahampath G. 1997. Mitochondrial mRNA 3′ cleavage/polyadenylation and RNA editing in Trypanosoma brucei are independent events. Mol. Biochem. Parasitol. 90:81–94 [DOI] [PubMed] [Google Scholar]
- 14.Hashimi H, Cicova Z, Novotna L, Wen YZ, Lukes J. 2009. Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA 15:588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grams J, Morris JC, Drew ME, Wang Z, Englund PT, Hajduk SL. 2002. A trypanosome mitochondrial RNA polymerase is required for transcription and replication. J. Biol. Chem. 277:16952–16959 [DOI] [PubMed] [Google Scholar]
- 16.Klingbeil MM, Motyka SA, Englund PT. 2002. Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol. Cell 10:175–186 [DOI] [PubMed] [Google Scholar]
- 17.Saxowsky TT, Choudhary G, Klingbeil MM, Englund PT. 2003. Trypanosoma brucei has two distinct mitochondrial DNA polymerase beta enzymes. J. Biol. Chem. 278:49095–49101 [DOI] [PubMed] [Google Scholar]
- 18.Bruhn DF, Sammartino MP, Klingbeil MM. 2011. Three mitochondrial DNA polymerases are essential for kinetoplast DNA replication and survival of bloodstream form Trypanosoma brucei. Eukaryot. Cell 10:734–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen RE, Englund PT. 2012. Network news: the replication of kinetoplast DNA. Annu. Rev. Microbiol. 66:473–491 [DOI] [PubMed] [Google Scholar]
- 20.Wirtz E, Leal S, Ochatt C, Cross GAM. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99:89–101 [DOI] [PubMed] [Google Scholar]
- 21.Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M. 2007. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol. Biochem. Parasitol. 151:28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panigrahi AK, Schnaufer A, Ernst NL, Wang B, Carmean N, Salavati R, Stuart K. 2003. Identification of novel components of Trypanosoma brucei editosomes. RNA 9:484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Morris JC, Drew ME, Englund PT. 2000. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275:40174–40179 [DOI] [PubMed] [Google Scholar]
- 24.Morris JC, Wang Z, Drew ME, Paul KS, Englund PT. 2001. Inhibition of bloodstream form Trypanosoma brucei gene expression by RNA interference using the pZJM dual T7 vector. Mol. Biochem. Parasitol. 117:111–113 [DOI] [PubMed] [Google Scholar]
- 25.Schnaufer A, Panigrahi AK, Panicucci B, Igo RP, Jr, Salavati R, Stuart K. 2001. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science 291:2159–2162 [DOI] [PubMed] [Google Scholar]
- 26.Panigrahi AK, Schnaufer A, Stuart KD. 2007. Isolation and compositional analysis of trypanosomatid editosomes. Methods Enzymol. 424:3–24 [DOI] [PubMed] [Google Scholar]
- 27.Panigrahi AK, Zikova A, Dalley RA, Acestor N, Ogata Y, Anupama A, Myler PJ, Stuart KD. 2008. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol. Cell. Proteomics 7:534–545 [DOI] [PubMed] [Google Scholar]
- 28.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature 440:631–636 [DOI] [PubMed] [Google Scholar]
- 29.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032 [DOI] [PubMed] [Google Scholar]
- 30.Panigrahi AK, Ogata Y, Zikova A, Anupama A, Dalley RA, Acestor N, Myler PJ, Stuart KD. 2009. A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 9:434–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392 [DOI] [PubMed] [Google Scholar]
- 32.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75:4646–4658 [DOI] [PubMed] [Google Scholar]
- 33.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claros MG, Vincens P. 1996. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241:779–786 [DOI] [PubMed] [Google Scholar]
- 35.Carnes J, Trotter JR, Ernst NL, Steinberg AG, Stuart K. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 102:16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acestor N, Panigrahi AK, Carnes J, Zikova A, Stuart KD. 2009. The MRB1 complex functions in kinetoplastid RNA processing. RNA 15:277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339:62–66 [DOI] [PubMed] [Google Scholar]
