

Abstract
Tau is a microtubule-associated protein thought to help modulate the stability of neuronal microtubules. In tauopathies, including Alzheimer’s disease and several frontotemporal dementias, tau is abnormally modified and misfolded resulting in its disassociation from microtubules and the generation of pathological lesions characteristic for each disease. A recent surge in the population of people with neurodegenerative tauopathies has highlighted the immense need for disease-modifying therapies for these conditions, and new attention has focused on tau as a potential target for intervention. In the current work we summarize evidence linking tau to disease pathogenesis and review recent therapeutic approaches aimed at ameliorating tau dysfunction. The primary therapeutic tactics considered include kinase inhibitors and phosphatase activators, immunotherapies, small molecule inhibitors of protein aggregation, and microtubule-stabilizing agents. Although the evidence for tau-based treatments is encouraging, additional work is undoubtedly needed to optimize each treatment strategy for the successful development of safe and effective therapeutics.
Keywords: Tau, Alzheimer’s disease, Tauopathy, Kinase inhibitors, Phosphatase activators, Immunotherapy, Aggregation inhibitors, Microtubule stabilization
I. Introduction
Tau is a microtubule-associated protein (MAP) believed to help regulate microtubule dynamics and promote their stability. In the disease state, however, tau undergoes post-translational modifications and conformational shifts that result in its dissociation from microtubules (Lee et al., 2001). This dissociation releases tau, allowing it to aggregate into both filamentous and non-filamentous inclusions, such as neurofibrillary tangles (NFTs), neuropil threads, and neuritic plaques. These inclusions constitute the characteristic tau pathology that can be detected in Alzheimer’s disease (AD) and in several other neurodegenerative tauopathies (Binder et al., 2005). Additionally, certain forms of these intracellular tau aggregates are thought to be toxic to various cellular processes, leading to the hypotheses that preventing aggregate formation, promoting tau aggregate clearance and/or stabilizing microtubules would constitute positive steps in the treatment of these diseases. An examination of these suppositions is what constitutes the impetus for this review article.
II. Properties and Functional Involvement of Tau in the CNS
Tau was discovered in the mid-seventies and described as a protein factor that stoichiometrically drives the assembly of brain tubulin into microtubules in vitro (Weingarten et al., 1975). Tau was further determined to have somewhat unique physical properties, as it is heat stable and soluble in perchloric acid, attributes that it shares with histones (Lindwall and Cole, 1984). Subsequent studies indicated that tau exerts its effect on microtubules by stabilizing the formed polymer (Drubin and Kirschner, 1986), reducing microtubule dynamics in vitro (Drechsel et al., 1992, Panda et al., 1995, Trinczek et al., 1995). Early antibody work led to the discovery that tau is largely found in the nervous system, present predominantly in axons (Binder et al., 1985) but also residing in somatodendritic and glial compartments (Papasozomenos and Binder, 1987). Moreover, tau is also present in the testes where it appears as a part of the Manchette, the microtubule organelle that helps shape the nucleus during spermiogenesis (Ashman et al., 1992).
Tau is the product of a single RNA transcript from a gene located on chromosome 17 (Neve et al., 1986). Alternative splicing of this transcript produces predominantly 6 isoforms in the central nervous system containing either 3 or 4 repeat domains involved in microtubule binding (MTBRs) and zero, one or two amino terminal inserts (Goedert et al., 1989) (Fig. 1).
Figure 1. Schematic representation of tau.
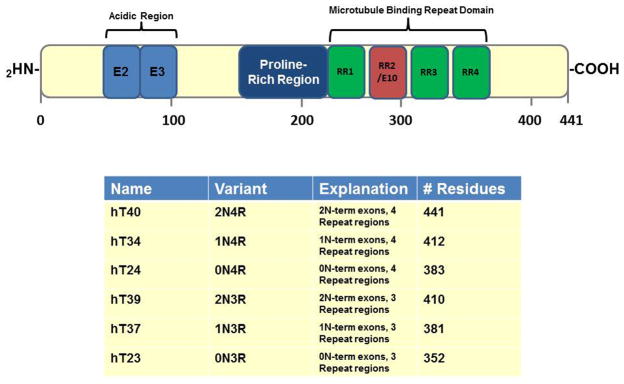
Diagram indicating the organization of the longest human tau isoform hT40 (2N4R). The primary transcript of tau contains 16 exons with 3 exons that can be alternatively spliced (exon 2, exon 3 and exon 10). This leads to 6 major human tau isoforms in the Central Nervous System (CNS), 2N4R, 1N4R, 0N4R, 2N3R, 1N3R and 0N3R. The repeat regions reside towards the C-terminal end and this is the area of the protein involved in microtubule binding. Within the center of the protein there is a proline-rich domain that is highly phosphorylated in the AD brain. The table outlines each of the six isoforms, listing number of N-terminal inserts, repeat regions and number of residues present.
In addition to its cytoplasmic involvements, tau was also discovered to be a nuclear protein, initially seen associated with the nucleolus (Loomis et al., 1990, Wang et al., 1993). Although for years no real nuclear function was assigned to tau, recently it was shown to bind to the minor grove in DNA and protect DNA from heat stress-induced damage (Sultan et al., 2011). While certainly an interesting and somewhat enigmatic protein, tau has come to prominence due to its extensive involvement in neurodegenerative disease such as AD and other “tauopathies”.
III. Tau in Neurodegenerative Disease
AD pathology is classically characterized by the extracellular accumulation of senile plaques composed of amyloid β (Aβ) and the intracellular accumulation of tau. Although autosomal dominant mutations in the amyloid precursor protein and presenilins result in increased production of Aβ and cause familial forms of AD (Hardy et al., 1998), certain experimentation suggests that Aβ toxicity requires the presence of tau (Rapoport et al., 2002, Roberson et al., 2007, Vossel et al., 2010, Roberson et al., 2011). Neurons in culture exposed to toxic Aβ do not degenerate if they lack the tau gene (Rapoport et al., 2002). An Aβ-producing mouse crossed into a tau null background demonstrates that although amyloid plaques can form as expected, behavioral deficits do not develop (Roberson et al., 2007). Both of these studies suggest that Aβ is somehow working through tau to induce neurodegeneration. Furthermore, unlike Aβ pathology, the progression of tau pathology in AD closely follows the spatial and temporal clinical progression of the disease (Braak and Braak, 1991, Arriagada et al., 1992).
Tau’s involvement in the neurodegenerative process is further supported by its pathological presence in several other tauopathies that lack Aβ pathology. This group of diseases includes Pick’s disease (PiD), corticobasal degeneration (CBD), and progressive supranuclear palsy (PSP) (for reviews, see (Spillantini et al., 1998, Spillantini and Goedert, 1998)). These tauopathies are all characterized by filamentous tau pathology, but can be differentiated by the subcellular compartments containing this pathology and the specific brain regions affected (Goedert et al., 1998, Spillantini and Goedert, 1998, Buee and Delacourte, 1999). In addition, autosomal dominant mutations in the tau gene cause frontotemporal dementia and Parkinsonism linked to chromosome-17 (FTDP-17) as well as a few hereditary forms of PiD (Hogg et al., 2003), which further demonstrates that tau dysfunction is sufficient to cause neurodegeneration (Murrell et al., 1999, Goedert and Spillantini, 2000, Lee, 2001). Significantly, many of these mutations have been shown to decrease tau’s affinity for microtubules (Goedert, 2005, Bunker et al., 2006) and/or increase its propensity to aggregate (Gamblin et al., 2000, von Bergen et al., 2001).
III.A. Phosphorylation as a Mechanism of Tau Toxicity
Although mounting evidence clearly links tau to neurodegeneration, the precise mechanism of tau toxicity remains to be determined. Given that tau is a cytoskeleton-associated protein, a loss of its physiological function due to its dissociation from microtubules may lead to toxicity. Conversely, it is also likely that tau aggregates themselves are neurotoxic. Some of the first work describing tau’s accumulation into fibrillar pathologies in AD noted that the protein is abnormally phosphorylated (Grundke-Iqbal et al., 1986, Wood et al., 1986). It is not surprising that tau is a substrate for several kinases as 17% of the macromolecule is composed of serine and threonine residues (Lee et al., 1998). Interestingly, phosphorylation has been shown to decrease the affinity of tau for microtubules (Biernat et al., 1993, Alonso et al., 1996) and decrease the flexibility of the protein (Hagestedt et al., 1989). Therefore, the tau hyperphosphorylation observed in AD and other tauopathies suggests that phosphorylation may play a role in tau aggregation and subsequent neurodegeneration. Since these initial descriptions, however, much controversy has arisen. Some phosphorylation events appear to inhibit in vitro tau aggregation while others in fact seem to stimulate the process (Schneider et al., 1999, Abraha et al., 2000, Alonso et al., 2001, Eidenmuller et al., 2001). Modifications to the structure of tau’s molecular backbone would be expected to affect its ability to associate into filaments and may also inhibit its ability to dissociate, depending on the nature and sites of the alterations. Therefore the idea that multiple phosphorylation events drive and/or maintain changes in tau’s conformation is not unexpected; nor is it surprising that such modifications, depending on their location, could have opposite effects.
III.B. Tau Structure
Initially, tau was considered a “natively unfolded protein” with limited secondary structure (Schweers et al., 1994). More recently, NMR based structural studies on both a short fragment of phospho-tau (208–324) and full-length phospho-tau (phosphorylated by the proline-directed CDK2/CysA3 kinase) have revealed that a short alpha-helical secondary structure runs from pSer235 to the beginning of the MTBR binding region (Thr245-Ser324). Further, phosphorylation of this same fragment of tau (208–324) resulted in rapid depolymerization of tubulin assembly in an in vitro experiment which highlights the idea that structural changes on tau due to phosphorylation affects its ability to associate with MTs (Sibille et al., 2011). Additionally, FRET studies of tau in solution have revealed that monomeric tau appears to adopt a “paperclip” structure, whereby the C-terminus of tau folds over to interact with the MTBRs and the N-terminus folds over to interact with the C-terminus (Jeganathan et al., 2006). However, this may not be the only conformation that tau adopts in the soluble state as others have suggested that tau transitions rapidly between several conformations (Minoura et al., 2004, Minoura et al., 2005) (Figure 2).
Figure 2. Hypothesized conformations of tau.
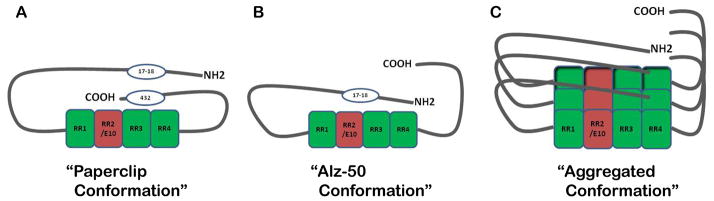
(A) The “paperclip conformation” was proposed using FRET analysis outlined by Jegenathan et al., 2006. The C-terminus folds in close proximity to the MTBRs while the N-terminus folds back towards the C-terminus but at a distance from the MTBRs. (B) The “Alz-50 conformation” is assumed when tau aggregation begins. When the C-terminus vacates its position near the MTBRs, it allows room for the N-terminus to interact with this region. This structure is recognized by the Alz-50 antibody and is associated with early tau polymerization as outlined by Carmel et al., 1996. (C) Putative stacking of tau monomers represents an aggregated tau conformation.
As outlined in figure 2, the idea that tau could assume a more fixed conformation was discovered by mapping the discontinuous Alz-50 antibody epitope (Carmel et al., 1996, Jicha et al., 1997). Within this work, it was revealed that in order to bind Alz-50 avidly, the extreme amino terminus of tau has to fold upon the microtubule binding domain. This conformation, although possible in soluble tau, seems to occur most readily in tau aggregates in vitro and in paired helical filaments (PHFs) purified from AD brain. Tau filaments are now realized to be highly ordered structures that possess β-pleated sheets typical of amyloidogenic proteins (Barghorn et al., 2004, Eliezer et al., 2005, Tokimasa et al., 2005). Tau fibrils can exist in the form of straight filaments (SFs) or PHFs consisting of two subfilaments seemingly twisted together with a regular periodicity (Crowther, 1991).
The appearance and accumulation of fibrillar tau structures is clearly disease related. As previously mentioned, tau aggregation is considered by most in the field to be toxic to the neuron or glial cell in which it occurs. Certainly a popular notion of the mechanism of toxicity involves the creation of a physical barrier within the neuron by the intracellular tau filaments (PHFs, SFs, etc.). Presumably, this barrier would impede transport and intracellular communication causing dysfunction and cell death (Ishihara et al., 1999). Therefore, both immunotherapy and anti-aggregation approaches to preventing tau toxicity are being explored by a number of laboratories in hopes of “clearing” or reversing the tau aggregation. Additionally, phosphorylation has been implicated in the induction and stabilization of numerous conformational changes in the tau molecule. “Abnormal phosphorylation” and “hyperphosphorylation” are terms used to describe tau’s pathological state when part of AD pathology (Grundke-Iqbal et al., 1986). Since phosphorylation can greatly reduce tau’s binding affinity for microtubules and tau is thought to stabilize these structures, its dissociation could possibly result in microtubule instability and vesicular transport anomalies. Therefore it is not surprising that other approaches being explored involve compounds known to stabilize microtubules, as well as agents that would slow or prevent tau phosphorylation by inhibiting key protein kinases. All of these therapeutic tactics will be discussed below followed by a discussion of future tau-based treatments derived from novel findings regarding tau’s aggregation and toxicity.
IV. Modulation of Phosphorylation
Tau can be phosphorylated by numerous kinases from several different classes both in vitro and in vivo; however, the exact functional implications of individual phosphorylation events are obscure. Tau becomes hyperphosphorylated in AD and related tauopathies, and as mentioned above, this is thought by many to influence tau’s pathological state by causing it to aggregate, become toxic, and/or dissociate from microtubules leading to their destabilization. Hence, considerable effort has been expended by the scientific community to discover and improve specific kinase inhibitors with the express goal of curtailing tau phosphorylation in vitro and in vivo. Numerous protein kinases have been implicated in tau’s hyperphosphorylation during disease including both serine/threonine kinases and tyrosine kinases. The more relevant appear to be the serine/threonine protein kinases; of these, most attention has been focused on designing inhibitors against the proline-directed kinases glycogen synthase kinase 3 beta (GSK3β), cyclin-dependent kinase 5 (CDK5), and extracellular signal-regulated kinase1/2 (ERK1/2), as tau contains 13 possible serine/threonine phosphorylation sites targeted by these kinases. This effort has proved a daunting task due to problems with inhibitor specificity and the promiscuity of these kinases due to site overlap. Below, we consider the current state of kinase inhibition in attempts to mitigate the hyperphosphorylation of tau.
IV.A. GSK3β and CDK5 Inhibitors
GSK3β has been a key target site for several research groups who design kinase inhibitors that regulate tau phosphorylation. However, since this enzyme plays a major role in many biological processes from metabolism to cardiac hypertrophy, inhibitors of this kinase could exhibit negative effects on non-disease-impacted pathways while also inducing unintended toxic side effects. Lithium was initially proposed as a potential therapeutic agent due to its inhibition of GSK3β. However interestingly, lithium only inhibits GSK3β with a modicum of selectivity when compared with its effects on other kinases. Further, in clinical trials lithium produced no change in global cognitive performance in patients with mild AD (Hampel et al., 2009). AR-A014418 is a specific inhibitor of GSK3β, (Bhat et al., 2003) and has been shown to inhibit this enzyme (IC50 = 104 ± 27nM) in an in vitro kinase functional assay carried out alongside 26 other kinases. Further experiments in cells expressing tau indicated an inhibition of tau phosphorylation at a specific GSK3β site, Ser-396. Additionally, this same inhibitor prevented Aβ-induced neurodegeneration in hippocampal slices. This was the first inhibitor shown to specifically target GSK3β without affecting the closely related cyclin-dependent kinases CDK2 and CDK5. Since the discovery of AR-A014418 Eli Lilly and Company developed the bisarylmaleimides, a class of potent and selective inhibitors of GSK3, analogues of which exhibited 160–10,000 fold selectivity for GSK3β over CDK2/4 and PKCBII kinases in a cell-based assay (Engler et al., 2005).
Of the cyclin-dependent kinases (CDKs), CDK5’s activity has been shown to be at its highest levels in the brain which is also where its activators, p35 and p39, predominately reside (Tsai et al., 1994, Mazanetz and Fischer, 2007). CDKs are important for cell cycle regulation and require that a regulatory subunit be associated in order for them to be activated. These regulatory subunits, p35 and p39, can be cleaved in the presence of elevated calcium levels producing p25 and p29, causing constitutive activation of CDK5. Interestingly, p25 accumulation is observed in AD brains (Tseng et al., 2002). Largely because of its putative role in tau hyperphosphorylation in AD, several CDK5 inhibitors have been developed. A high-throughput-screen (HTS) aimed at identifying lead compounds for the CDK5/p25 complex identified an active compound that proved to be equally inhibitory of CDK5/p25 and CDK2/cyclin E; this same compound is only weakly inhibitory to 24 other kinases. Based on this lead compound a library of analogous compounds, generally termed 2-aminothiazole inhibitors, was screened and a ligand was obtained that inhibits CDK5 with selectivity of 12-fold over CDK2 in in vitro assays (Helal et al., 2004). More recently, certain analogues proved to have protective effects in human neuroblastoma cells expressing P301L mutant tau which is implicated in tauopathies (Lagoja et al., 2011). A CDK inhibitory peptide (CIP) was discovered that inhibits CDK5 but shows little activity against other CDKs (CDC2, CDK2, CDK4 and CDK6) (Zheng et al., 2005). This peptide inhibitor consists of 125 residues (p35154–279), and suppresses CDK5 activity in vitro and in HEK293 cells (Zheng et al., 2002). In addition, CIP inhibits CDK5/p25 mediated phosphorylation of tau in cortical neurons but not CDK5/p35 when expression constructs individually producing these proteins are used.
Cross-inhibition of more than one kinase with a single compound has also been attempted using paullones, which are potent inhibitors of both GSK3β and CDKs. Specifically, alsterpaullone is able to reduce GSK3β phosphorylation of hT23 both in vitro and in insect cells (Leost et al., 2000). Similarly, indirubins, which are normally used to treat leukemias, were shown to be potent inhibitors of CDKs (IC 50 = 50–100nm) (Leclerc et al., 2001) and GSK3β (IC 50 = 5–50 nm). One indirubin, indirubin-3′-monoxime, has been shown to reduce tau phosphorylation in vitro and in insect cells. It must be noted, however, that although inhibiting more than one kinase may enhance tau dephosphorylation, the risk of pleiotropic negative effects is heightened should this be used in patients.
IV.B. Erk2 Inhibitors
Erk2 is involved in many biological processes including proliferation, differentiation, transcription regulation and development. In AD neurons that contain hyperphosphorylated tau and neurofilaments, Erk2 is activated, perhaps signifying a link between this kinase and tau hyperphosphorylation (Mazanetz and Fischer, 2007). In general, inhibitors that have been developed toward Erk2 also inhibit other kinases. For example, a natural product, K252a was identified as an Erk2 inhibitor but this molecule has only moderate selectivity as it also targets GSK3β and other kinases in vitro. However, K252a does mitigate against tau hyperphosphorylation in both human neuroblastoma cells and rat hippocampal brain slices (Hubinger et al., 2008). Despite these promising results, pharmacokinetically, K252a proved to have low oral bioavailability and poor blood brain barrier penetration. To improve on these characteristics an analogue of K252a, SRN-003-556, was developed that is able to cross the blood brain barrier and is more orally bioavailable (Bell and Claudio Cuello, 2006, Le Corre et al., 2006). Unfortunately, this analogue is even less selective than K252a as it also inhibits other kinases (CDC2, GSK3B, PKA and PKC) in vitro. SRN-003-556 is able to prevent okadaic acid-induced tau hyperphosphorylation in rat hippocampal slices and delay the onset of motor deficits in P301L-tau transgenic mice (Le Corre et al., 2006); this is an interesting finding since there are few studies demonstrating both the reduction in tau phosphorylation and functional motor improvements in mouse models. In addition to inhibiting hyperphosphorylation, SRN-003-556 was shown to reduce the amount of insoluble aggregated tau in the same mouse model, although even long term dosing did not result in a decrease in NFT numbers.
IV.C. Phosphatase activators
Another strategy directed at regulating tau hyperphosphorylation is to activate phosphatases that are directly responsible for removing phosphate groups from tau. One key phosphatase that has been shown to regulate tau phosphorylation is protein phosphatase 2A (PP2A). PP2A is a serine/threonine phosphatase with broad substrate specificity and reduced activity in the AD brain (Li et al., 2004). Several groups have targeted PP2A activation in attempts to reverse tau hyperphosphorylation in vitro and in vivo (Chohan et al., 2006, Kickstein et al., 2010). The Iqbal laboratory found that the N-methyl-D-aspartate receptor antagonist memantine is able to both inhibit and reverse abnormal tau hyperphosphorylation (Li et al., 2004). In the presence of okadaic acid (OA), which decreases PP2A’s activity, memantine restores PP2A function in rat hippocampal slices and reduces tau phosphorylation at Ser-262 to near normal levels. This same research group also used PC12 cells transfected with hT40 to demonstrate that, in the presence of memantine and a known inhibitor of PP2A, I2PP2A, tau hyperphosphorylation is reduced when compared to inhibitor alone (Chohan et al., 2006). Although its mechanism of action is still poorly understood, Memantine is FDA approved and is currently used to treat Alzheimer’s disease.