- 39.Stuart KD. 1971. Evidence for the retention of kinetoplast DNA in an acriflavin-induced dyskinetoplastic strain of Trypanosoma brucei which replicates the altered central element of the kinetoplast. J. Cell. Biochem. 49:189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart K, Gelvin SR. 1980. Kinetoplast DNA of normal and mutant Trypanosoma brucei. Am. J. Trop. Med. Hyg. 29:1075–1081 [DOI] [PubMed] [Google Scholar]
- 41.Acestor N, Panigrahi AK, Ogata Y, Anupama A, Stuart KD. 2008. Protein composition of Trypanosoma brucei mitochondrial membranes. Proteomics 2009:5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziková A, Panigrahi AK, Dalley RA, Acestor N, Anupama A, Ogata Y, Myler PJ, Stuart K. 2008. Trypanosoma brucei mitochondrial ribosomes: affinity purification and component identification by mass spectrometry. Mol. Cell. Proteomics 7:1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruhn DF, Mozeleski B, Falkin L, Klingbeil MM. 2010. Mitochondrial DNA polymerase POLIB is essential for minicircle DNA replication in African trypanosomes. Mol. Microbiol. 75:1414–1425 [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Drew ME, Morris JC, Englund PT. 2002. Asymmetrical division of the kinetoplast DNA network of the trypanosome. EMBO J. 21:4998–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Lindsay ME, Motyka SA, Englund PT, Wang CC. 2008. Identification of a bacterial-like HslVU protease in the mitochondria of Trypanosoma brucei and its role in mitochondrial DNA replication. PLoS Pathog. 4:e1000048. 10.1371/journal.ppat.1000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acestor N, Zikova A, Dalley RA, Anupama A, Panigrahi AK, Stuart KD. 2011. Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell Proteomics 10:M110.006908. 10.1074/mcp.M110.006908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Z, Lindsay ME, Roy CA, Robinson DR, Englund PT. 2008. p166, a link between the trypanosome mitochondrial DNA and flagellum, mediates genome segregation. EMBO J. 27:143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R. 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol. Cell 32:198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hashimi H, Zimmer SL, Ammerman ML, Read LK, Lukes J. 2013. Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol. 29:91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pelletier M, Read LK. 2003. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA 9:457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Molina H, Kalume D, Pandey A, Griffith JD, Englund PT. 2006. Role of p38 in replication of Trypanosoma brucei kinetoplast DNA. Mol. Cell. Biol. 26:5382–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu B, Yildirir G, Wang J, Tolun G, Griffith JD, Englund PT. 2010. TbPIF1, a Trypanosoma brucei mitochondrial DNA helicase, is essential for kinetoplast minicircle replication. J. Biol. Chem. 285:7056–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, Motyka SA, Englund PT. 2005. Effects of RNA interference of Trypanosoma brucei structure-specific endonuclease-I on kinetoplast DNA replication. J. Biol. Chem. 280:35513–35520 [DOI] [PubMed] [Google Scholar]
- 54.Ernst NL, Panicucci B, Carnes J, Stuart K. 2009. Differential functions of two editosome ExoUases in Trypanosoma brucei. RNA 15:947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carnes J, Ernst NL, Wickham C, Panicucci B, Stuart KD. 2012. KREX2 is not essential for procyclic or bloodstream form Trypanosoma brucei. PLoS One 7:e33405. 10.1371/journal.pone.0033405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rettig J, Wang Y, Schneider A, Ochsenreiter T. 2012. Dual targeting of isoleucyl-tRNA synthetase in Trypanosoma brucei is mediated through alternative trans-splicing. Nucleic Acids Res. 40:1299–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li F, Herrera J, Zhou S, Maslov DA, Simpson L. 2011. Trypanosome REH1 is an RNA helicase involved with the 3′-5′ polarity of multiple gRNA-guided uridine insertion/deletion RNA editing. Proc. Natl. Acad. Sci. U. S. A. 108:3542–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu B, Liu Y, Motyka SA, Agbo EC, Englund PT. 2005. Fellowship of the rings: the replication of kinetoplast DNA. Trends Parasitol. 21:363–369 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.