Another phosphatase activator that acts on the PP2A phosphatase is biguanide metformin, a compound currently approved for use in diabetes. This drug was shown to reduce tau phosphorylation by inducing PP2A activity in primary cortical neurons from mice expressing human tau on a tau null background (Kickstein et al., 2010). Furthermore, in the same study, metformin decreased tau phosphorylation in wild-type mice.
IV.D. Summary of Phosphorylation Modulators
In summary, while kinase inhibitors and phosphatase activators hold promise as effective therapeutics for AD and other tauopathies, there are significant specificity limitations. This is a real concern because inhibiting or activating multiple kinases or phosphatases could have undesirable off target effects especially since most kinases/phosphatases have multiple protein substrates in numerous organ systems. Due to this fact, the implications of long-term dosing remain a significant concern.
V. Tau-directed immunotherapeutic approaches to tauopathies
A promising approach in the treatment of tauopathies involves using the immune system to clear pathological protein aggregates. There are currently a number of ongoing immunotherapy clinical trials aimed at targeting Aβ. However, the clearance of Aβ plaques has been shown to have limited effect attenuating cognitive decline once Alzheimer symptoms are already underway (Holmes et al., 2008). Furthermore, Aβ immunotherapies have little to no influence on the accumulation of tau pathology. Work reviewed below suggests that targeting pathological tau aggregates may prove to be a more effective therapeutic tactic.
Two main immunotherapeutic approaches directed at tau have been employed: Active and passive immunization. Active immunization involves introducing foreign antigen into the subject, such as a tau peptide, which induces a T-cell response and results in the generation of antibodies against the immunogen. In passive immunization, antibodies against pathological tau forms are administered directly. Recent studies using both active and passive tau immunization successfully diminished tau aggregates and mitigated against cognitive impairments in tauopathy mouse models. These reports are summarized below.
The initial investigation regarding the safety and feasibility of tau immunotherapies (Rosenmann et al., 2006) was of great importance, as many of the Aβ immunotherapy clinical trials were halted due to encephalitis (Schenk, 2002, Orgogozo et al., 2003). Indeed, when wild type mice were immunized with recombinant human tau protein, the animals developed tau pathology and had severe astrogliosis and microgliosis with clinical signs of encephalomyelitis (Rosenmann et al., 2006). Most mice also displayed impaired cognitive performance. However, the authors suggest that the induction of tau pathology and cognitive symptoms could be explained by the use of full-length unphosphorylated tau (Rosenmann et al., 2006). This may also explain the adverse autoimmune reactions, as all of the more recent studies discussed below use smaller phosphorylated tau peptides and do not elicit such strong immunogenic responses.
In 2010, this same group evaluated the efficacy and safety of an active immunization protocol using a mixture of three tau peptides; each containing phosphorylated sites that are prominent in tauopathies (Tau195-213[P202/205], Tau207-220[P212/214], Tau224-238[P231]) (Boimel et al., 2010). Immunization was carried out on a double mutant transgenic tau mouse model (K257T/P301S) that develops NFT-like inclusions beginning at 6 months of age. Using antibodies harvested from these immunizations, it was demonstrated that they react only with neurons and glia of the transgenic mice but are negative within the wild-type controls. By 8 months post-immunization, there was a marked reduction in tau pathology in the cortex, hippocampus and brain stem. Interestingly, despite an increase in microglial number, there was no change in the number of activated microglia or astrocytes and no evidence of encephalitogenicity. While this study shows some promise as an effective immunotherapy, the increase in microglia and the use of complete Freund’s adjuvant (CFA) and pertussis toxin (PT), which are very strong adjuvants that have been linked to encephalomyelitis in Aβ immunization trials (Furlan et al., 2003), still raise toxicity concerns.
A more encouraging active immunization approach was undertaken using a phosphorylated tau immunogen emulsified in Alum-adjuvant without pertussis toxin (Asuni et al., 2007). The peptide immunogen used, Tau379-408, contains two phosphorylated serine residues (396 and 404) that are also prominent in tauopathies and form part of the PHF-1 epitope (Otvos et al., 1994). These investigators (Asuni et al., 2007) utilized a P301L tau tangle mouse model, referred to as JNPL3, (Lewis et al., 2000) which displays classic tau pathology, neuronal loss, and sensorimotor abnormalities driven by the P301L FTDP-17 mutation. Vaccines were administered monthly in mice between 2–5 months or between 2–8 months of age. Antibodies purified from immunized animals detected pathological tau aggregates in brain sections from both JNPL3 mice and AD patients. Immunoreactivity using antibodies against conformational and phosphorylated tau epitopes (MC1 and PHF1, respectively) was significantly reduced in the dentate gyrus, motor cortex, and brainstem of immunized mice compared to controls. Immunized mice performed significantly better on multiple sensorimotor tests relative to non-treated mice. This group also displayed enhanced clearance of pathological tau aggregates. As expected, a greater therapeutic effect was observed when immunizations were initiated at an earlier stage (5 months postnatal) than later in disease progression (8 months postnatal), impacting both behavioral performance and tau clearance (Asuni et al., 2007).
Subsequently, when FITC-tagged antibodies purified from a JNPL3 mouse were injected into the carotid artery of another JNPL3 mouse, the labeled IgG was detected in the brain and shown to colocalize with PHF1 and MC1 antibodies. This suggests that not only can injected anti-tau antibodies enter the brain, but they can also bind to pathological tau (Asuni et al., 2007). This result was further supported in a subsequent study where an FITC-labeled phospho-tau antibody (Tau379-408 [P-Ser396, 404]) purified from a JNPL3 immunized mouse was added to an ex vivo JNPL3 mouse brain slice model (Krishnamurthy et al., 2011). Not only was the antibody internalized by neurons and shown to co-localize with phosphorylated pathological tau, but it was also detected in late endosomes and lysosomes, suggesting a potential lysosome-mediated mechanism as a possible explanation of antibody-mediated clearance of tau aggregates.
The same tau immunogen in the aforementioned study (Tau379-408 [P-Ser396, 404]) was assessed in another active immunization approach using a novel model of tau pathology with accelerated tangle development (Boutajangout et al., 2010). This mouse model displays early onset tau pathology and is generated by crossing hTau mice (Andorfer et al., 2005) with mice carrying the M146L human presenilin 1 (PS1) mutation (hTau/PS1) (Duff et al., 1996). hTau/PS1 mice are better suited than JNPL3 mice for cognitive testing as they lack the latter’s motor impairments, which precludes the ability of the JNPL3 mice to be accurately tested for cognitive deficits. hTau/PS1 mice display extensive tau pathology in hippocampal and cortical regions, with onset of tau pathology and behavioral symptoms prior to 2 months of age. Mice were immunized beginning at 3–4 months of age and immunization was continued at monthly intervals up to 7–8 months postnatal, at which time the mice were subjected to behavioral testing and sacrificed for analysis. Importantly, not only did this immunotherapy reduce tau pathology throughout the brain, but it prevented cognitive impairments in three separate cognitive tasks. Since the hTau/PS1 mouse model is a more appropriate paradigm for human AD than other mouse models mentioned which primarily display motor deficits (JNPL3), the efficacy of Tau379-408[P-Ser396, 404] immunization better supports the choice of this peptide to attenuate cognitive decline in human clinical trials. Moreover, the fact that there was no significant difference between treated and control mice in the extent of microglial response strongly suggest that this immunogen exhibits low neurotoxicity (Boutajangout et al., 2010).
Although the active immunization approach certainly has advantages, there is a higher chance of deleterious autoimmune effects that can be avoided with passive immunization. As a follow-up study to their active immunization protocol, the Boutajangout and colleagues investigated the feasibility of a passive immunization technique using the PHF1 antibody, which targets the same phospho-epitope as Tau379-408[P-Ser396, 404] (Boutajangout et al., 2011). In this study, JNPL3 transgenic mice received antibody injections once a week for 13 weeks. Western analysis of brain tissue from the immunized mice revealed a reduction in the ratio of pathological tau to total tau in comparison to non-treated controls. Immunohistochemical analysis showed significantly less PHF1 staining in the dentate gyrus of the hippocampus in the treated group than in untreated JNPL3 controls. The reduction in tau pathology in the immunized mice correlated positively with improved motor performance. Similar to this group’s previous results employing active immunization approaches, (Asuni et al., 2007, Boutajangout et al., 2010) the degree of microgliosis and astrogliosis did not appear to differ among treatment groups.
A second passive immunotherapy approach (Chai et al., 2011) was carried out employing anti-tau antibodies PHF1 and also MC1, which detects tau in an early pathological conformation (Jicha et al., 1997). Antibodies were peripherally administered 2–3 times a week for 2 months and their effects were explored using both JNPL3 and P301S transgenic tau mouse models. The P301S mouse is a transgenic model expressing human tau with a P301S mutation under the control of the Thy1 promoter. These mice develop a neurological phenotype characterized by a severe paraparesis by 5 months of age (Allen et al., 2002). Western blot and ELISA analyses revealed that treatment with both PHF1 and MC1 antibodies reduced the 64kD biochemical correlate of tau pathology in both mouse models. Treatment with both anti-tau antibodies also reduced the presence of tau tangles and neuropil threads. Furthermore, treated mice had a reduction of neurofilament-positive axonal spheroids in the spinal cord, which indicates reduced neurodegeneration, and this correlated with an improved motor test performance (Chai et al., 2011).
One major criticism of many of the immunotherapy studies mentioned above is that although they target phosphorylated tau epitopes found in AD, these phosphorylated sites are also present in normal physiological conditions. However, using a different mouse model, immunizations were performed with peptides containing phospho-Ser422, a residue that is specific to pathological tau and is found in many neurodegenerative tauopathies (Troquier et al., 2012). The mouse model employed, HY-Tau22, develops late-stage hippocampal pathology and cognitive defects but lacks motor symptoms. Mice immunized for 18 weeks with the pS422 peptide displayed a reduction in pathological tau immunoreactivity that correlated positively with an improvement in spatial memory. Interestingly, an increase in tau concentration was observed in the blood sera of the vaccinated group over time, suggesting that immunization facilitates tau clearance from the brain to the blood (Troquier et al., 2012).
V.A. Mechanism of Antibody-Mediated Clearance
The observation of increased tau within blood as opposed to the brain (Troquier et al., 2012) could be explained by the “peripheral sink hypothesis”. In this hypothesis, peripheral administration and therefore circulation of antibodies effectively sequesters tau away from the brain toward the blood (Troquier et al., 2012). This hypothesis functions under the assumption that exogenous antibodies do not have the ability to cross the Blood Brain Barrier (BBB). However, peripherally-injected FITC-labeled antibodies were shown to enter the brain, suggesting that antibodies can in fact cross the BBB (Troquier et al., 2012). This is not entirely implausible, as the BBB is known to be compromised in many neurological diseases including AD (Zlokovic, 1997). Once inside the brain, antibodies could potentially target intracellular tau through the endosome-lysosome pathway and facilitate the clearance of extracellular tau through microglia activation (Morgan, 2009).
V.B. Summary of Immunotherapeutic Approaches
Based on mouse studies, immunotherapies targeting tau appear to provide a viable potential treatment of AD and other tauopathies. Although preliminary evidence on the safety and efficacy of this approach appears promising, as it stands, several questions still remain. In the future, key information on the ideal adjuvant for active immunization, the mechanism of passive immunization antibody entry, and epitope specificity of antibody-mediated tau clearance will prove invaluable.
VI. Small molecule inhibitors of protein aggregation
Aggregates of tau have long been considered the key elements of cellular toxicity in AD. Therefore, a fundamental aspect of research into tauopathies is to prevent tau aggregates from forming and/or to disassemble pre-formed species. To accomplish these goals, most attention has been given to the repeat regions of the tau molecule. This is understandable as these MTBR’s and surrounding sequences facilitate tau’s binding to microtubules, and it is this region of the molecule that forms the core of the PHF or SF polymers present in AD and other tauopathies (Wischik et al., 1988a, Wischik et al., 1988b). There are two hexapeptide motifs located in the 2nd and 3rd MTBRs, “VQIINK” and “VQIVYK” respectively, which demonstrate the greatest propensity for β sheet formation. In fact, the motif in the 3rd MTBR is absolutely essential for polymerization (von Bergen et al., 2000, Mukrasch M. D., 2005). In addition, numerous phosphorylation domains are located in and around the MTBRs and most of the mutations causing FTDP-17 tauopathies are also found in this region of the tau molecule. Our current understanding of tau aggregates, including attempts to interdict and reverse their formation, are outlined below.
VI.A. Methodologies and approaches utilized in anti-aggregant screening
Most tau aggregation inhibitors appear to disrupt tau’s β sheet structure (von Bergen et al., 2001). Many studies have utilized constructs like K19, which contains only one hexapeptide motif and is equivalent to the repeat region of 3R tau isoform, or K18, which contains two hexapeptide motifs and is equivalent to the repeat region of 4R tau isoforms (Figure 3).
Figure 3. Diagram of recombinant tau constructs.
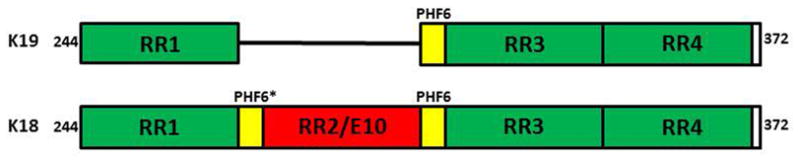
(A) Construct K19 consisting of only three repeats in the Repeat Regions (RR) from residue 244–372 missing residues 308–326 corresponding to RR2/E10. (B) Construct K18 consisting of four repeats in the RR from residues 244–372. The hexapeptide motifs PHF6 (VQIVYK) and PHF6* (VQIINK) that promote the assembly of β structure are highlighted in yellow in both constructs.
Additional studies employed full length recombinant isoforms, hT23 (3R, 0N) and hT24 (4R, 0N) (see Fig. 1) (Pickhardt et al., 2005, Pickhardt et al., 2007). Utilizing each of these constructs, a 200,000 compound screen was carried out in order to identify chemicals that can inhibit and reverse non-phosphorylated recombinant tau aggregates from forming paired helical filaments (Pickhardt et al., 2005). The compound library contained many different structurally diverse chemical classes in order to sufficiently span the potential spectrum of inhibitors of tau aggregation. Any compounds that did not both inhibit tau aggregation and disassemble pre-formed aggregates were excluded from the study, which eliminated 99.96% of the compounds. The remaining 0.04% which were considered “hits” from the compound screen were then investigated further. The efficacy of inhibition/disassembly was determined using Electron Microscopy and several different biochemical assays: Thioflavin-S fluorescence assay, Tryptophan Fluorescence Spectroscopy, Filter Trap Assay, Pelleting Assay, and Lactate Dehydrogenase Assay. Cellular models of tauopathy were also employed to screen for prevention of neurotoxicity (Honson et al., 2007, Pickhardt et al., 2007, Chang and Kuret, 2008). Lead “hits” identified from this initial screen are briefly described in the work below. The chemical structure of each of the five main classes of inhibitors summarized herein, are represented in Figure 4.
Figure 4. Chemical structures showing examples of tau aggregation inhibitors.
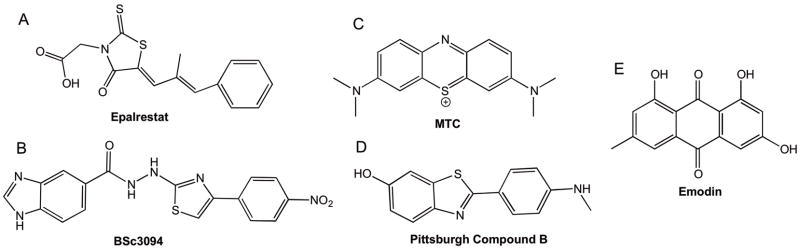
(A) Epalrestat: A rhodanine inhibitor containing a 5-membered heterocyclic compound. (B) BSc3094: A phenylthiazolyl-hydrazide inhibitor containing two separate functional groups- the thiazolyls and the hydrazides. (C) MTC: a phenothiazine tricyclic structure with sulfur and nitrogen incorporated into its heterocycle. Phenothiazines contain two aromatic rings connected by a nitrogen atom and have a much more planar structure. (D) PiB: a benzothiazole is also a heterocyclic compound with aromatic conjugation. There are many examples of these compounds that act as tau aggregation inhibitors. (E) Emodin: an anthraquinone is the building block of many dyes and exhibits a tricyclic structure with at least one phenolic moiety.
VI.B. Rhodanines
Rhodanine–based inhibitors are an interesting class of cyclic compounds that are used in a wide array of clinical applications and have no apparent side effects. The promising properties of rhodanines include their permeability in vitro, their bioavailability, and the fact that they are well tolerated. The basic rhodanine structure that displays optimal inhibition is a 5-membered heterocyclic compound which was shown to be more potent than homocyclic compounds (e.g. Epalrestat) (Figure 4a). Within the rhodanines the thioxo group is of utmost importance for functional inhibition. Variations in the length of the linker between the carboxylic acid and the core heterocycle show that increasing the distance by up to two carbon bonds increases inhibition. These still remain a novel class of compounds in relation to tau aggregation, with more optimization necessary in respect to Absorption, Distribution, Metabolism and Excretion (ADME) parameters. In addition to their anti-aggregant properties, rhodanines may potentially target GSK3 in AD (Bulic et al., 2010).
VI.C. Phenylthiazolyl-hydrazides
Another class of compounds, the Phenylthiazolyl-hydrazide (PTH) inhibitors, exhibit a higher cytotoxicity compared with the rhodanines although they appear to have a similar level of activity in cells (Bulic et al., 2010). This is a very intriguing compound as it appears to have two separate functional groups – the thiazolyls and the hydrazides. The increased level of cytotoxicity observed is not surprising due to the presence of the hydrazide-moiety, since many substances containing such a motif appear to have some level of antibacterial or antifungal properties (Hafez and El-Gazzar, 2008). Conversely, the hydrazide motif displays a somewhat ambiguous behavior in vivo since it produces a reactive oxygen species (ROS) from the oxidative cleavage of the N-N hydrazine bond. The side-effects observed likely result from the formation of reactive acyl radicals as illustrated by the anti-tuberculosis compound isoniazid (Preziosi, 2007). The hydrazides also appear to induce liver toxicity and carcinogenic activity due to their targeting of the enzyme monoamine oxidase (MAO).
The thiazole moiety is also a common heterocyclic compound that is often used in medicinal chemistry. Previously it was reported that the thiazole motif could produce irreversible inhibition of MAO (Raciti et al., 1995). Therefore although the hydrazide motif causes hepatotoxicity when targeting MAO’s, the thiazole motif may be protective against Alzheimer’s disease through its inhibition of MAOs. This could be an important value-added effect of thiazoles should they be used as tau aggregation inhibitors in patients (Bulic et al., 2010). Although the targets and mode of action of PTH’s are not completely understood, it is known that they have some anti-malarial activity by inhibiting the protease cruzain (Leite et al., 2006). A Lactate Dehydrogenase (LDH) Assay was performed to assess cell toxicity using five novel PTH compounds (BSc2998, BSc3000, BSc3016, BSc3551 and BSc3094) in an inducible cell culture model of N2a cells. Only compounds that displayed low cytotoxicity in un-induced cells were included in further studies. Subsequently, these same cells were utilized with the K18Δ280 construct, since this readily aggregates in solution in the absence of any inducer, in order to investigate the effects of each of these PTH’s as aggregation inhibitors. It was determined that the compound BSc3094 (Figure 4b) was most efficient at reducing tau aggregation while still retaining low levels of cytotoxicity (Pickhardt et al., 2007, Bulic et al., 2010). The authors stressed the point that the efficiencies of inhibition and depolymerization appear to be within the same range for all of the five novel compounds mentioned. This is noteworthy since it suggests that PHF inhibition and disassembly may work on similar principles (Pickhardt et al., 2007).
VI.D Phenothiazines
Phenothiazines are tricyclic structures with sulfur and nitrogen incorporated into their heterocycle. They contain two aromatic rings connected by a nitrogen atom, have a cationic charge, and have a much more planar structure with a high level of aromatic conjugation. It is these properties that are believed to enable this compound to inhibit tau aggregation (Hattori et al., 2008). Methylthioninium chloride (MTC) (Figure 4c), also known as Methylene Blue, is an example of a phenothiazine. MTC treatment was demonstrated to reduce tau levels in ex vivo slice cultures from transgenic tauopathy mice (Congdon et al., 2012). There are studies which strongly suggest that this compound has the ability to penetrate the BBB (Oz et al., 2009); and therefore, its potential as a tau aggregation inhibitor increases greatly. In a phase II clinical trial, this compound significantly reduced cognitive decline relative to a placebo. Therefore, MTC is effective at blocking the tau-tau binding interaction that is known to occur through the repeat domain in vitro (Wischik et al., 1996). Interestingly, the method of administration of this compound affects its distribution throughout the body and how toxic it will become. For instance, if MTC is given orally, it appears to distribute to the liver and intestines. Administered in this manner, the concentrations necessary for tau aggregation inhibition would be toxic to humans, since such high levels can lead to accumulation within the liver. Conversely, when administered intravenously, the compound appears to localize and accumulate in the brain. When used at an effective dosage in humans, MTC appears to have few side effects. However, The National Toxicology Program demonstrated that long term usage of this compound (1–24 months) in rodents led to lymphomas and some malignancies within the intestines (Oz et al., 2009). In addition to its anti-aggregant properties, it appears that MTC, like the phenylthiazolyl-hydrazides, can behave as an MAO inhibitor or a NO-synthase inhibitor (Oz et al., 2009). Furthermore its redox potential may rescue mitochondrial dysfunction. Hence, its mode of action, in vivo, remains obscure yet promising.
VI.E. Benzothiazoles
Benzothiazoles resemble the phenothiazines in that they have a positive charge and aromatic conjugation. The benzothiazoles are also heterocyclic compounds which are advantageous for supporting the hydrophobic interactions observed with many aggregation inhibitors. There are already many examples of these compounds that act as tau aggregation inhibitors, such as Pittsburgh Compound B (PiB) (N-methyl-[11C] 2-(4′-methylaminophenyl)-6-hydroxybenzothiazole) (Figure 4d). (Klunk et al., 2004, Bulic et al., 2010). This is a hydroxylated derivative of the prototypical benzothiazole termed 2-(4′ methylamino-phenyl) benzothiazole or BTA-1. It is an analog of Thioflavin T which has long been used as a dye to bind and quantify amyloid beta-pleated sheet structures within protein aggregates. Similarly, PiB can also bind to aggregated amyloid at low concentrations both in vitro and in vivo and does so only in the cortex and striatum (Klunk et al., 2004). In fact, a study performed by Klunk et al. demonstrated that PiB was found at 2-fold greater concentration in the cortical regions of AD patients compared to healthy controls, which suggests high levels of amyloid deposition in the cortices of Alzheimer’s patients (Klunk et al., 2004). This study did outline some initial problems, however, illustrating individual cases where interpretation of data was difficult. For example, PiB binding was observed in the cortical region of an asymptomatic healthy control. This could be due to a ‘false positive’ result where PiB binds to non-amyloid structures, or perhaps due to the presence of amyloid deposits in healthy individuals who lack the accompanying cognitive decline. For these reasons, the authors suggest that dyes such as the benzothiazoles be used as a tool to detect and quantify pathogenesis, but should not be considered a reliable method of diagnosis (Klunk et al., 2004).
Another example of a well-known benzothiazole is N744 (3, 3′-bis (β-hydroxyethyl)-9-ethyl-5, 5′ –dimethoxythiacarbo-cyanine iodide) (Congdon et al., 2007). N744 is a thiacarbocyanine dye that appears to have a biphasic function. At high concentrations it forms columnar stacks followed by H-aggregates which lead to a decrease in its inhibitory action. Conversely, this compound exerts its greatest inhibitory effect when it is in its monomeric state, and this only occurs at lower concentrations (Congdon et al., 2007, Bulic et al., 2010). It is believed that the mechanism of inhibition occurs via increasing the ‘critical concentration’ of tau. Therefore, at low levels of N744, increasing levels of tau are needed to complete an in vitro aggregation reaction, which illustrates that this compound acts as anti-aggregant (Necula et al., 2005).
Additional work on the role of the benzothiazole class of inhibitors on tau aggregation has been carried out with special emphasis on thiacarbocyanines. In particular, Honson and colleagues show that cyclic N, N′-alkylene bis thiacarbocyanine, which is referred to by the authors as compound 2, has potent effects on tau fibrillization (Honson et al., 2007). This study complemented the previously mentioned thiacarbocyanine study due to the fact that compound 2 is essentially a multi-valent form of N744. The authors successfully demonstrated that compound 2 reproduces the inhibition of tau aggregation that is observed with N744, but with greater potency. This is most likely due to the fact the compound 2 contains two binding sites within one ligand (Honson et al., 2007). It was hypothesized that compound 2 binds to tau oligomers through one of its cyanine groups, which leads to a conformation where the second cyanine group can disrupt the addition of more monomers, and this subsequently prevents elongation of the tau fibril. Alternatively, these small molecule inhibitors could be recruiting extraneous proteins into small aggregates at the site of filament extension which are not part of the normal aggregation pathway (Honson et al., 2007).
Subsequent studies on other thiacarbocyanine dyes demonstrated that this class of compounds also functions in a biphasic manner. When 3, 3′-diethyl-9-methylthiacarbocyanine iodide, referred to by the authors as compound 11, was tested on organotypic slices from the JNPL3 mouse model, it was discovered that at low levels (1–10nM) it demonstrates greater than 50% efficacy at inhibiting tau aggregation (Chang et al., 2009). Interestingly, however, when used at much higher levels, compound 11 instead has the ability to induce tau aggregation in vitro. Levels of aggregation were assessed by extracting and analyzing amounts of sarkosyl-insoluble tau present in slices that were incubated with increasing concentrations of compound 11 versus the vehicle DMSO (Chang et al., 2009). As outlined above, there is much evidence that benzothiazoles, and perhaps thiacarbocyanines in particular, have exceptional potential as tau aggregation inhibitors. These dyes are very readily taken up by cells and tissues and they usually congregate near membranes which are thought to be the sites of tau nucleation (Gray et al., 1987). They appear to be very potent as inhibitors of aggregation and therefore provide an excellent starting point for further studies.
VI.F. Anthraquinones
Anthraquinones are the building blocks of many dyes and exhibit a tricyclic structure with at least one phenolic moiety. These compounds have been used successfully in cancer treatment due to their ability to halt cell growth (Pickhardt et al., 2005). However, for this same reason they cannot be used as a long term treatment for AD. The sugars (glycones) generally have low oral bioavailability and therefore are usually administered intravenously. The non-sugars (aglycones) have better bioavailability but still seem to be insufficient. The aforementioned screen of 200,000 compounds led the investigators to focus on five main Anthraquinone compounds, namely Emodin (Figure 4e). Daunorubicin, Adriamycin, PHF005 and PHF016. Each one was successful in inhibiting aggregation of each construct tested, and they were all able to initiate PHF disassembly, albeit to different degrees (Pickhardt et al., 2005). Across the five compounds, Emodin appeared to be the most effective at PHF disassembly, producing the most robust effect in both K18 and hT23 constructs. However, PHF016 was the only compound that displayed a high ability to effectively inhibit aggregation and disassemble pre-formed aggregates within the same construct – K19 (Pickhardt et al., 2005).
Using the filter trap assay and the inducible cell line N2a, under a “tet-on” transactivator that was transfected with the construct K18ΔK280/Y310W, Emodin proved most efficacious in inhibiting tau aggregation and disassembly of pre-formed aggregates. Electron microscopy demonstrated that overnight incubation of PHF’s with any of the five compounds leads to breaks and shortening of the filaments (Pickhardt et al., 2005).
VI.G. Summary of Tau Aggregation Inhibitors
In summary, much work has been performed on tau aggregation inhibitors with a moderate level of success. Researchers have demonstrated that the inhibitors described above can be used to prevent the aggregation of tau, although there is still much refinement required before this method of therapy can be used. The ability to dissolve pre-formed aggregates has also been demonstrated. However, this usually leads to the generation of small oligomers which are now hypothesized to be harmful (Bulic et al., 2010, Patterson et al., 2011a, Lasagna-Reeves et al., 2012). Therefore, this method of therapy may not be as beneficial as once thought.
VII. Microtubule-stabilization as a treatment for tauopathies
It has long been postulated that tau plays a role in stabilizing microtubules within neuronal cell populations (Gamblin et al., 2003a, Dickey et al., 2006, Brunden et al., 2011, Zhang et al., 2012). Clearly, events such as hyperphosphorylation or certain mutations can lower tau’s affinity for the microtubule (Brunden et al., 2010), which most likely leads to destabilization. Certainly, microtubule destabilization could have dire consequences for the neuron, causing structural disruption and impaired axonal transport (Brunden et al., 2010). Therefore it is not surprising that microtubule-stabilizing drugs have recently received much attention as potential treatments for tauopathies. Fortunately, known microtubule stabilizing drugs, such as those derived from Taxol, have been approved and are commonly used in the treatment of cancer as they have the ability to affect mitotic spindle formation and dynamicity leading to cell death (Long and Fairchild, 1994, Yvon et al., 1999). The effects of a microtubule-stabilizer on tau regulation has been studied in a transgenic mouse model of human tauopathies with the shortest isoform of tau expressed under the control of the mouse prion promoter (PrPT44) (Ishihara et al., 1999). When 9-month old transgenic mice were treated with paclitaxel delivered in a micelle vehicle (Paxceed, Angiotech Pharmaceuticals), the compound rescued the previously impaired Fast Axonal Transport (FAT), increased microtubule numbers, and improved motor skills compared to the mice treated with a placebo control (Zhang et al., 2005).
Although paclitaxel is a well-known taxane normally used in the treatment of cancers, it has been deemed unsuitable for tauopathies as it does not effectively penetrate the Blood-Brain-Barrier and its side effects render it ineffective for long-term use (Fellner et al., 2002, Brunden et al., 2011). Therefore, the therapeutic potential of the known microtubule-stabilizing agent Epothilone D (EpoD) was recently investigated (Brunden et al., 2010). Not only was EpoD successful in crossing the blood brain barrier, but it continued to have a good brain: plasma ratio throughout the trial. EpoD was shown to improve both microtubule density and axonal integrity in 3 month old transgenic mice expressing human tau containing a P301S mutation, or PS19 mice, without any apparent toxic side effects. Interestingly, by 6 months of age it seemed to reduce cognitive defects that had already developed in the mice. However, it must be noted that the effects of EpoD did not lead to a decrease in tau pathology in the brain (Brunden et al., 2010). More recently, an investigation using aged mice treated with EpoD demonstrated its success in improving microtubule density, FAT, and cognitive performance. An unexpected difference from the younger mice in the previous study was the fact that EpoD caused a decrease in tau pathology in the aged mice. The actual pathway responsible for this remains unclear. An important aspect of this study was that the doses used were approximately 30–100 times lower than those needed to treat cancer. During the entire course of an extended treatment, the aged PS19 mice sustained their body weight and maintained high blood cell counts while displaying no immune deficiencies. The lack of perceivable side effects was thought to be due to EpoD maintaining a higher brain: plasma ratio. Hence, EpoD most likely remains in the brain longer and can be cleared from the blood in a timely manner thus minimizing undesirable negative consequences. These data indicate that EpoD may have potential as a therapeutic agent for human tauopathies (Zhang et al., 2012).
VII. A. Summary of Microtubule-Stabilization as a Treatment Strategy
A decrease in the number and length of MT’s and reduction in the levels of MT-binding found in the AD brain (Bramblett et al., 1992, Cash et al., 2003) can be clearly linked to a disruption in tau function following its abnormal phosphorylation and folding. Therefore, molecules aimed at stabilizing microtubules should prove to be effective therapeutic strategies to compensate for this tau loss-of-function. MT-stabilizing drugs currently in use for cancer treatments appear to be a good place to start, as their metabolism and toxicity effects have already been well established. However, due to its ability to penetrate the BBB and cross into the brain, EpoD now appears to be the most promising candidate. This molecule also holds more potential as its effective doses are much lower than those needed for cancer treatments. These dosing and distribution properties are extremely valuable considering chronic use is likely required for the treatment of tauopathies. However, additional analyses of both EpoD and other brain-penetrant MT-stabilizing agents are necessary to provide more information regarding the safety and efficacy of these molecules for therapeutic use in AD and other tauopathies.
VIII. Non-canonical Targets of Tau Function
Recent discoveries regarding tau alterations, interactions and toxicity in tauopathies have been documented although drug discovery efforts have yet to be put forth in these areas. Below, we briefly discuss more recent tau involvements and suggest potential new tau and tau-related drug targets.
VIII.A. Role of the N- and C-termini
Mapping of the Alz-50 antibody revealed that conformational shifts involving the N-terminus are important for tau filament formation. Work from our laboratory found that removal of a small portion of the extreme N-terminus of tau (residues 2–18) results in shorter filaments (Gamblin et al., 2003b), indicating that the amino terminus is crucial to efficient aggregation of the full-length tau molecule. FTDP-17-associated N-terminal mutations R5L and R5H further attest to the importance of this region of tau. When tested, the R5L mutation appeared to efficiently drive tau into small aggregates in vitro while the removal of amino acids 2–18 greatly inhibited the rate of tau association. Earlier work indicated that the amino terminus of tau was capable of associating with membranes (Brandt et al., 1995). Interestingly, primate tau is somewhat unique in that it diverges from rodent, bovine and porcine tau containing a unique tyrosine and flanking amino acids around N-terminal residue 18 (Y18). This site has been demonstrated to be readily phosphorylated by fyn kinase, and phosphorylation at this site may be important for regulation of aspects of tau function. Interestingly, the tau src tyrosine kinase-interacting domains are in and around the MTBRs, again suggesting an intramolecular folding event may be responsible for efficient phosphorylation at Y18 (Lee et al., 1998).
In contrast to the role of the N-terminus in stabilizing filaments, removal of the C-terminus (residues 421–441) has been shown to accelerate the rate and extent of in vitro aggregation, and adding this fragment back inhibits filament formation (Abraha et al., 2000, Berry et al., 2003). Notably, removal of the C-terminal tail occurs in vivo via truncation at a canonical caspase-cleavage site (Berry et al., 2003). This suggests that caspase cleavage could be a contributing factor of enhanced filamentation seen in the diseased state in the formation of NFTs and NFT-like structures. Association of the tau pathology with the specific regions of neurodegeneration in AD would appear to indicate that these aggregates are toxic.
VIII.B. Evidence for Prefibrillar Tau as the Toxic Species
Although NFTs have traditionally been considered the main pathological tau component in AD and other tauopathies, recent evidence suggests that they may not in fact be the main neurotoxic tau species (Morsch et al., 1999, Vana et al., 2011). Although tau overexpression leads to neurodegeneration in animal models, memory deficits and cell loss precede detectable NFT-like tau pathology. Moreover, memory deficits can occur in the absence of any visible neurofibrillary pathology in animal models (Wittmann et al., 2001, Andorfer et al., 2003, Oddo et al., 2003, Spires et al., 2006). In a neurodegenerative mouse model, suppression of the tau transgene improves memory function and halts further cell loss without lessening the NFT burden (Santacruz et al., 2005, Sydow et al., 2011). Together, these data suggest that NFTs may actually be somewhat inert and that toxicity may be primarily conferred by some other pathological state of the tau molecule.
Recent evidence suggests that a prefibrillar form of aggregated tau may in fact be the toxic species. In a transgenic tau mouse model, memory deficits correlate with early tau multimeric aggregates that precede NFTs (Berger et al., 2007). Human AD brain studies revealed that tau oligomers are found at a 4-fold higher concentration in AD brains than in healthy controls (Lasagna-Reeves et al., 2012). An in vitro crosslinking experiment demonstrated tau filament formation is preceded by tau dimerization (Patterson et al., 2011a). A novel tau antibody generated in our lab, which selectively recognizes tau dimers and oligomers but not filaments, localizes in pretangle neurons, neuropil threads and dystrophic neurites in vulnerable regions of MCI and AD brain and co-localizes with early stage markers of tau pathology (Patterson et al., 2011a). The existence of prefibrillar tau aggregates has also been demonstrated in AD brain homogenates using atomic force microscopy (AFM) (Maeda et al., 2006, Berger et al., 2007, Sahara et al., 2007). AFM analysis of fractionated prefrontal cortex homogenates revealed that granular tau oligomers are elevated in Braak I brains over non-AD (Braak 0) controls (Maeda et al., 2006). Braak-stage I is typically characterized by a paucity of NFT tau pathology and corresponds with a time prior to the manifestation of most clinical symptoms. In addition, the formation of similar granular oligomers was shown to precede PHF formation in vitro (Maeda et al., 2007). Also, several groups have reported the existence of SDS-stable tau multimers in AD that are similar to the ones associated with cognitive decline in a tau transgenic mouse model (Berger et al., 2007, Sahara et al., 2007, Lasagna-Reeves et al., 2010).
Due to difficulties isolating and characterizing tau intermediates, direct evidence linking tau oligomers to neuronal dysfunction is still limited. Recently, we have utilized extruded axoplasm from the squid giant axon to study the effects of tau monomers and aggregates on axonal transport. The addition of recombinant human tau monomers has no effect on transport when physiological concentrations of tau are used. However, addition of the same amounts of a mixture of recombinant tau oligomers plus filaments caused a selective inhibition of anterograde FAT (LaPointe et al., 2009, Kanaan et al., 2011a, Kanaan et al., 2011b). Furthermore, addition of the tau-binding protein Hsp70 to the oligomer plus filament mixture prevents FAT inhibition in the squid axoplasm assay. Since Hsp-70 has been demonstrated to bind to tau oligomers and not filaments, it can be deduced that oligomers are the primary cause of toxicity in this assay (Patterson et al., 2011b).
VIII.C. Role of Tau in Axonal Transport
Inhibitor studies in the squid axoplasm indicate that the aggregated tau works through a signal transduction cascade in which protein phosphatase 1 (PP1) is activated; this in turn activates GSK3 which phosphorylates kinesin light chain, causing it to release its cargo (LaPointe et al., 2009). Internal deletions of the recombinant tau molecule narrowed the “toxic region” to amino acids 2–18 on the amino end of the tau molecule, referred to as the Phosphatase Activation Domain (PAD). The use of the small, non-canonical isoforms of tau (6D & 6P) which lack the proline rich region, MTBR, and the carboxy terminus present in full length CNS tau (Luo et al., 2004) further demonstrated that the toxic cascade is dependent only on the amino end of the molecule. This was confirmed by introduction of a synthetic peptide consisting of residues 2–18 into the axoplasm which inhibited anterograde transport as well as or better than the aggregated tau filaments or the 6D/6P monomers (LaPointe et al., 2009, Kanaan et al., 2011b). A control scrambled peptide had no effect. Together, this suggests that the folding of tau during oligomer formation displays the toxic domain while it is obviously unavailable in the monomer. These data further indicate that this toxic domain of tau is necessary and sufficient for initiating this phosphatase-dependent inhibitory cascade and that targeting oligomers or the N-terminal toxic region of tau could have promising therapeutic potential.
Tau’s availability as a substrate for phosphatases can be controlled in a site-specific manner by peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) (Lu et al., 1999, Lim et al., 2008). Pin1 isomerizes only phospho-Serine/Threonine-Proline motifs and has been shown to be responsible for doing so at the Thr231-Pro position (Lu et al., 1999). The change from cis to trans at this position promotes dephosphorylation of tau by PP2A (Zhou et al., 2000). It has been demonstrated that when Pin1 is knocked out in mice, tau filament formation occurs and neuronal degeneration is observed suggesting its protective role in age-dependent neurodegeneration (Liou et al., 2003). To understand the conformation of tau present in tauopathies, Nakamura et. al. recently reported, using antibodies specific to either cis or trans phosphorylated tau conformers, that the cis and not the trans conformation was found in AD and MCI brains (Nakamura et al., 2012). This research group also demonstrated that Pin1 is able to restore phospho-tau’s ability to promote microtubule assembly, suggesting that the conversion of cis to trans is important for maintaining tau’s microtubule stabilizing function. Further, in double transgenic mice (tau and Pin1) the presence of Pin1 aids in the conversion of cis to trans pThr231-tau when compared to tau transgenic mice samples. Conversely, when Pin1 is knocked out (KO) of the tau transgenic mice the level of cis pThr231-tau is the more abundant species when compared to tau transgenic controls. The role prolyl isomerases play among tauopathies can differ, as evidenced in P301L mutant mice (Lim et al., 2008). Specifically, Lim et. al. demonstrated that when Pin1 is either knocked down or KO, wild type tau protein maintains stability in vitro and in 3R transgenic mice. Further, when Pin1 is overexpressed in transgenic mice containing wild type tau there is an observed reduction in the amount of total hyperphosphorylated tau. Conversely, in P301L mice when Pin1 is overexpressed the opposite effect is observed as there is an increase in the amount of total hyperphosphorylated tau (Lim et al., 2008). Collectively, these results suggest that Pin1 is a possible target for therapeutic approaches designed to intervene in tauopathies and that the interventions used may need to be disease-specific.
There are few Pin1 inhibitors and even fewer that have been tested for effectiveness in tau stability in model systems. One example is the Pin1 inhibitor, juglone, which is both noncompetitive and irreversible (Rudrabhatla and Pant, 2010). Although juglone inhibits Pin1 it is also able to inhibit other enzymes and block transcription. It has, however, been demonstrated that juglone inhibition changes tau localization which ultimately affects the microtubule network and eventually results in neuronal death (Atabay and Karabay, 2012). Although the P301L mutant was not used in this experiment, it appears that more efforts need to be exerted toward studying the regulation of Pin1 in relationship to non-AD tauopathies vs. AD.
VIII.D. Cysteine Proteases
Another potential target for therapeutic intervention involves the calpain family of calcium-dependent cysteine proteases. Hippocampal neurons incubated with pre-aggregated Aβ were shown to induce the activation of calpain-1 and the subsequent cleavage of tau into a 17 kDa fragment (Park and Ferreira, 2005). Transfecting this 17kDa tau fragment into CHO cells and cultured hippocampal neurons appears cytotoxic, and enhanced levels of this cleaved fragment can be detected in cortical brain samples of people with AD and other neurodegenerative tauopathies (Park and Ferreira, 2005, Ferreira and Bigio, 2011). In addition, incubating hippocampal neurons with calpain inhibitors prevents the generation of the cleaved tau fragment and blocks Aβ-induced neurodegeneration. Together this work suggests a novel pathway that may impact tau creating a toxic species (Park and Ferreira, 2005). A recent report appeared to refute these initial findings by demonstrating that this tau fragment is not toxic; however, these investigators used different culture conditions, younger hippocampal neurons, and a smaller tau fragment generated by the cleavage of calpain 2, not calpain 1 (Garg et al., 2011). While additional studies are clearly needed to better characterize the role of calpain and tau cleavage in neurodegeneration, targeting calpain pathways to mediate tau toxicity may provide yet another therapeutic approach.
VIII.E. Summary of Non-canonical Targets of Tau Function
Together these novel findings suggest new tau targets and sites of intervention in disease initiation and progression of tauopathies. Potential points of intervention include the PAD domain on tau, prevention of tau oligomer formation, inhibition of PP1, calpain inhibition, regulation of Pin1, and for different reasons than given above, inhibition of GSK3 isoforms. Targeting the PAD peptide may be the most straightforward of these choices as this could be approached by either immunotherapy or discovery of small molecule inhibitors. Binding of an inhibitor to PAD would likely result in less pleiotrophic toxicity than inhibition or activation of a major enzyme system. Additionally, anti-aggregants may provide another approach by focusing on prevention of the formation of oligomeric tau species rather than on prevention of NFT formation.
IX. Therapeutic Outlook
We have examined ongoing efforts to inhibit processes thought to be involved in producing the tau pathologies in AD and other tauopathies that include antibody immunotherapy, discovery of pharmacological inhibitors to mitigate tau hyperphosphorylation/aggregation, and discovery of activators of PP2A. There is ample evidence that suggests that any one of these processes provides a viable target for intervention in the disease process. However, each also has major drawbacks. Kinases and phosphatases are present in nearly every cell in the body and it has proven difficult to find inhibitors that alter activity of one enzyme without also affecting the other members of the same or closely related class. Even if successful in discovering compounds that would target the brain specifically, the pathology present in tauopathies is region-specific. What would be the effects on non-involved brain regions? Anti-aggregants at least potentially have the advantage of directly interacting with the abnormal form of toxic tau in question, but this form is still undergoing definition.
Immunotherapy has shown excellent results in mouse models, however the success of this approach in humans has yet to be vetted and the results of active and passive immunization trials using Aβ raises real concerns regarding induction of inflammatory events (Schenk, 2002, Orgogozo et al., 2003). However, there are currently multiple Aβ immunotherapy compounds being investigated which display strong promise to have disease-modifying potential in AD patients (Delrieu et al., 2012). This leads us to hope that we can then gain knowledge from these studies and use it to facilitate a similar approach to clear the tau pathologies from AD and FTD brains.
Of all the therapeutic paradigms examined within this review, the microtubule stabilization approach appears the most advanced and ready for human trials. This is largely due to the previous work done on these compounds for use in cancer therapy. It would seem that at the very least, microtubule stabilization may prove useful in combination therapy even if it doesn’t develop into a stand-alone approach. This of course assumes that EpoD is non-toxic after long-term human use. Whereas cancer can often be treated as an acute insult in which short term therapies can be employed with some degree of success in eradicating the disease, AD and other tauopathies are likely to be chronic, requiring long-term treatment that lacks serious adverse side-effects. Clearly, much work is left to be done in order to translate the therapies highlighted in this review toward effective use in the clinic, but the therapeutic advances discussed herein demonstrate that significant progress is being made.
Acknowledgments
This work was supported by NIH grants AG014449 and AG09466 to L.I.B. Diana Himmelstein was supported in part by NIA Training Grant T32 AG20506.
Abbreviations
- MAP
microtubule associated protein
- NFT
neurofibrillary tangle
- AD
Alzheimer’s disease
- MTBR
microtubule binding repeat
- Aβ
amyloid beta
- PiD
Pick’s disease
- CBD
corticobasal degeneration
- PSP
progressive supranuclear palsy
- FTDP-17
frontotemporal dementia and Parkinsonism linked to chromosome-17
- PHF
paired helical filaments
- SF
straight filaments
- CDK
cyclin dependent kinase
- GSK3β
glycogen synthase kinase 3 beta
- ERK1/2
extracellular signal-regulated kinase 1/2
- HTS
high throughput screen
- CIP
CDK inhibitory peptide
- PP2A
protein phosphatase 2A
- OA
okadaic acid
- CFA
complete freund’s adjuvant
- PT
pertussis toxin
- PS1
presenilin 1
- JNPL3
P301L tau tangle mouse model
- BBB
blood brain barrier
- hT23
recombinant human tau with 3 MTBRs and 0 N-terminal inserts
- hT24
recombinant human tau with 4 MTBRs and 0 N-terminal inserts
- ADME
absorption, distribution, metabolism, and excretion
- PTH
phenylthiazolyl-hydrazide inhibitors
- ROS
reactive oxygen species
- MOA
monoamine oxidase
- LDH
lactate dehydrogenase assay
- MTC
methylthioninium chloride
- PiB
pittsburgh compound B
- PrPT44
transgenic mouse model with the shortest form of tau expressed under the control of mouse prion promoter
- FAT
fast axonal transport
- EpoD
epothiolone D
- AFM
atomic force microscopy
- PP1
protein phosphatase 1
- Pin1
peptidyl-prolyl cis-trans isomerase NIMA-interacting 1
Footnotes
XI. Conflict of Interest: The authors have no conflicts of interest.
XII. References
- Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LI. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci. 2000;113:3737–3745. doi: 10.1242/jcs.113.21.3737. [DOI] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of neurochemistry. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Ashman JB, Hall ES, Eveleth J, Boekelheide K. Tau, the neuronal heat-stable microtubule-associated protein, is also present in the cross-linked microtubule network of the testicular spermatid manchette. Biol Reprod. 1992;46:120–129. doi: 10.1095/biolreprod46.1.120. [DOI] [PubMed] [Google Scholar]
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atabay KD, Karabay A. Pin1 inhibition activates cyclin D and produces neurodegenerative pathology. J Neurochem. 2012;120:430–439. doi: 10.1111/j.1471-4159.2011.07259.x. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–1703. doi: 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- Bell KF, Claudio Cuello A. Altered synaptic function in Alzheimer’s disease. European journal of pharmacology. 2006;545:11–21. doi: 10.1016/j.ejphar.2006.06.045. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, Binder LI. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325–8331. doi: 10.1021/bi027348m. [DOI] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. The Journal of cell biology. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Experimental neurology. 2010;224:472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. Journal of neurochemistry. 2011;118:658–667. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathology. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Trojanowski JQ, Lee VM. Regions with abundant neurofibrillary pathology in human brain exhibit a selective reduction in levels of binding-competent tau and accumulation of abnormal tau-isoforms (A68 proteins) Laboratory investigation; a journal of technical methods and pathology. 1992;66:212–222. [PubMed] [Google Scholar]
- Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. The Journal of cell biology. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan AM, Trojanowski JQ, Smith AB, 3rd, Lee VM. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacological research: the official journal of the Italian Pharmacological Society. 2011;63:341–351. doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, 3rd, Lee VM, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;9:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology. 2010;59:276–289. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem. 2006;281:11856–11863. doi: 10.1074/jbc.M509420200. [DOI] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. The American journal of pathology. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, Hutton ML, Citron M. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Congdon EE, Honson NS, Duff KE, Kuret J. Structure-activity relationship of cyanine tau aggregation inhibitors. Journal of medicinal chemistry. 2009;52:3539–3547. doi: 10.1021/jm900116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Kuret J. Detection and quantification of tau aggregation using a membrane filter assay. Anal Biochem. 2008;373:330–336. doi: 10.1016/j.ab.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chohan MO, Khatoon S, Iqbal IG, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006;580:3973–3979. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Necula M, Blackstone RD, Kuret J. Potency of a tau fibrillization inhibitor is influenced by its aggregation state. Archives of biochemistry and biophysics. 2007;465:127–135. doi: 10.1016/j.abb.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff K. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8 doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delrieu J, Ousset PJ, Caillaud C, Vellas B. ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches. Journal of neurochemistry. 2012;120(Suppl 1):186–193. doi: 10.1111/j.1471-4159.2011.07458.x. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Ash P, Klosak N, Lee WC, Petrucelli L, Hutton M, Eckman CB. Pharmacologic reductions of total tau levels; implications for the role of microtubule dynamics in regulating tau expression. Molecular neurodegeneration. 2006;1:6. doi: 10.1186/1750-1326-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Molecular biology of the cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Kirschner MW. Tau protein function in living cells. The Journal of cell biology. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Eidenmuller J, Fath T, Maas T, Pool M, Sontag E, Brandt R. Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochem J. 2001;357:759–767. doi: 10.1042/0264-6021:3570759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliezer D, Barre P, Kobaslija M, Chan D, Li X, Heend L. Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry. 2005;44:1026–1036. doi: 10.1021/bi048953n. [DOI] [PubMed] [Google Scholar]
- Engler TA, Malhotra S, Burkholder TP, Henry JR, Mendel D, Porter WJ, Furness K, Diefenbacher C, Marquart A, Reel JK, Li Y, Clayton J, Cunningham B, McLean J, O’Toole JC, Brozinick J, Hawkins E, Misener E, Briere D, Brier RA, Wagner JR, Campbell RM, Anderson BD, Vaughn R, Bennett DB, Meier TI, Cook JA. The development of potent and selective bisarylmaleimide GSK3 inhibitors. Bioorg Med Chem Lett. 2005;15:899–903. doi: 10.1016/j.bmcl.2004.12.063. [DOI] [PubMed] [Google Scholar]
- Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhanel M, Spruss T, Bernhardt G, Graeff C, Farber L, Gschaidmeier H, Buschauer A, Fricker G. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. The Journal of clinical investigation. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17:676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan R, Brambilla E, Sanvito F, Roccatagliata L, Olivieri S, Bergami A, Pluchino S, Uccelli A, Comi G, Martino G. Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain. 2003;126:285–291. doi: 10.1093/brain/awg031. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI. Modeling tau polymerization in vitro: a review and synthesis. Biochemistry. 2003a;42:15009–15017. doi: 10.1021/bi035722s. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI. Tau polymerization: role of the amino terminus. Biochemistry. 2003b;42:2252–2257. doi: 10.1021/bi0272510. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI. In vitro polymerization of tau protein monitored by laser light scattering: Method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of Tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiology of aging. 2011;32:1–14. doi: 10.1016/j.neurobiolaging.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Goedert M. Tau gene mutations and their effects. Movement disorders: official journal of the Movement Disorder Society. 2005;20(Suppl 12):S45–52. doi: 10.1002/mds.20539. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Hasegawa M, Smith MJ, Spillantini MG. Intraneuronal filamentous tau protein and alpha-synuclein deposits in neurodegenerative diseases. Biochem Soc Trans. 1998;26:463–471. doi: 10.1042/bst0260463. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:110–121. doi: 10.1016/s0925-4439(00)00037-5. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Gray EG, Paula-Barbosa M, Roher A. Alzheimer’s disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol. 1987;13:91–110. doi: 10.1111/j.1365-2990.1987.tb00174.x. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafez HN, El-Gazzar AB. Design and synthesis of 3-pyrazolyl-thiophene, thieno[2,3-d]pyrimidines as new bioactive and pharmacological activities. Bioorg Med Chem Lett. 2008;18:5222–5227. doi: 10.1016/j.bmcl.2008.08.071. [DOI] [PubMed] [Google Scholar]
- Hagestedt T, Lichtenberg B, Wille H, Mandelkow EM, Mandelkow E. Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. The Journal of cell biology. 1989;109:1643–1651. doi: 10.1083/jcb.109.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, Frolich L, Schroder J, Schonknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Moller HJ, Kurz A, Basun H. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–931. [PubMed] [Google Scholar]
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau [published erratum appears in Nat Neurosci 1998 Dec;1(8):743] Nature neuroscience. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Hattori M, Sugino E, Minoura K, In Y, Sumida M, Taniguchi T, Tomoo K, Ishida T. Different inhibitory response of cyanidin and methylene blue for filament formation of tau microtubule-binding domain. Biochemical and biophysical research communications. 2008;374:158–163. doi: 10.1016/j.bbrc.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Helal CJ, Sanner MA, Cooper CB, Gant T, Adam M, Lucas JC, Kang Z, Kupchinsky S, Ahlijanian MK, Tate B, Menniti FS, Kelly K, Peterson M. Discovery and SAR of 2-aminothiazole inhibitors of cyclin-dependent kinase 5/p25 as a potential treatment for Alzheimer’s disease. Bioorg Med Chem Lett. 2004;14:5521–5525. doi: 10.1016/j.bmcl.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, Herzog LL, Weintraub S, Mesulam MM, LaPointe NE, Gamblin TC, Berry RW, Binder LI, de Silva R, Lees A, Espinoza M, Davies P, Grover A, Sahara N, Ishizawa T, Dickson D, Yen SH, Hutton M, Bigio EH. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta neuropathologica. 2003;106:323–336. doi: 10.1007/s00401-003-0734-x. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Honson NS, Jensen JR, Darby MV, Kuret J. Potent inhibition of tau fibrillization with a multivalent ligand. Biochemical and biophysical research communications. 2007;363:229–234. doi: 10.1016/j.bbrc.2007.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubinger G, Geis S, LeCorre S, Muhlbacher S, Gordon S, Fracasso RP, Hoffman F, Ferrand S, Klafki HW, Roder HM. Inhibition of PHF-like tau hyperphosphorylation in SH-SY5Y cells and rat brain slices by K252a. J Alzheimers Dis. 2008;13:281–294. doi: 10.3233/jad-2008-13306. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. Journal of neuroscience research. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Morfini G, Pigino G, Lapointe NE, Andreadis A, Song Y, Leitman E, Binder LI, Brady ST. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiology of aging. 2011a doi: 10.1016/j.neurobiolaging.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, Lapointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011b;31:9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H, Schneider R, Sutherland C, Schweiger S. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A. 2010;107:21830–21835. doi: 10.1073/pnas.0912793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic Studies of Antibody-Mediated Clearance of Tau Aggregates Using an ex vivo Brain Slice Model. Front Psychiatry. 2011;2:59. doi: 10.3389/fpsyt.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagoja I, Pannecouque C, Griffioen G, Wera S, Rojasdelaparra VM, Van Aerschot A. Substituted 2-aminothiazoles are exceptional inhibitors of neuronal degeneration in tau-driven models of Alzheimer’s disease. Eur J Pharm Sci. 2011;43:386–392. doi: 10.1016/j.ejps.2011.05.014. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. Journal of neuroscience research. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012 doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisenbrand G, Meijer L. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Annual Review of Neuroscience. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Leite AC, de Lima RS, Moreira DR, Cardoso MV, Gouveia de Brito AC, Farias Dos Santos LM, Hernandes MZ, Kiperstok AC, Soares MB. Synthesis, docking, and in vitro activity of thiosemicarbazones, aminoacyl-thiosemicarbazides and acyl-thiazolidones against Trypanosoma cruzi. Bioorganic & medicinal chemistry. 2006;14:3749–3757. doi: 10.1016/j.bmc.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM, Sausville EA, Kunick C, Meijer L. Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur J Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–269. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. The Journal of clinical investigation. 2008;118:1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwall G, Cole RD. The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J Biol Chem. 1984;259:12241–12245. [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- Long BH, Fairchild CR. Paclitaxel inhibits progression of mitotic cells to G1 phase by interference with spindle formation without affecting other microtubule functions during anaphase and telephase. Cancer research. 1994;54:4355–4361. [PubMed] [Google Scholar]
- Loomis PA, Howard TH, Castleberry RP, Binder LI. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci U S A. 1990;87:8422–8426. doi: 10.1073/pnas.87.21.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Luo MH, Tse SW, Memmott J, Andreadis A. Novel isoforms of tau that lack the microtubule-binding domain. Journal of neurochemistry. 2004;90:340–351. doi: 10.1111/j.1471-4159.2004.02508.x. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A. Granular tau oligomers as intermediates of tau filaments. Biochemistry. 2007;46:3856–3861. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neuroscience Research. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Minoura K, Mizushima F, Tokimasa M, Hiraoka S, Tomoo K, Sumida M, Taniguchi T, Ishida T. Structural evaluation of conformational transition state responsible for self-assembly of tau microtubule-binding domain. Biochemical & Biophysical Research Communications. 2005;327:1100–1104. doi: 10.1016/j.bbrc.2004.12.129. [DOI] [PubMed] [Google Scholar]
- Minoura K, Yao TM, Tomoo K, Sumida M, Sasaki M, Taniguchi T, Ishida T. Different associational and conformational behaviors between the second and third repeat fragments in the tau microtubule-binding domain. European Journal of Biochemistry. 2004;271:545–552. doi: 10.1046/j.1432-1033.2003.03956.x. [DOI] [PubMed] [Google Scholar]
- Morgan D. The role of microglia in antibody-mediated clearance of amyloid-beta from the brain. CNS Neurol Disord Drug Targets. 2009;8:7–15. doi: 10.2174/187152709787601821. [DOI] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. Journal of Neuropathology & Experimental Neurology. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Mukrasch MDBJ, von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M. Sites of tau important for aggregation populate beta structure and bind to microtubules and polyanions. J Biol Chem. 2005;280:24978–24986. doi: 10.1074/jbc.M501565200. [DOI] [PubMed] [Google Scholar]
- Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999;58:1207–1226. doi: 10.1097/00005072-199912000-00002. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell. 2012;149:232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necula M, Chirita CN, Kuret J. Cyanine dye N744 inhibits tau fibrillization by blocking filament extension: implications for the treatment of tauopathic neurodegenerative diseases. Biochemistry. 2005;44:10227–10237. doi: 10.1021/bi050387o. [DOI] [PubMed] [Google Scholar]
- Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986;387:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. Journal of neuroscience research. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Petroianu GA. Methylene blue and Alzheimer’s disease. Biochemical pharmacology. 2009;78:927–932. doi: 10.1016/j.bcp.2009.04.034. [DOI] [PubMed] [Google Scholar]
- Panda D, Goode BL, Feinstein SC, Wilson L. Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry. 1995;34:11117–11127. doi: 10.1021/bi00035a017. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil Cytoskeleton. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- Park S-Y, Ferreira A. The Generation of a 17 kDa Neurotoxic Fragment: An Alternative Mechanism by which Tau Mediates {beta}-Amyloid-Induced Neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011a;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Ward SM, Combs B, Voss K, Kanaan NM, Morfini G, Brady ST, Gamblin TC, Binder LI. Heat Shock Protein 70 Prevents both Tau Aggregation and the Inhibitory Effects of Preexisting Tau Aggregates on Fast Axonal Transport. Biochemistry. 2011b;50:10300–10310. doi: 10.1021/bi2009147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007;46:10016–10023. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- Preziosi P. Isoniazid: metabolic aspects and toxicological correlates. Current drug metabolism. 2007;8:839–851. doi: 10.2174/138920007782798216. [DOI] [PubMed] [Google Scholar]
- Raciti G, Mazzone P, Raudino A, Mazzone G, Cambria A. Inhibition of rat liver mitochondrial monoamine oxidase by hydrazine-thiazole derivatives: structure-activity relationships. Bioorganic & medicinal chemistry. 1995;3:1485–1491. doi: 10.1016/0968-0896(95)00137-6. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006;63:1459–1467. doi: 10.1001/archneur.63.10.1459. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Pant HC. Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1: implications for therapeutics in neurodegeneration. J Alzheimers Dis. 2010;19:389–403. doi: 10.3233/JAD-2010-1243. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. The European journal of neuroscience. 2007;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nature reviews Neuroscience. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- Sibille N, Huvent I, Fauquant C, Verdegem D, Amniai L, Leroy A, Wieruszeski JM, Lippens G, Landrieu I. Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins. 2011 doi: 10.1002/prot.23210. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. The American journal of pathology. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buee L, Galas MC. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566–4575. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D’Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokimasa M, Minoura K, Hiraoka S, Tomoo K, Sumida M, Taniguchi T, Ishida T. Importance of local structures of second and third repeat fragments of microtubule-binding domain for tau filament formation. FEBS Letters. 2005;579:3481–3486. doi: 10.1016/j.febslet.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Molecular biology of the cell. 1995;6:1887–1902. doi: 10.1091/mbc.6.12.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troquier L, Caillierez M, Burnouf S, Fernandez-Gomez FJ, Grosjean MJ, Zommer N, Sergeant N, Schraen-Maschke S, Blum D, Buee L. Targeting phospho-Ser422 by active Tau immunotherapy in the THY-Tau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res. 2012 doi: 10.2174/156720512800492503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Tseng HC, Zhou Y, Shen Y, Tsai LH. A survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett. 2002;523:58–62. doi: 10.1016/s0014-5793(02)02934-4. [DOI] [PubMed] [Google Scholar]
- Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder LI. Progression of Tau Pathology in Cholinergic Basal Forebrain Neurons in Mild Cognitive Impairment and Alzheimer’s Disease. The American journal of pathology. 2011 doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming beta structure. Proc Natl Acad Sci U S A. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Loomis PA, Zinkowski RP, Binder LI. A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. The Journal of cell biology. 1993;121:257–267. doi: 10.1083/jcb.121.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988a;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988b;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) [published erratum appears in Proc Natl Acad Sci U S A 1986 Dec;83(24):9773] Proc Natl Acad Sci U S A. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Molecular biology of the cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB, 3rd, Lee VM, Brunden KR. The microtubule-stabilizing agent, epothilone d, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:3601–3611. doi: 10.1523/JNEUROSCI.4922-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Li BS, Amin ND, Albers W, Pant HC. A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur J Biochem. 2002;269:4427–4434. doi: 10.1046/j.1432-1033.2002.03133.x. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- Zlokovic B. Can blood-brain barrier play a role in the development of cerebral amyloidosis and Alzheimer’s disease pathology. Neurobiol Dis. 1997;4:23–26. doi: 10.1006/nbdi.1997.0134. [DOI] [PubMed] [Google Scholar]